

Abstract
Oxidative stress acutely increases the permeability of the vascular endothelium to large molecules that would not otherwise cross the barrier. Ascorbic acid is an antioxidant that tightens the endothelial permeability barrier, so we tested whether it might also prevent the increase in endothelial permeability due to cellular oxidative stress. Treatment of EA.hy926 endothelial cells cultured on filter inserts with H2O2, menadione, and buthionine sulfoximine increased endothelial permeability to radiolabeled inulin. Short-term ascorbate loading of the cells to what are likely physiologic concentrations of the vitamin by treating them with dehydroascorbate prevented the increase in endothelial permeability due to these agents. The non-physiologic antioxidants dithiothreitol and Tempol also prevented increases in endothelial barrier permeability induced by the agents. These results suggest that oxidative stress induced directly by oxidants or indirectly by GSH depletion impairs endothelial barrier function and that intracellular ascorbate may serve to prevent this effect.
Keywords: paracellular transport, H2O2, menadione, buthionine sulfoximine, oxidative stress, endothelial barrier permeability
1. Introduction
Dysfunction of the vascular endothelium is an early feature of diseases associated with oxidative stress, including atherosclerosis [1,2], smoking [3], and diabetes [4,5]. A marker of such dysfunction is increased vascular permeability to serum proteins, leading to tissue edema [6]. Increased permeability to large molecules due to oxidative stress has also been documented at the cellular level. For example, treatment of endothelial cells in culture on semi-permeable filter supports with the oxidants H2O2 [7–9] and menadione [10] increases endothelial permeability by causing a cytoskeleton-dependent contraction of the endothelial cells and opening of gaps between adjacent cells, which then allow large molecules such as albumin to diffuse across the endothelial barrier [11]. At least for H2O2, the increase in endothelial permeability is preceded by release of calcium from intracellular stores [7,8].
Whereas oxidative stress increases endothelial permeability, the mechanisms by which cells might defend themselves against such stress are less clear. Clearly, the enzymes catalase (for H2O2) and superoxide dismutase (for superoxide generated due to menadione-induced redox cycling) should play a role [12]. Less certain is whether physiologically relevant small molecule antioxidants such as GSH and ascorbate are protective. Increases in endothelial barrier permeability due to menadione were associated with decreases in cellular GSH, leading to the conclusion that GSH was involved in barrier failure [10]. We recently showed that intracellular ascorbate fairly rapidly (over 15–60 min) decreased the permeability endothelial cells cultured on porous membrane supports [13]. Therefore, in the present work we tested whether intracellular ascorbate can ameliorate increases in endothelial permeability induced H2O2 and menadione, as well as by GSH depletion with buthionine sulfoximine. The results showed that what are likely to be physiologic endothelial cell ascorbate concentrations prevented the increase in barrier permeability induced by each of the agents.
2. Materials and Methods
2.1. Materials
Reagent chemicals, including ascorbate, L-buthionine-S,R-sulfoximine (BSO), dehydroascorbic acid (DHA), N-2-hydroxyethylpiperazine N′-2-ethanesulfonic acid (Hepes), and menadione were supplied by Sigma-Aldrich Chemical Co. (St. Louis, MO). Menadione was initially dissolved in a small amount of dimethylsulfoxide, and then diluted with culture medium such that the maximal dimethylsulfoxide concentration was 0.8%. Perkin-Elmer Life and Analytical Sciences, Inc. (Boston, MA) supplied the [carboxyl-14C]inulin (molecular weight range 5000–5500, 2 mCi/g, ~10 Ci/mol).
2.2. Cell Culture
EA.hy926 cells were initially provided by Dr. Dr. Cora Edgell (University of North Carolina, Chapel Hill, NC, USA). They were cultured to confluence at 37 °C in humidified air containing 5% CO2 in Dulbecco’s minimal essential medium and 10% (v/v) heat-inactivated fetal bovine serum, 25 mM D-glucose, and HAT media supplement (Sigma-Aldrich Chemical Co., St. Louis, MO).
2.3. Assay of trans-endothelial ascorbate and inulin transfer
EA.hy926 cells were cultured either on standard 6-well culture plates or on polyethylene terephthalate cell culture inserts (6-wells, 0.4 micron pores at a density of 2 ± 0.2 × 106 pores per cm2, Falcon BD Biosciences, Franklin Lakes, NJ). Cells were cultured to confluence and thereafter for 6 days with 1.7 ml of medium in the upper well and 2.8 ml of medium in the lower well. Medium was changed every 3 days. After incubations as described, either ascorbate (0.3 mM) or [carboxyl-14C]inulin (10 μM), was added above the cells/filter and incubation was carried out at 37 °C for 1 h, except where noted. For assay of marker transfer, medium from above and below the cells/filter was sampled taken for counting of radioactivity or ascorbate assay as described below.
Assay of [carboxyl-14C]inulin permeability was determined as previously described [14] with minor modifications [13]. The permeability coefficients for [carboxyl-14C]inulin were calculated, including adjustment for the rate of [carboxyl-14C]inulin transfer across filters after removal of cells [15]. This corrects in each well for any changes in permeability due to deposition of the matrix laid down by the cells during culture and the experiment.
2.4. Assay of ascorbate, GSH and lipid peroxidation
For determination of ascorbate transferred across the cells and filter, ascorbate was measured in culture medium as follows. An aliquot of medium (0.1 ml) taken from below the cells was added to 0.1 ml of 25% metaphosphoric acid (w/v), mixed, neutralized with 0.35 ml of the above phosphate/EDTA buffer, and centrifuged to remove any precipitated solids before assay of ascorbate as described below.
For assay of intracellular ascorbate and GSH, after removal of the medium, cells on the 6-well plate or filter were rinsed 3 times with Krebs-Ringer Hepes buffer (KRH) that consisted of 20 mM Hepes, 128 mM NaCl, 5.2 mM KCl, 1 mM NaH2PO4, 1.4 mM MgSO4, and 1.4 mM CaCl2, pH 7.4. After removal of the last rinse, the cell monolayer was treated with 0.1 ml of 25 % (w/v) metaphosphoric acid, detached from the filter with a rubber spatula, and then treated with 0.35 ml of 0.1M Na2HPO4 and 0.05 mM EDTA, pH 8.0. The lysate was removed and centrifuged at 3 °C for 1 min at 13,000 × g and the supernatant was taken for assay of ascorbate. Assay of ascorbic acid was performed in duplicate by high performance liquid chromatography as previously described [16]. Cellular GSH was measured in duplicate by the method of Hissin and Hilf [17] on the same samples as ascorbate. Intracellular concentrations of ascorbate and GSH were calculated based on the intracellular distribution space of 3-O-methylglucose in EA.hy926 cells, which was previously measured to be 3.6 ± 1.2 μl/mg protein [18].
Lipid peroxidation was assessed by measurement of thiobarbituric acid reactive-substances (TBARS) as previously described [19].
2.5. Data Analysis
Results are shown as mean + standard error. Statistical comparisons were made using SigmaStat 2.0 software (Jandel Scientific, San Rafael, CA). Differences between treatments were assessed by two-way analysis of variance with post-hoc testing using Tukey’s test.
3. Results
Endothelial cells cultured to confluence on trans-well filter inserts for 6 days allowed passage of 5500 kDa radiolabeled inulin from above to below the cell layer and filter, expressed here as a permeability (Fig. 1). Treatment of cells with H2O2, menadione, and BSO was carried out in separate experiments for each agent, which is shown as different sets of bars in the figure. Each of the agents increased endothelial barrier permeability (Fig. 1, compare first and second sets of bars). Treatment of the cells for 30 min with either 0.5 mM H2O2 or 50 μM menadione increased transfer of radiolabeled inulin across the endothelial barrier by 64% and 60%, respectively. An 18 h treatment with 1 mM BSO also increased endothelial permeability to inulin by 56%. The effect of BSO required >3 h to become significant and was complete after 18 h of treatment (results not shown). To ascertain whether the observed increases in permeability due to each of the agents was due to changes in the cytoskeleton, cells incubated as described for each agent in Fig. 1 were treated for 30 min with 10 μM colchicine just before the transfer assay. The effect of each agent to increase endothelial permeability was abolished by colchicine (results not shown), indicating that their effects were related to changes in the cytoskeleton.
Figure 1. Prevention of oxidative stress-induced increases in endothelial permeability by ascorbate.
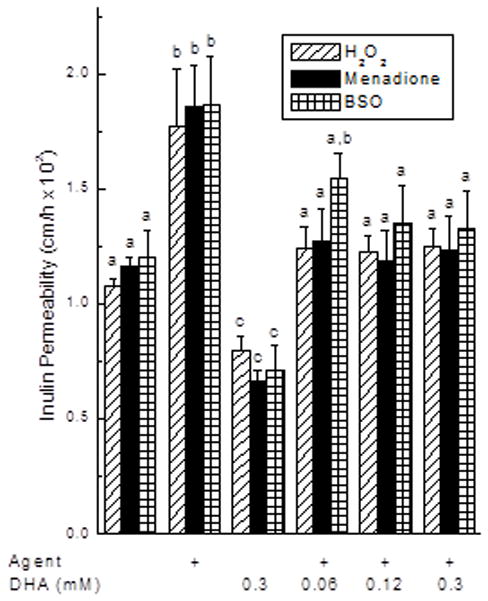
EA.hy926 cells in culture were incubated for 30 min with 1 mM H2O2 or 50 μM menadione, or were cultured 18 h with 0.5 mM BSO. Where noted, DHA was then added at the concentration noted for an additional 15 min without changing the culture medium before addition of 10 μM [carboxyl-14C]inulin and the transfer assay as described under Materials and Methods. Results are shown for separate experiments with H2O2 (N = 6), menadione (N = 4) and BSO (N = 5). Bars with different letters indicate p < 0.05 compared to other treatments in the same experiment.
To assess the effect of intracellular ascorbate on oxidant-induced endothelial permeability, cells were loaded with ascorbate by incubation for 15 min with DHA. DHA is taken up by the cells on glucose transporters and rapidly reduced to ascorbate [20]. The advantage of this approach, rather than incubation with ascorbate, is that extracellular ascorbate concentrations are undetectable, thus avoiding extracellular reactions with the oxidants or components of the medium. DHA loading over 15 min decreased barrier permeability in the 1 h transfer assay by about 40% compared to controls for each of the 3 treatments (Fig. 1, compare first and third sets of bars). Loading with 120 μM or higher DHA for an additional 15 min prevented the increase in endothelial permeability induced by each of the agents back to control levels (Fig. 1, last 3 sets of bars).
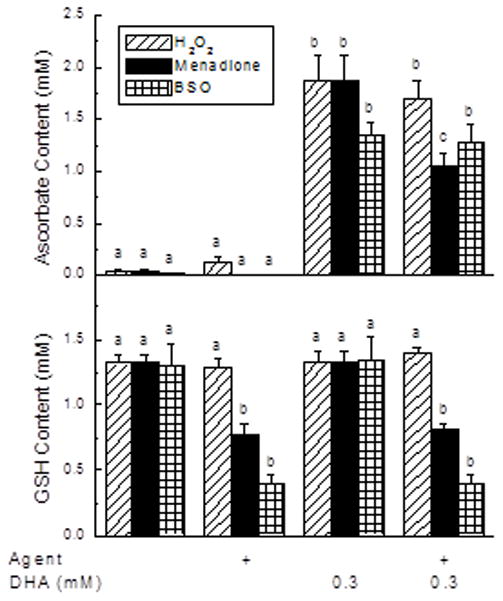
To assess the oxidative stress induced by each agent, their effects on cellular ascorbate and GSH concentrations were measured and are shown in Fig. 2. In the absence of loading with DHA, no ascorbate was detected in the cells (Fig. 2A, first 2 sets of bars). When cells were loaded with 0.3 mM DHA under conditions of the transfer assay (a total of 115 min of exposure), intracellular ascorbate increased to 1.4–1.8 mM (Fig. 2A, third set of bars). These levels were similar to those observed when cells were treated with 50 μM ascorbate for 18 h (1.4 ± 0.2 mM, N = 7). Although H2O2 did not affect intracellular ascorbate, menadione significantly decreased it by about 40%. An 18 h pretreatment with BSO did not affect the ability of the cells to take up and reduce DHA to ascorbate. Intracellular GSH concentrations were similar to ascorbate concentrations achieved by DHA loading and not affected by DHA uptake and reduction to ascorbate (Fig. 2B, compare first and third sets of bars). Treatment of the cells with H2O2 was without effect on the cell content of GSH, but GSH was decreased about 50% by menadione. As expected, an 18 h treatment with BSO (an inhibitor of glutathione synthesis) decreased cellular GSH to one third of control, and this was also not affected by DHA treatment. Lipid peroxidation was assessed at TBARS generation under the agent concentrations and times used in Figs. 1 and 2. However, none of the agents significantly increased TBARS generation (results not shown).
Figure 2. Ascorbate and GSH concentrations following agent treatments.
EA.hy926 cells in culture on 6-well plates were incubated with 0.5 mM H2O2 or 50 μM menadione for 30 min or cultured for 18 h with 0.5 mM BSO. Where indicated, DHA was added to 0.3 mM, followed in 15 min by 3 rinses in KRH and removal of the cells for assay of ascorbate (Panel A) or GSH (Panel B). Results are shown from 6 experiments, with bars having different letters indicating p < 0.05 compared to other samples treated with the same stressor agent.
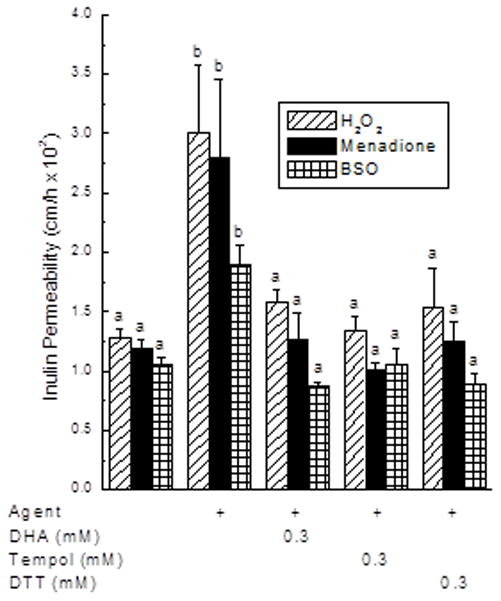
To determine if the ability of intracellular ascorbate to prevent the increase in endothelial permeability due to the oxidants relates to a decrease in oxidative stress, the effects of two non-physiologic antioxidants with different mechanisms of action were tested. As shown in Fig. 3, the increase in endothelial permeability due to H2O2, menadione, and BSO was prevented by treatments with DHA, with the free radical scavenger Tempol, and with the cell-penetrant dithiothreitol.
Figure 3. Prevention of oxidant-induced increases in endothelial barrier permeability by antioxidants.
EA.hy926 cells in culture were incubated for 30 min with 1 mM H2O2 or 50 μM menadione, or were cultured 18 h with 0.5 mM BS0. The different agents were then added to concentrations of 0.3 mM as noted, followed in 15 min by the [carboxyl-14C]inulin transfer assay. Different letters above the bars indicate p < 0.05 compared to other treatments for each stressor.
To examine the effect of GSH depletion on endothelial barrier function more closely, the concentration dependence of BSO to deplete GSH was compared with its induction of increased barrier permeability. As shown in Fig. 4, GSH depletion was quite sensitive to low BSO concentrations, whereas increases in endothelial barrier permeability were approximately linear and became significant only at BSO concentrations above 100 μM.
Figure 4. Concentration dependence of BSO to decrease cell GSH and increase endothelial permeability.
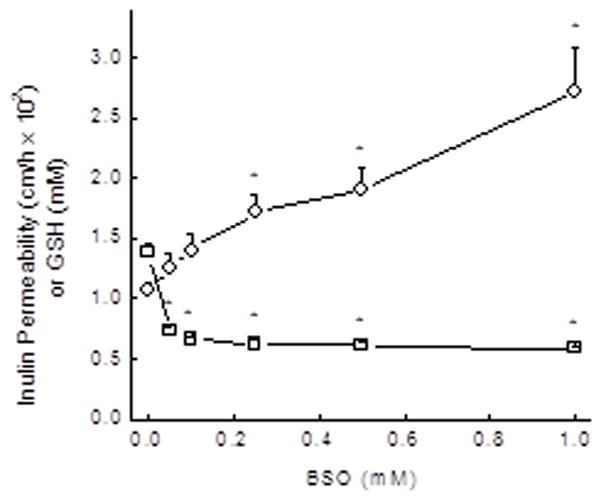
EA.hy926 cells were cultured on filters for 18 h with the indicated concentrations of BSO, followed by assay of GSH (circles) and the inulin transfer assay (squares). Results are shown from 7 experiments, with an “*” indicating p < 0.05 compared to the sample not treated with BS
4. Discussion
The presence of ascorbate in endothelial cells prevented the increase in endothelial barrier permeability induced by the oxidants H2O2 and menadione, as well as that caused by GSH depletion with BSO. The oxidative stress induced by these agents was not severe enough to increase lipid peroxidation as measured by TBARS. Several different types of low molecular weight antioxidants also prevented the increase in permeability in this and in previous studies [10,21]. Together, these results implicate oxidative stress in the cells as the causative factor. Since ascorbate is a natural cellular antioxidant, the present results suggest that it could have this function in vivo. The intracellular concentration of ascorbate in endothelial cells in vivo has not been determined. However, based on our finding that EA.hy926 endothelial cells took up and retained ascorbate to low millimolar intracellular concentrations when cultured overnight with a physiologic blood ascorbate concentration (50 μM), it is likely that the concentrations achieved in this study with DHA loading mirror those in vivo. To the extent that they do, then ascorbate could delay or even prevent endothelial barrier function induced by oxidative stress. Intracellular ascorbate also tightened the endothelial barrier, even in the absence of added oxidative stress. Regarding the latter, endothelial cells under standard culture conditions are known to be under some oxidative stress and that is ameliorated by addition of ascorbate [22]. Prevention of this oxidative stress may well have contributed to the ability of ascorbate to tighten basal endothelial barrier permeability.
Endothelial barrier function was also responsive to changes in GSH. For example, although the increase in endothelial permeability caused by menadione was associated with a 50% decrease in intracellular GSH, this was not observed with H2O2, despite a similar increase in endothelial barrier permeability in this and in an earlier study [10]. Second, the gradual increase in endothelial permeability due to GSH depletion with BSO did not reflect the abrupt decrease in intracellular GSH to about 50% of basal after 18 h of culture with BSO concentrations as low as 50 μM. A previous study showed that BSO concentrations of 1 mM or greater over 48 h of culture were required to significantly increase endothelial cell barrier permeability [21]. Given the high specificity of BSO for depleting GSH by inhibiting its synthesis [23], these results suggest that endothelial barrier permeability does not reflect intracellular GSH concentrations.
These results show that intracellular ascorbate prevents the increase in endothelial barrier permeability induced by oxidants and by GSH depletion with BSO. BSO depletion of GSH by more than 50% was required for a significant increase, however. Protection against oxidative stress-induced increases in endothelial barrier permeability by what are likely physiologic ascorbate concentrations in endothelial cells suggests that ascorbate may have this function in vivo. A corollary of this conclusion is that to mimic the physiologic situation, endothelial cells used in barrier function studies should contain ascorbate.
Acknowledgments
This work was supported by NIH grant DK050435 and by the Cell Culture Core of the Vanderbilt Diabetes Research and Training Center (DK020593).
Abbreviations
- BSO
L-buthionine-S,R-sulfoximine
- DHA
dehydroascorbic acid
- Hepes
N-2-hydroxyethylpiperazine-NN-2-ethanesulfonic acid
- KRH
Krebs-Ringer Hepes
References
- 1.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 3.Cross CE, Van der Vliet A, Eiserich JP. Cigarette smokers and oxidant stress: a continuing mystery. Am J Clin Nutr. 1998;67:184–185. doi: 10.1093/ajcn/67.2.184. [DOI] [PubMed] [Google Scholar]
- 4.Baynes JW. Role of oxidative stress in development of complications of diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- 5.Ceriello A. Oxidative stress and glycemic regulation. Metabolism. 2000;49:27–29. doi: 10.1016/s0026-0495(00)80082-7. [DOI] [PubMed] [Google Scholar]
- 6.Plante GE, Alfred J, Chakir M. The blood vessel, linchpin of diabetic lesions. Metabolism. 1999;48:406–409. doi: 10.1016/s0026-0495(99)90094-x. [DOI] [PubMed] [Google Scholar]
- 7.Holman RG, Maier RV. Oxidant-induced endothelial leak correlates with decreased cellular energy levels. Am Rev Respir Dis. 1990;141:134–140. doi: 10.1164/ajrccm/141.1.134. [DOI] [PubMed] [Google Scholar]
- 8.Siflinger-Birnboim A, Lum H, del Vecchio PJ, Malik AB. Involvement of Ca2+ in the H2O2-induced increase in endothelial permeability. Am J Physiol. 1996;270:L973–L978. doi: 10.1152/ajplung.1996.270.6.L973. [DOI] [PubMed] [Google Scholar]
- 9.Lee HS, Namkoong K, Kim DH, Kim KJ, Cheong YH, Kim SS, Lee WB, Kim KY. Hydrogen peroxide-induced alterations of tight junction proteins in bovine brain microvascular endothelial cells. Microvasc Res. 2004;68:231–238. doi: 10.1016/j.mvr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 10.McAmis WC, Schaeffer RC, Jr, Baynes JW, Wolf MB. Menadione causes endothelial barrier failure by a direct effect on intracellular thiols, independent of reactive oxidant production. Biochim Biophys Acta Mol Cell Res. 2003;1641:43–53. doi: 10.1016/s0167-4889(03)00063-6. [DOI] [PubMed] [Google Scholar]
- 11.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 12.Shasby DM, Lind SE, Shasby SS, Goldsmith JC, Hunninghake GW. Reversible oxidant-induced increases in albumin transfer across cultured endothelium: alterations in cell shape and calcium homeostasis. Blood. 1985;65:605–614. [PubMed] [Google Scholar]
- 13.May JM, Qu ZC, Qiao H. Transfer of ascorbic acid across the vascular endothelium: mechanism and self-regulation. Am J Physiol Cell Physiol. 2009;297:C169–C178. doi: 10.1152/ajpcell.00674.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siflinger-Birnboim A, del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol. 1987;132:111–117. doi: 10.1002/jcp.1041320115. [DOI] [PubMed] [Google Scholar]
- 15.Utoguchi N, Ikeda K, Saeki K, Oka N, Mizuguchi H, Kubo K, Nakagawa S, Mayumi T. Ascorbic acid stimulates barrier function of cultured endothelial cell monolayer. J Cell Physiol. 1995;163:393–399. doi: 10.1002/jcp.1041630219. [DOI] [PubMed] [Google Scholar]
- 16.May JM, Qu ZC, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 17.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 18.Jones W, Li X, Perriott LM, Whitesell RR, May JM. Uptake, recycling, and antioxidant functions of α-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 19.Harrison FE, Hosseini AH, Dawes SM, Weaver S, May JM. Ascorbic acid attenuates scopolamine-induced spatial learning deficits in the water maze. Behav Brain Res. 2009;205:550–558. doi: 10.1016/j.bbr.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May JM, Asard H. Ascorbate Recycling. In: Asard H, May JM, Smirnoff N, editors. Vitamin C Functions and biochemistry in animals and plants. Bios Scientific Publishers; London: 2004. pp. 139–158. [Google Scholar]
- 21.Hui YY, McAmis WC, Baynes JW, Schaeffer RC, Jr, Wolf MB. Effect of advanced glycation end products on oxidative stress in endothelial cells in culture: a warning on the use of cells studied in serum-free media. Diabetologia. 2001;44:1310–1317. doi: 10.1007/s001250100646. [DOI] [PubMed] [Google Scholar]
- 22.Smith AR, Visioli F, Hagen TM. Vitamin C matters: increased oxidative stress in cultured human aortic endothelial cells without supplemental ascorbic acid. FASEB J. 2002;16:1102–1104. doi: 10.1096/fj.01-0825fje. [DOI] [PubMed] [Google Scholar]
- 23.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]