

Abstract
Vitamin C, or ascorbic acid, is important as an antioxidant and participates in numerous cellular functions. Although it circulates in plasma in micromolar concentrations, it reaches millimolar concentrations in most tissues. These high ascorbate cellular concentrations are thought to be generated and maintained by the SVCT2 (Slc23a2), a specific transporter for ascorbate. The vitamin is also readily recycled from its oxidized forms inside cells. Neurons in the central nervous system (CNS) contain some of the highest ascorbic acid concentrations of mammalian tissues. Intracellular ascorbate serves several functions in the CNS, including antioxidant protection, peptide amidation, myelin formation, synaptic potentiation, and protection against glutamate toxicity. The importance of the SVCT2 for CNS function is supported by the finding that its targeted deletion in mice causes widespread cerebral hemorrhage and death on post-natal day one. Neuronal ascorbate content as maintained by this protein also has relevance for human disease, since ascorbate supplements decrease infarct size in ischemia-reperfusion injury models of stroke, and since ascorbate may protect neurons from the oxidant damage associated with neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s. The aim of this review is to assess the role of the SVCT2 in regulating neuronal ascorbate homeostasis and the extent to which ascorbate affects brain function and antioxidant defenses in the CNS.
Keywords: ascorbate transport, brain, dehydroascorbate, glutamate, neurons, SVCT2
1. Ascorbic acid physiology and recycling
Ascorbic acid is a vitamin because humans, higher primates, and a few other vertebrates such as guinea pigs have lost the ability to synthesize it from glucose. These species lack the enzyme L-gulono-1,4-lactone oxidase (EC 1.1.3.8), which carries out the last step in ascorbate biosynthesis (Chatterjee et al. 1960). Species that cannot synthesize ascorbate must therefore have efficient mechanisms for both absorption of the vitamin and for its recycling. Absorption of vitamin C occurs largely in the distal ileum of humans and guinea pigs, where uptake is mediated by a saturable, sodium- and energy-dependent transporter (Stevenson and Brush 1969, Bianchi et al. 1986) that is located in the apical brush border membrane (Bianchi et al. 1986; Rose 1988). This transporter has been cloned (Tsukaguchi et al. 1999) and is termed SVCT1 (sodium-dependent vitamin C transporter-1, Slc23a1). The SVCT1 helps to generate plasma ascorbate concentrations of 40–60 µM in humans (Table 1), which can double with typical levels of dietary ascorbate supplementation (e.g., 250–1000 mg daily) (Levine et al. 1996). In contrast to plasma ascorbate concentrations, tissue concentrations are typically 1 mM or higher, as exemplified by leukocytes and platelets in human blood (Table 1). High intracellular ascorbate concentrations are achieved by two mechanisms. The most important is probably unidirectional transport of the vitamin into cells on a transporter very closely related to the SVCT1, termed the SVCT2 (Slc23a2) (Tsukaguchi et al. 1999). These transporters share 65% sequence homology, substrate saturability, high affinity for ascorbate (20–60 µM), as well as energy- and sodium-dependence. Intracellular ascorbate tends to exit cells only slowly (May et al. 1998a), probably because of its hydrophilic structure and negative charge at physiologic pH. The second route by which ascorbate can enter cells is by uptake of its two-electron-oxidized form, dehydroascorbic acid (DHA, Figure 1). DHA is taken up on the ubiquitous GLUT-type glucose transporters (Vera et al. 1993). Although DHA, a triketone with a strained lactone ring, has little resemblance to glucose, it forms a hydrated bicyclic hemiketal (Pastore et al. 2001) for which these transporters have affinity (Bigley & Stankova 1974). Since the GLUTs mediate facilitated diffusion, transport of DHA by this mechanism is bi-directional. This also means that any DHA formed within cells by ascorbate oxidation will rapidly efflux and be lost. This is prevented by efficient cellular mechanisms of DHA reduction or recycling to ascorbate.
Table 1.
Ascorbate concentrations in plasma and blood cells from normal humans
| Tissue/Fluid | Ages; # of Subjects | Diet (mg/day) |
Concentration (mM) |
Reference |
|---|---|---|---|---|
| Plasma | >60 years; 226 | < 120 | 0.057 ± 0.003 | (Jacques et al. 1995) |
| Plasma | 73 years; 161 | 257 | 0.072 ± 0.027 | (Taylor et al. 1997) |
| Aqueous humor | 73 years; 161 | 257 | 1.64 ± 0.58 | (Taylor et al. 1997) |
| Lens | 73 years; 161 | 257 | 3.74 ± 1.4 | (Taylor et al. 1997) |
| Neutrophils | 19–27 years; 13 | 100 | 1.17 | (Levine et al. 2001) |
| Monocytes | 17–55 years; 41 | Unknown | 3.8 | (Evans et al. 1982) |
| Platelets | 17–55 years; 41 | Unknown | 1.9 | (Evans et al. 1982) |
| Erythrocytes | 17–55 years; 41 | Unknown | 0.043 | (Evans et al. 1982) |
| Plasma | 17–55 years; 41 | Unknown | 0.045 | (Evans et al. 1982) |
Figure 1.
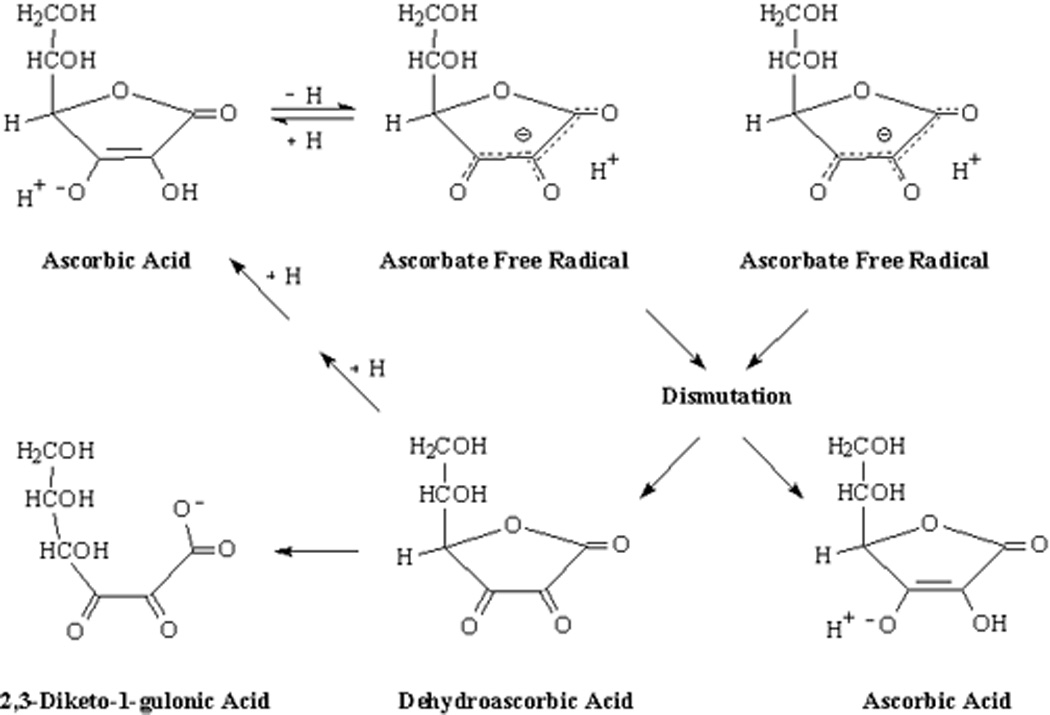
Ascorbate oxidation and recycling.
Ascorbate recycling from both of its oxidized forms occurs largely within cells, although cell-surface reduction of its radical one electron-oxidized form has been described (May et al. 2000, VanDuijn et al. 2000). As shown in Figure 1, ascorbate is oxidized when it donates a hydrogen atom (proton + electron) to enzyme reactions or to scavenge free radicals. The one-electron oxidized form of ascorbate, commonly termed the ascorbate free radical (AFR) or monodehydroascorbate, is surprisingly stable and can be detected at 10 nM concentrations in biological fluids by EPR (Mehlhorn 1991; Coassin et al. 1991; Buettner & Jurkiewicz 1993). lthough the AFR has a lower mid-point reduction potential than ascorbate (Buettner 1993), it is not very reactive. Instead of donation of another hydrogen and electron, the preferred reaction shown in Figure 1 is for two molecules of the AFR to dismutate, forming ascorbate and dehydroascorbate (Bielski et al. 1981). DHA is unstable at physiologic pH, with a half-life of about 6 min (Drake et al. 1942; Winkler 1987) (Tolbert & Ward 1982). Hydrolysis of the lactone ring irreversibly converts DHA to 2,3-diketo-1-gulonic acid (Figures 1) (Chatterjee 1970; Bode et al. 1990). Ascorbate loss by DHA decomposition is obviously wasteful of the vitamin, and DHA reduction to ascorbate is rapid and efficient within cells.
Mechanisms of GSH-dependent DHA reduction include direct chemical reduction (Winkler et al. 1994) and enzyme-dependent reduction with GSH serving as electron donor (Maellaro et al. 1994; Winkler et al. 1994; Washburn & Wells 1999). DHA is also reduced to ascorbate by NADPH-dependent enzymes, including thioredoxin reductase (May et al. 1997) and a liver 3α-hydroxysteroid dehydrogenase (Del Bello et al. 1994). The AFR is also reduced to ascorbate with high affinity by both NADH-dependent reductases (Nazemi & Staudinger 1968; Ito et al. 1981; Villalba et al. 1993) and by the NADPH-dependent enzyme thioredoxin reductase (May et al. 1998a). As reviewed previously (May & Asard 2004), these cellular systems provide redundant and efficient mechanisms for recycling of ascorbate from its oxidized forms.
2. Functions of ascorbate
As noted above, ascorbate provides electrons for crucial enzyme reactions in cells and acts as a primary antioxidant capable of scavenging radicals generated within cells or plasma. The chapter by Dr. deTulio details the non-antioxidant functions of ascorbate, which are crucial in preventing scurvy and for maintaining cell integrity and health. The main antioxidant function of the vitamin involves direct scavenging of radical species before they can damage DNA, proteins, or lipids. Another mechanism by which ascorbate functions as an antioxidant is to recycle other antioxidants. For example, ascorbate can reduce the α-tocopheroxyl radical at the surface of biological membranes, thus contributing to the ability of α-tocopherol to break the chain of lipid peroxidation in lipid bilayers (Buettner 1993). Another example relates to the finding that ascorbate spares tetrahydrobiopterin in cultured endothelial cells, thus allowing continued action of endothelial nitric oxide synthase (Heller et al. 2001; Baker et al. 2001). As with α-tocopherol, the mechanism of this effect also appears to be due to reduction of the tetrahydrobiopterin radical by ascorbate (Patel et al. 2002). Consideration of both non-antioxidant and antioxidant functions of the vitamin shows that it has multiple sites of action in cells. Although it may not be absolutely required for any specific action in a cell, ascorbate is clearly necessary for proper function of many organs. Perhaps most important is its requirement in the brain and central nervous system (CNS), as discussed next.
3. Importance of vitamin C in the CNS
Ascorbic acid is an essential micronutrient in the CNS. Whereas whole brain ascorbate concentrations are one to two mM, intracellular neuronal concentrations have been calculated to be much higher (Rice & Russo-Menna 1998; Rice 2000), as discussed below. Neurons are also especially sensitive to ascorbate deficiency, perhaps because they have 10-fold higher rates of oxidative metabolism than supporting glia (Wilson 1997; Hediger 2002). This heightened neuronal sensitivity is most apparent in states of ascorbate deficiency and in conditions in which there is excess oxidant stress. A neuroprotective role for ascorbate is also suggested by the existence of homeostatic mechanisms that maintain high concentrations of ascorbate in cerebrospinal fluid (CSF) and in neurons. A key feature in this regard is the ability to sustain steep ascorbate concentration gradients: a) from plasma to the CSF across the choroid plexus and b) from the CSF and interstitium into neurons (Figure 2). In both instances, this gradient is generated for the most part by the SVCT2 protein. As discussed next, intracellular ascorbate and thus the SVCT2 protects the CNS in syndromes of oxidant stress.
Figure 2.
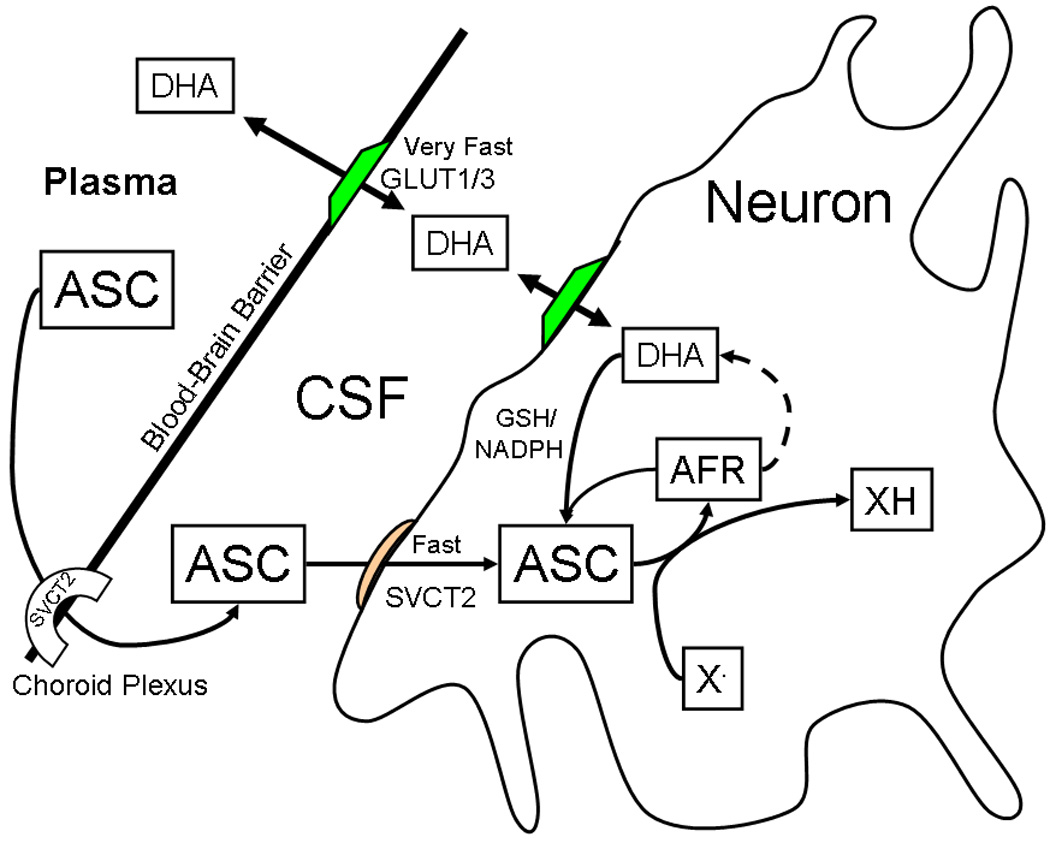
Ascorbate uptake and metabolism in the CNS. ASC, ascorbate; AFR, ascorbate free radical; DHA, dehydroascorbate; X•, oxidizing free radical species. AFR generated by X• dismutates to form ascorbate and dehydroascorbate. Both the AFR and DHA are recycled back to ascorbate by cellular metabolism.
Scurvy causes severe lassitude and asthenia in humans. Although the disease has been associated with paraparesis in humans, death appears to be due more to complications of systemic collagen dysfunction and not to a distinct neurologic syndrome (Hirschmann & Raugi 1999). This likely relates to the fact that ascorbate is avidly retained by the CNS during ascorbate deficiency (Hornig 1975). For example, 14 days after removal of ascorbate from their diet, guinea pig brains still had 24% of their original ascorbate content, compared to only 4% for adrenal gland and 3% for spleen (Hughes et al. 1971). Whereas this could indicate that decreases in CNS ascorbate do not play a major role in generalized scurvy, the argument can also be made that the strong retention of ascorbate in the CNS reflects its importance to neuronal function. This hypothesis is supported by our own studies in guinea pigs with moderate vitamin E deficiency in which an acute deficiency of ascorbate was superimposed (Hill et al. 2003). After two weeks of vitamin E deprivation in male guinea pigs, plasma and brain α-tocopherol concentrations were modestly decreased by 65% and 32%, respectively. These animals gained weight and appeared completely normal without evidence of abnormal gait or ataxia (signs of vitamin E deficiency). However, within fife to six days of removing vitamin C from their diet, the animals typically developed a progressive ascending paralysis and died within 24 hours. No neurologic signs were apparent in animals with single deficiencies of either vitamin C or E alone. Death occurred in the doubly deficient animals despite only 37% and 67% decreases in brain and spinal cord ascorbate concentrations, respectively. Despite over 95% decreases in plasma and liver ascorbate in the animals on the vitamin C-deficient diet, neither the C-deficient nor the doubly deficient animals had signs of scurvy (no skin or hair changes, no hemarthroses). The doubly deficient animals had only small increases in liver and brain F2-isoprostanes (a marker of oxidant stress). Standard hematoxylin and eosin staining of the brains and spinal cords showed no specific abnormalities. However, subsequent studies using Nissl and silver degeneration stains revealed widespread neuronal loss and degeneration in the pons and long tracts of the spinal cord (Burk et al. 2006). Thus, even a modest decrease in CNS ascorbate accelerated signs of vitamin E deficiency in this model, indicative of a crucial role for ascorbate in protecting the brain against oxidant stress.
If the SVCT2 transporter is responsible for maintaining high CSF and neuronal ascorbate concentrations, what happens when the protein is knocked out in the mouse? Targeted deletion of the SVCT2 protein results in homozygotes that die shortly after birth: although they appear to develop normally in utero, they never take a breath and have diffuse cerebral hemorrhage (Sotiriou et al. 2002). Whereas the latter likely reflects increased capillary fragility due to defects in basement membrane type IV collagen (Gould et al. 2005), the animals do not show hemorrhage elsewhere and lack signs of generalized scurvy. Further, hydroxyproline levels in skin are normal (Sotiriou et al. 2002). The cause of death is unknown. Catecholamine synthesis is decreased, but this is apparently not the cause of death (Bornstein et al. 2003). Although the lungs fail to expand, their architecture is normal, as are surfactant levels (Sotiriou et al. 2002). Brain ascorbate concentrations are decreased 30–50% in the heterozygotes, and are very low to undetectable in the homozygous mice (Sotiriou et al. 2002). It seems likely that diffuse cortical capillary hemorrhage along with neuronal dysfunction contributed significantly to the death of these mice, although further studies are needed to assess the role of the lung and metabolic defects.
Perhaps the most dramatic acute oxidant stress in the CNS is the ischemia-reperfusion injury that occurs with stroke. It is generally accepted that ischemia depletes intracellular GSH and ascorbate in brain, and that subsequent reperfusion generates reactive oxygen species (ROS) that further deplete these two antioxidants and extend tissue damage to areas with decreased oxidant defenses (Rice et al. 1995). Ascorbate supplements decrease infarct size in animal studies. For example, in monkeys given one g/day of ascorbate parenterally for six days before middle cerebral artery occlusion, brain ascorbate was increased by 50%. This indicates that ascorbate had access to the CNS. Further, infarct size was decreased by 50% in the ascorbate-treated group compared to the control group (Ranjan et al. 1993). Similarly, in rats with middle cerebral artery occlusion, high dose dehydroascorbate given by intraperitoneal injection just before, 15 min after, or three h after occlusion markedly decreased infarct volume, mortality, and neurological deficits in mice (Huang et al. 2001). In contrast, ascorbate itself showed no protective effect in these studies. As shown in Figure 2, dehydroascorbate is rapidly transported across the blood-brain barrier by glucose transporters (Agus et al. 1997; Huang et al. 2001; Mack et al. 2006), taken up by glucose transporters in neurons, and reduced to ascorbate (May & Asard 2004). The inability of acute treatment with ascorbate to prevent neuronal damage in the rat model is supported by other evidence that ascorbate does not cross the blood-brain barrier in its fully reduced form (Agus et al. 1997; Hosoya et al. 2004). This appears to be due to lack of SVCT2 in endothelial cells of the blood-brain barrier, as shown by in situ hybridization (Berger et al. 2003; García et al. 2005) and more recently by immunostaining for the SVCT2 protein (Mun et al. 2006).
The ability of ascorbate to decrease infarct size following ischemia-reperfusion likely relates to scavenging of ROS, both within and around cells. Ascorbate at the concentrations present in CSF and neurons in vivo (i.e., >100 µM) will effectively scavenge superoxide (Jackson et al. 1998) and spare or recycle α-tocopherol in the lipid bilayer (Niki et al. 1995; May et al. 1998b). For example, an increase in intracellular (but not extracellular) ascorbate content of (rat) brain slices decreased the swelling induced by oxidant stress (Brahma et al. 2000). We reported that ascorbate prevented loss of α-tocopherol and lipid peroxidation induced by culture in oxygenated medium (Li et al. 2003). Finally, in our recent studies, primary cultures of hippocampal neurons prepared from homozygous SVCT2 knockout mice showed increased susceptibility to cell death induced by N-methyl-D-aspartate (NMDA) and H2O2 compared to cultures from wild-type mice (Qiu et al. 2006). The improved survival of neurons containing the SVCT2 was likely due to small amounts of endogenous ascorbate (0.1–0.2 mM) retained by the cells after 2 weeks in culture, since the culture medium did not contain ascorbate.
Regarding extracellular ascorbate, it has long been known that ischemia releases large amounts of ascorbate from brain cells (Hillered et al. 1988). Since this release was associated with glutamate uptake by neurons and glia (Miele et al. 1994, Rice 2000), these two processes were thought to be linked in mechanism involving heteroexchange across one or more transporters (Grünewald 1993; Rebec and Pierce 1994; Yusa 2001). Removal of extracellular glutamate by such heteroexchange would decrease excitotoxicity caused by activation of cell surface and synaptic glutamate receptors (Grünewald 1993; Rebec & Pierce 1994; Yusa 2001). In addition, intracellular ascorbate would be expected to scavenge ROS generated during to glutamate-induced excitotoxicity. In this regard, ascorbate added to the culture medium was found to prevent glutamate-induced cell damage and death in cultured cerebellar granule cells (Ciani et al. 1996; Atlante et al. 1997). Ascorbate also has been shown to protect neurons from excitotoxicity induced by activation of the NMDA receptor (MacGregor et al. 1996; Majewska et al. 1990; Majewska & Bell 1990). Whereas intracellular ascorbate protects cells against glutamate toxicity, recent evidence suggests that it does not do so via a heteroexchange mechanism. Wilson and colleagues confirmed that glutamate increases ascorbate efflux from cerebral astrocytes, but found no effect of intracellular ascorbate on glutamate uptake (Wilson et al. 2000). The release of ascorbate was attributed to glutamate-induced cell swelling followed by opening of organic ion channels through which ascorbate can pass (Wilson et al. 2000). We found the analogous results in SH-SY5Y neuroblastoma cells: glutamate caused release of intracellular ascorbate, but neither intra- nor extracellular ascorbate affected glutamate uptake (May et al. 2006). Together, the results of these studies strongly support a protective role for ascorbate and possibly for the SVCT2 in brain during acute ROS generation.
4. Ascorbate function in the CNS
Whereas ascorbate is well known to be involved in neuronal biochemistry (e.g., peptide amidation, myelination, and catecholamine synthesis), evidence for a direct role in neuronal maturation and function has only recently been forthcoming. Culture of embryonic cortical precursor cells in medium containing ascorbate was first shown to induce their maturation into neurons and astrocytes by Lee and colleagues (Lee et al. 2003). We confirmed and extended these findings in primary cultures of hippocampal neurons (Qiu et al. 2006). Key to these experiments was the use of cells prepared from late-stage embryos (gestational day 18–19) that either expressed or completely lacked the SVCT2. After 10–14 days in culture in serum-free medium in the absence of ascorbate, no ascorbate was present in cells prepared from Slc23a2 (−/−) mice, whereas the ascorbate content of cells from wild-type littermates was 0.1–0.2 mM. The SVCT2 was not detected by immunostaining in cells from the Slc23a2 (−/−) mice, whereas it was clearly present in cells from wild-type mice. Although the SVCT2 was previously reported to be restricted to neuronal cell bodies in sections prepared from adult rat brain (Mun et al. 2006), we found that it was present in punctate pattern almost exclusively in neuronal axons and not in dendrites or cell bodies. The presence of the SVCT2 enhanced neuronal maturation in culture. Compared to neurons from wild-type mice, neurons from Slc23a2 (−/−) mice showed slower neuronal growth, as indicated by fewer dendritic branches and reduced total dendritic length. In addition, the numbers of α-amino-5-hydroxy-3-methyl-4-isoxazole propionic acid (AMPA) receptor subunit GluR1 clusters were also significantly reduced in cells lacking the SVCT2. The mechanism for reduced GluR1 clustering was not established, but it may be due to ascorbate-dependent abnormalities in neuronal activity, since neuronal activity can induce neurotransmitter receptor clustering (Rao et al. 2000). Perhaps related to the abnormalities in AMPA receptor subunit clustering, cells lacking the SVCT2 also had decreased neuronal activity. In patch-clamp experiments, cells from Slc23a2 (−/−) mice showed both decreased amplitude and frequency of miniature excitatory post-synaptic currents. It seems likely that these findings are due to the ability of cells containing the SVCT2 to retain intracellular ascorbate in culture, rather than to the presence of the SVCT2 per se.
5. Ascorbate accumulation and maintenance in the CNS: Role of SVCT2
As shown in Figure 2, there are two mechanisms by which ascorbate can enter neurons and glia: transport of ascorbate on the SVCT2 and uptake and reduction of dehydroascorbate. The SVCT2 mediates transport of ascorbate both from plasma across the choroid plexus to the CSF and across the neuronal cell plasma membrane. The plasma-CSF ascorbate gradient is about 4-fold in humans (Reiber et al. 1993; Lönnrot et al. 1996) and animals (Spector and Lorenzo 1973; Spector 1977), resulting in CSF ascorbate concentrations of about 200 µM, compared to plasma concentrations of 50 µM or less (Table 7.1). Although DHA enters the CNS more rapidly than ascorbate, the latter does readily penetrate the central nervous system after oral administration (Kontush et al. 2001). For example, supplements of one g vitamin C a day to volunteers already replete with vitamin C increased plasma levels from 50 to 75 µM, and increased CSF ascorbate from 200 µM to 250 µM (Lönnrot et al. 1996). Neuronal ascorbate uptake further accentuates the ascorbate concentration gradient from plasma to neurons, and brain neurons are said to have the highest ascorbate concentrations in the body (Rose 1988). In this regard, neuronal ascorbate concentrations in young rats have been calculated to be as high as 10 mM, whereas glial ascorbate concentrations are only 0.9 mM (Rice a& Russo-Menna 1998; Rice 2000). Such high ascorbate concentrations in young rats agree with the finding that SVCT2 mRNA is increased in fetal mouse brain compared to brain from adult animals (Castro et al. 2001).
It also appears that much of the ascorbate in brain is in neurons. This was first shown over four decades ago in histochemical studies (Shimizu et al. 1960). More recent investigations have localized high ascorbate concentrations to neuron-rich areas of the hippocampus and neocortex in normal human brain, where the ascorbate content is as much as two-fold higher in than in other brain regions (Mefford et al. 1981; Milby et al. 1982). The higher ascorbate contents of neurons in comparison to glia, as alluded to above, may well account for varying ascorbate contents of different brain regions. In post-natal rat brain, for example, areas rich in glia have ascorbate levels only about 10–20% those of areas with high neuronal density (Rice & Russo-Menna 1998). Indeed, whereas GSH is the major low molecular weight cytosolic reducing agent in most cells (e.g., in glia the ascorbate/GSH ratio is 0.24), in neurons ascorbate may take that role, since neuron-rich brain areas have an ascorbate/GSH ratio of 4.0 (Rice & Russo-Menna 1998). This differential in ascorbate content is probably due to the SVCT2, since in situ hybridization studies in rat brain show that SVCT2 mRNA is present only in neurons and not in astrocytes (Castro et al. 2001; Berger et al. 2003). More recent studies show that SVCT2 mRNA is expressed in certain glial elements in the hypothalamus (García et al. 2005).
Although astrocytes may not express the SVCT2 in vivo, oxidant stress can induce SVCT2 expression in astrocytes and increase expression in neurons. For example, ischemia-reperfusion injury following middle cerebral artery occlusion in rats increased SVCT2 mRNA over several hours in the peri-infarct penumbra, both in neurons and in glia (Berger et al. 2003). Also, astrocytes in culture develop high affinity ascorbate transport and SVCT2 mRNA (Wilson et al. 1990), an effect that has been attributed to exposure to high oxygen concentrations in culture (Berger et al. 2003).
As noted previously, the second route by which ascorbate can enter the CNS is via transport of dehydroascorbate across the blood-brain barrier on the ubiquitous GLUT1 glucose transporter. Any DHA generated in the brain interstitium would also be taken up by GLUT1 and GLUT3 in glia and neurons (Figure 2). Since dehydroascorbate concentrations in plasma are usually only 0–2% of ascorbate concentrations (Dhariwal et al. 1991), this route of entry across the blood-brain barrier may not contribute substantially to total brain ascorbate concentrations under normal conditions. However, during oxidant stress in the CNS, ascorbate in the CSF and interstitial space will be oxidized to the AFR, which will then dismutate to form ascorbate and DHA (Figure 1). The latter can then enter both glia and neurons on glucose transporters in substantial amounts, where it will be rapidly reduced to ascorbate. This likely serves as a back-up mechanism to recover oxidized ascorbate before it is irretrievably lost with degradation of DHA. A similar mechanism in cultured HL-60 cells has recently been termed the “bystander” effect (Nualart et al. 2003).
The SVCT2 thus appears to be crucial in maintaining ascorbate homeostasis in the CNS and in neurons. However, little known about SVCT2 structure as it relates to function or about specific mechanisms by which SVCT2 expression or function are regulated.
6. Structure-function and regulation of the SVCT2 in neurons
The SVCT2 transporter was one of two SVCT-type transporters cloned in 1999 by Hediger’s group (Tsukaguchi et al. 1999), both of which turned out to be members of the solute carrier (Slc) family of transporters. As noted previously, the two SVCT proteins are quite homologous, with only minor differences between species (Takanaga et al. 2004). There has been confusion in the literature regarding official designations of the ascorbate transporter genes, but currently accepted usage is that the Slc23a1 gene encodes the SVCT1 protein, and that Slc23a2 encodes the SVCT2 protein (Takanaga et al. 2004). The distributions of the two SVCT proteins differ in that mRNA for the SVCT1 is found in epithelial tissues involved in ascorbate absorption (intestine) and re-absorption (kidney). The SVCT2 (Figure 3) is the major isoform in most other tissues in the mouse, with highest expression in brain and in areas of high neuronal density in particular (Tsukaguchi et al. 1999).
Figure 3.
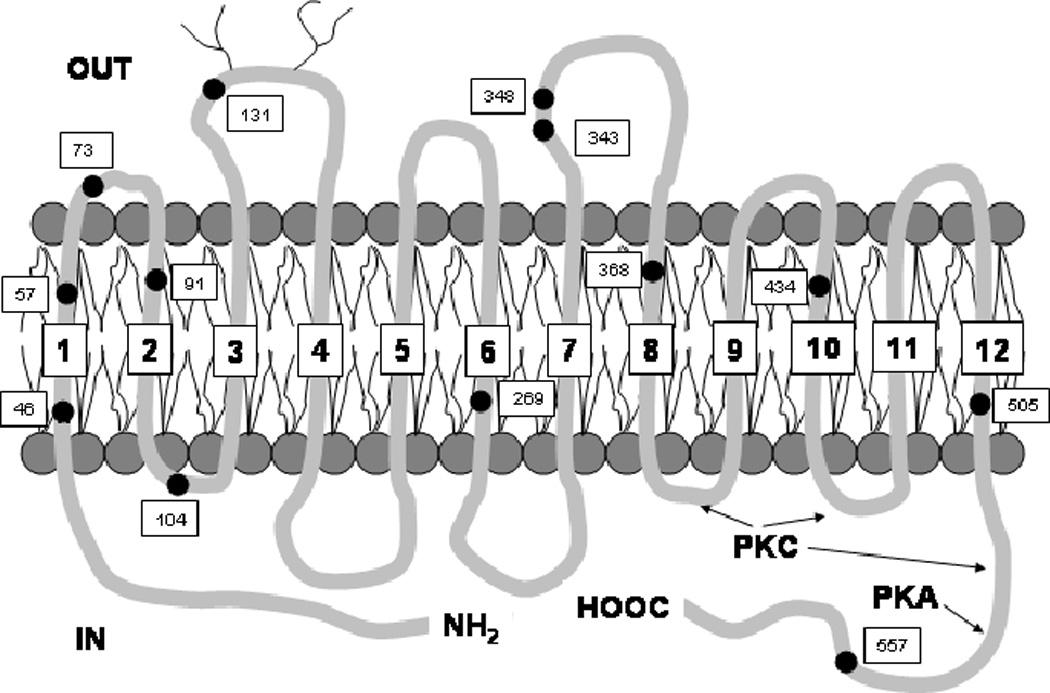
Trans-membrane structure of rat brain SVCT2 (Tsukaguchi et al. 1999). Numbered dots show the location of the 13 cysteines in the sequence.
Neuronal and most other cells have long been known to accumulate ascorbate against a concentration gradient via a saturable, sodium-dependent mechanism (Wilson 2004). With the cloning of the protein, it became apparent that this sodium- and energy-dependent transport is mediated largely by SVCT2 (Tsukaguchi et al. 1999). The SVCT2 has a high affinity for ascorbate (Km = 20–40 µM) that corresponds well to plasma ascorbate concentrations (30–50 µM). In neuronal cells the apparent Km for ascorbate appears to be somewhat higher (113 µM (May et al. 2006)), but even with this modest decrease in affinity, the SVCT2 will still be nearly saturated at the ascorbate concentrations present in CSF (200–400 µM). If the SVCT2 is saturated, then acute regulation of its function by changes in substrate affinity will be minimal in contrast to changes in transporter Vmax or transporter expression in the membrane. It seems likely that transporter number and ability to function rather than affinity are the major mechanisms involved in regulation of the SVCT2.
Rat brain SVCT2 cDNA codes for a 647-amino acid protein that hydropathy analysis shows to cross the cell membrane 12 times, with both the N- and C-termini in the cytoplasm (Figure 3) (Tsukaguchi et al. 1999). Assuming an average amino acid molecular weight of 121, this predicts that it will migrate as a 78 kDa protein on electrophoretic gels. This electrophoretic migration on Western blotting has been reported by two laboratories (Lutsenko et al. 2004; García et al. 2005). Although there have been reports of the SVCT2 from various tissues migrating in the 50–65 kDa region on Western blots using detection by polyclonal antibodies (Li et al. 2003; Wu et al. 2003; Jin et al. 2005; Mun et al. 2006), these results could be explained by proteolysis during sample preparation, by expression of short forms of the transporter (Lutsenko et al. 2004), or by variable glycosylation of the protein. Regarding the latter, the SVCT2 does have potential N-glycosylation sites on an exofacial loop between trans-membrane segments three and four (Tsukaguchi et al. 1999). Confirmation that the protein is glycosylated was provided by Liang and co-workers (Liang et al. 2002), who showed that endoglycosidase digestion of extracted protein decreased its apparent migration on electrophoretic gels from about 80 kDa to 65 kDa. This finding adds support to the trans-membrane loop topology originally projected by hydropathy analysis (Tsukaguchi et al. 1999). Moreover, the expression of non-glycosylated transporter precursors could explain the presence of bands migrating at lower apparent molecular weights on Western blots in the studies noted above. Shortening of the protein will also generate lower molecular weight bands. Lutsenko, et al. (Lutsenko et al. 2004) showed that a truncated form of the SVCT2 was present in cultured cells by Western blotting and in normal human brain by PCR. They hypothesized that this truncated SVCT2 corresponds to a non-functional variant that is missing 115 amino acids from domains five and six (Figure 3). Whereas a 75–80 kDa protein found on Western blots likely represents the SVCT2, shorter forms may also be present as well.
There are several plausible mechanisms for regulation of SVCT2 function. First, the SVCT2 protein has putative sites for protein kinase-A and protein kinase-C regulation on the cytoplasmic portions (Tsukaguchi et al. 1999) (Figure 3) Studies in cultured cells support the notion that these are functional in acute regulation of the protein, since ascorbate transport Vmax is decreased by both dibutyryl cyclic AMP (Siushansian and Wilson 1995, Korcok et al. 2000) and by protein kinase-C agonists (Daruwala et al. 1999; Liang et al. 2002) (Figures 4 and 5). Intracellular ascorbate may also regulate SVCT2 expression, based on the findings of Wilson, et al, that there is a time-dependent up-regulation of ascorbate transport in astrocytes depleted of the vitamin in culture (Wilson et al. 1990). A recent study (Seno et al. 2004) has also shown that function of the SVCT2 expressed in human umbilical vein endothelial cells is inhibited by tumor necrosis factor-α and by interleukin-1β. Since these inhibitor effects were maximal after 5 or more hours of exposure in culture, it seems likely that they were due to changes in protein expression rather than affinity. If so, these studies bring up the notion that the SVCT2 may be regulated by inflammatory cytokines and thus possibly by the level of oxidant stress experienced by the cells.
Figure 4.
Ascorbate preservation of nitric oxide sustains capillary perfusion.
Figure 5.
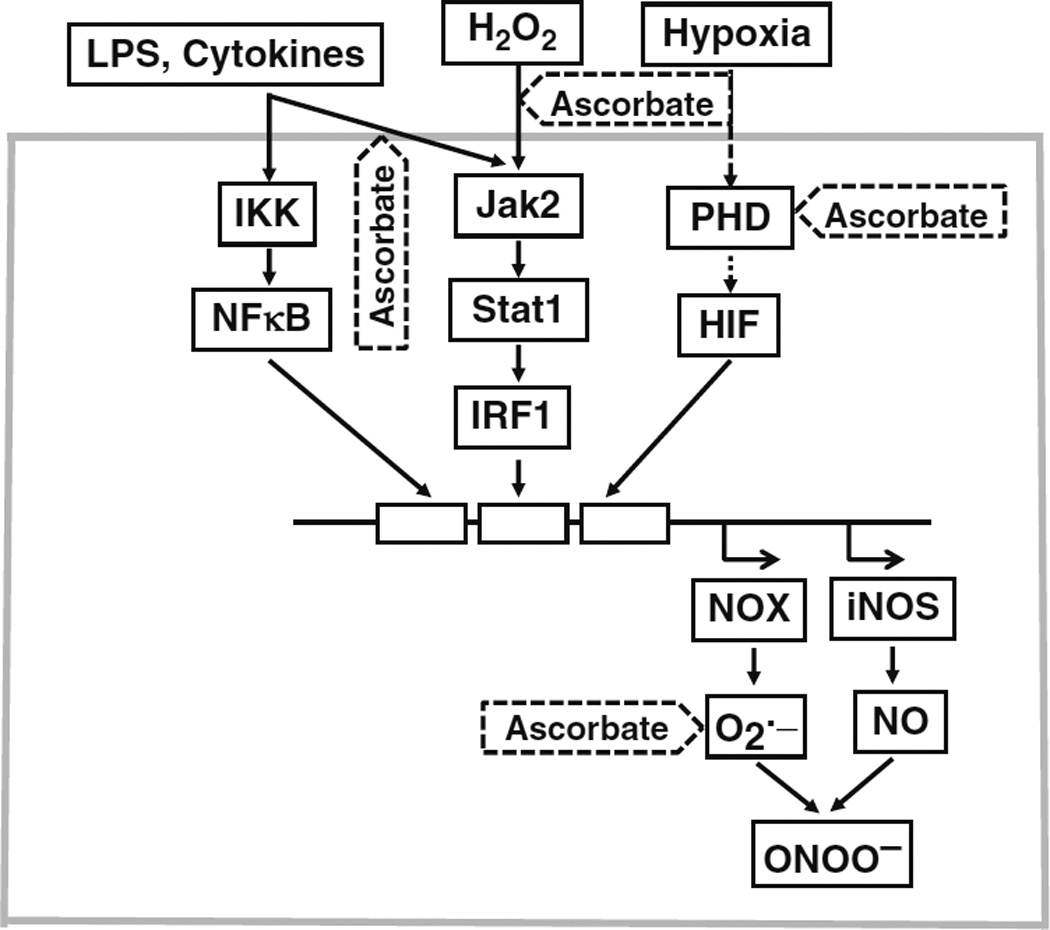
Sites of cellular protection by ascorbate against oxidative stress.
In addition to regulating SVCT2 transporter expression, oxidant stress could directly modify transporter function by oxidizing key amino acids in the protein. This would most likely involve formation of mixed disulfides or oxidation of cysteine sulfhydryls or methionine, thus causing changes in cell surface or protein redox status. For example, we found in cultured endothelial cells (May & Qu 2004) that rates of ascorbate transport on the SVCT2 are very sensitive to sulfhydryl reagents. Transport was inhibited by agents restricted to the extracellular space, suggesting that one or more of the four cysteines on the exofacial transporter (Figure 2) are involved in the transport mechanism. The presence of one or more exofacial sulfhydryls on the SVCT2 mirrors the situation with the GLUT1 glucose transporter. On that protein, modification of a single sulfhydryl (Cys-429) decreased affected transporter affinity for glucose (May 1988; May et al. 1990), although cysteine-scanning mutagenesis showed that its substitution with serine did not affect transporter function in Xenopus oocytes (Due et al. 1995). SVCT2 expression or function may also be regulated by changes in intracellular redox stress. For example, ascorbate transport in cultured endothelial cells is decreased in parallel with intracellular GSH concentrations in cultured endothelial cells (May & Qu 2004), which suggests that it is susceptible to redox regulation.
The presence of a non-functional truncated SVCT2 may also modify transporter function. In cultured cells truncated SVCT2 was present on the cell surface and expressed inversely in proportion to rates of ascorbate transport (Lutsenko et al. 2004). Further, transfection of the cells with truncated SVCT2 cDNA inhibited ascorbate transport. This dominant-negative inhibition was thought due to protein-protein interaction between functional and non-functional transporters. decreased ascorbate transport (Lutsenko et al. 2004).
Whether any of these potential mechanisms for SVCT2 regulation are important for the CNS protein remains to be determined. Other considerations regarding how neuronal ascorbate concentrations are regulated are the extent to which entry of ascorbate as DHA on the GLUT transporters supplies neuronal ascorbate, whether neurons and glia interact to maintain CNS ascorbate, and the extent to which glutamate-induced ascorbate efflux from neurons modulates intracellular ascorbate concentrations.
7. Conclusion
Lethality of the SVCT2 knockout on day one of life could reflect the need for ascorbate in the lung or the CNS. Indeed, any damage to the CNS in utero may simply show that ascorbate is required for neuronal maturation, not for maintenance and function of mature neurons. Nonetheless, differential retention of brain ascorbate in scurvy and the novel two-step concentrating mechanism used by the CNS to get ascorbate into neurons suggests that the importance of the vitamin for neuronal repair, myelination, and function may have been underestimated.
Acknowledgement
This work was supported by NIH grant AG023138.
References
- Agus DB, Gambhir SS, Pardridge WM, Speilholz C, Baselga J, Vera JC. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J. Clin. Invest. 1997;100:2842–2848. doi: 10.1172/JCI119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Minervini GM, Ciotti MT, Marra E, Calissano P. Glutamate neurotoxicity in rat cerebellar granule cells: a major role for xanthine oxidase in oxygen radical formation. J. Neurochem. 1997;68:2038–2045. doi: 10.1046/j.1471-4159.1997.68052038.x. [DOI] [PubMed] [Google Scholar]
- Baker RA, Milstien S, Katusic ZS. Effect of vitamin C on the availability of tetrahydrobiopterin in human endothelial cells. J. Cardiovasc. Pharmacol. 2001;37:333–338. doi: 10.1097/00005344-200103000-00012. [DOI] [PubMed] [Google Scholar]
- Berger UV, Lu XC, Liu W, Tang Z, Slusher BS, Hediger MA. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J. Neurochem. 2003;86:896–906. doi: 10.1046/j.1471-4159.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- Bianchi J, Wilson FA, Rose RC. Dehydroascorbic acid and ascorbic acid transport systems in the quinea pig ileum. Am. J. Physiol. 1986;250:G461–G468. doi: 10.1152/ajpgi.1986.250.4.G461. [DOI] [PubMed] [Google Scholar]
- Bielski BH, Allen AO, Schwarz HA. Mechanism of disproportionation of ascorbate radicals. J. Am. Chem. Soc. 1981;103:3516–3518. [Google Scholar]
- Bigley RH, Stankova L. Uptake and reduction of oxidized and reduced ascorbate by human leukocytes. J. Exp. Med. 1974;139:1084–1092. doi: 10.1084/jem.139.5.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Cunningham L, Rose RC. Spontaneous decay of oxidized ascorbic acid (dehydro-L-ascorbic acid) evaluated by high-pressure liquid chromatography. Clin. Chem. 1990;36:1807–1809. [PubMed] [Google Scholar]
- Bornstein SR, Yoshida-Hiroi M, Sotiriou S, Levine M, Hartwig HG, Nussbaum RL, Eisenhofer G. Impaired adrenal catecholamine system function in mice with deficiency of the ascorbic acid transporter (SVCT2) FASEB J. 2003;17:1928–1930. doi: 10.1096/fj.02-1167fje. [DOI] [PubMed] [Google Scholar]
- Brahma B, Forman RE, Stewart EE, Nicholson C, Rice ME. Ascorbate inhibits edema in brain slices. J. Neurochem. 2000;74:1263–1270. doi: 10.1046/j.1471-4159.2000.741263.x. [DOI] [PubMed] [Google Scholar]
- Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- Buettner GR, Jurkiewicz BA. Ascorbate free radical as a marker of oxidative stress: an EPR study. Free Radic. Biol. Med. 1993;14:49–55. doi: 10.1016/0891-5849(93)90508-r. [DOI] [PubMed] [Google Scholar]
- Burk RF, Christensen JM, Maguire MJ, Austin LM, Whetsell WO, Jr, May JM, Hill KE, Ebner FF. A combined deficiency of vitamins E and C causes severe central nervous system damage in guinea pigs. J. Nutr. 2006;136:1576–1581. doi: 10.1093/jn/136.6.1576. [DOI] [PubMed] [Google Scholar]
- Castro M, Caprile T, Astuya A, Millán C, Reinicke K, Vera JC, Vásquez O, Aguayo LG, Nualart F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 2001;78:815–823. doi: 10.1046/j.1471-4159.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee IB. Biosynthesis of L-ascorbate in animals. Methods Enzymol. 1970;18:28–34. [Google Scholar]
- Chatterjee IB, Chatterjee GC, Ghosh NC, Guha BC. Biological synthesis of L-ascorbic acid in animal tissues: conversion of L-gulonolactone into L-ascorbic acid. Biochem. J. 1960;74:193–203. doi: 10.1042/bj0740193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani E, Groneng L, Voltattorni M, Rolseth V, Contestabile A, Paulsen RE. Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Res. 1996;728:1–6. [PubMed] [Google Scholar]
- Coassin M, Tomasi A, Vannini V, Ursini F. Enzymatic recycling of oxidized ascorbate in pig heart: one- electron vs two-electron pathway. Arch. Biochem. Biophys. 1991;290:458–462. doi: 10.1016/0003-9861(91)90566-2. [DOI] [PubMed] [Google Scholar]
- Daruwala R, Song J, Koh WS, Rumsey SC, Levine M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999;460:480–484. doi: 10.1016/s0014-5793(99)01393-9. [DOI] [PubMed] [Google Scholar]
- Del Bello B, Maellaro E, Sugherini L, Santucci A, Comporti M, Casini AF. Purification of NADPH-dependent dehydroascorbate reductase from rat liver and its identification with 3a-hydroxysteroid dehydrogenase. Biochem. J. 1994;304:385–390. doi: 10.1042/bj3040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhariwal KR, Hartzell WO, Levine M. Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am. J. Clin. Nutr. 1991;54:712–716. doi: 10.1093/ajcn/54.4.712. [DOI] [PubMed] [Google Scholar]
- Drake BB, Smythe CV, King CG. Complexes of dehydroascorbic acid with three sulfhydryl compounds. J. Biol. Chem. 1942;143:89–98. [Google Scholar]
- Due AD, Cook JA, Fletcher SJ, Zhi-Chao Q, Powers AC, May JM. A "cysteineless" GLUT1 glucose transporter has normal function when expressed in Xenopus oocytes. Biochem. Biophys. Res. Commun. 1995;208:590–596. doi: 10.1006/bbrc.1995.1379. [DOI] [PubMed] [Google Scholar]
- Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br. J. Nutr. 1982;47:473–482. doi: 10.1079/bjn19820059. [DOI] [PubMed] [Google Scholar]
- García ML, Salazar K, Millán C, Rodríguez F, Montecinos H, Caprile T, Silva C, Cortes C, Reinicke K, Vera JC, Aguayo LG, Olate J, Molina B, Nualart F. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia. 2005;50:32–47. doi: 10.1002/glia.20133. [DOI] [PubMed] [Google Scholar]
- Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SW. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- Grünewald RA. Ascorbic acid in the brain. Brain Res. Rev. 1993;18:123–133. doi: 10.1016/0165-0173(93)90010-w. [DOI] [PubMed] [Google Scholar]
- Hediger MA. New view at C. Nat. Med. 2002;8:445–446. doi: 10.1038/nm0502-445. [DOI] [PubMed] [Google Scholar]
- Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J. Biol. Chem. 2001;276:40–47. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- Hill KE, Montine TJ, Motley AK, Li X, May JM, Burk RF. Combined deficiency of vitamins E and C causes paralysis and death in guinea pigs. Am. J. Clin. Nutr. 2003;77:1484–1488. doi: 10.1093/ajcn/77.6.1484. [DOI] [PubMed] [Google Scholar]
- Hillered L, Persson L, Bolander HG, Hallstrom A, Ungerstedt U. Increased extracellular levels of ascorbate in the striatum after middle cerebral artery occlusion in the rat monitored by intracerebral microdialysis. Neurosci. Lett. 1988;95:286–290. doi: 10.1016/0304-3940(88)90672-6. [DOI] [PubMed] [Google Scholar]
- Hirschmann JV, Raugi GJ. Adult scurvy. J. Am. Acad. Dermatol. 1999;41:895–906. doi: 10.1016/s0190-9622(99)70244-6. [DOI] [PubMed] [Google Scholar]
- Hornig D. Distributin of ascorbic acid, metabolites and analogues in man and animals. Ann. NY Acad. Sci. 1975;258:103–118. doi: 10.1111/j.1749-6632.1975.tb29271.x. [DOI] [PubMed] [Google Scholar]
- Hosoya K, Minamizono A, Katayama K, Terasaki T, Tomi M. Vitamin C transport in oxidized form across the rat blood-retinal barrier. Invest. Ophthalmol. Vis. Sci. 2004;45:1232–1239. doi: 10.1167/iovs.03-0505. [DOI] [PubMed] [Google Scholar]
- Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, Choudhri TF, Kim LJ, Mocco J, Pinsky DJ, Fox WD, Israel RJ, Boyd TA, Golde DW, Connolly ES., Jr Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc. Natl. Acad. Sci. USA. 2001;98:11720–11724. doi: 10.1073/pnas.171325998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RE, Hurley RJ, Jones PR. The retention of ascorbic acid by guinea-pig tissues. Br. J. Nutr. 1971;26:433–438. doi: 10.1079/bjn19710048. [DOI] [PubMed] [Google Scholar]
- Ito A, Hayashi S, Yoshida T. Participation of a cytochrome b5-like hemoprotein of outer mitochondrial membrane (OM cytochrome b) in NADH-semidehydroascorbic acid reductase activity of rat liver. Biochem. Biophys. Res. Commun. 1981;101:591–598. doi: 10.1016/0006-291x(81)91300-0. [DOI] [PubMed] [Google Scholar]
- Jackson TS, Xu AM, Vita JA, Keaney JF., Jr Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998;83:916–922. doi: 10.1161/01.res.83.9.916. [DOI] [PubMed] [Google Scholar]
- Jacques PF, Halpner AD, Blumberg JB. Influence of combined antioxidant nutrient intakes on their plasma concentrations in an elderly population. Am. J. Clin. Nutr. 1995;62:1228–1233. doi: 10.1093/ajcn/62.6.1228. [DOI] [PubMed] [Google Scholar]
- Jin SN, Mun GH, Lee JH, Oh CS, Kim J, Chung YH, Kang JS, Kim JG, Hwang DH, Hwang YI, Shin DH, Lee WJ. Immunohistochemical study on the distribution of sodium-dependent vitamin C transporters in the respiratory system of adult rat. Microsc. Res. Tech. 2005;68:360–367. doi: 10.1002/jemt.20255. [DOI] [PubMed] [Google Scholar]
- Kontush A, Mann U, Arlt S, Ujeyl A, Lührs C, Müller-Thomsen T, Beisiegel U. Influence of vitamin E and C supplementation on lipoprotein oxidation in patients with Alzheimer's disease. Free Radic. Biol. Med. 2001;31:345–354. doi: 10.1016/s0891-5849(01)00595-0. [DOI] [PubMed] [Google Scholar]
- Korcok J, Yan R, Siushansian R, Dixon SJ, Wilson JX. Sodium-ascorbate cotransport controls intracellular ascorbate concentration in primary astrocyte cultures expressing the SVCT2 transporter. Brain Res. 2000;881:144–151. doi: 10.1016/s0006-8993(00)02829-8. [DOI] [PubMed] [Google Scholar]
- Lee JY, Chang MY, Park CH, Kim HY, Kim JH, Son H, Lee YS, Lee SH. Ascorbate-induced differentiation of embryonic cortical precursors into neurons and astrocytes. J. Neurosci. Res. 2003;73:156–165. doi: 10.1002/jnr.10647. [DOI] [PubMed] [Google Scholar]
- Levine M, Conry-Cantilena C, Wang YH, Welch RW, Washko PW, Dhariwal KR, Park JB, Lazarev A, Graumlich JF, King J, Cantilena LR. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Wang YH, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA. 2001;98:9842–9846. doi: 10.1073/pnas.171318198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Huang J, May JM. Ascorbic acid spares alpha-tocopherol and decreases lipid peroxidation in neuronal cells. Biochem. Biophys. Res. Commun. 2003;305:656–661. doi: 10.1016/s0006-291x(03)00836-2. [DOI] [PubMed] [Google Scholar]
- Liang WJ, Johnson D, Ma LS, Jarvis SM. Regulation of the human vitamin C transporters and expressed in COS-1 cells by protein kinase C. Am. J. Physiol. Cell Physiol. 2002;283:C1696–C1704. doi: 10.1152/ajpcell.00461.2001. [DOI] [PubMed] [Google Scholar]
- Lönnrot K, Metsä-Ketelä T, Molnár G, Ahonen JP, Latvala M, Peltola J, Pietilä T, Alho H. The effect of ascorbate and ubiquinone supplementation on plasma and CSF total antioxidant capacity. Free Radic. Biol. Med. 1996;21:211–217. doi: 10.1016/0891-5849(95)02207-4. [DOI] [PubMed] [Google Scholar]
- Lutsenko EA, Carcamo JM, Golde DW. A human sodium-dependent vitamin C transporter 2 isoform acts as a dominant-negative inhibitor of ascorbic acid transport. Mol. Cell. Biol. 2004;24:3150–3156. doi: 10.1128/MCB.24.8.3150-3156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor DG, Higgins MJ, Jones PA, Maxwell WL, Watson MW, Graham DI, Stone TW. Ascorbate attenuates the systemic kainate-induced neurotoxicity in the rat hippocampus. Brain Res. 1996;727:133–144. doi: 10.1016/0006-8993(96)00362-9. [DOI] [PubMed] [Google Scholar]
- Mack WJ, Mocco J, Ducruet AF, Laufer I, King RG, Zhang Y, Guo W, Pinsky DJ, Connolly ES., Jr A cerebroprotective dose of intravenous citrate/sorbitol-stabilized dehydroascorbic acid is correlated with increased cerebral ascorbic acid and inhibited lipid peroxidation after murine reperfused stroke. Neurosurgery. 2006;59:383–388. doi: 10.1227/01.NEU.0000223496.96945.A7. [DOI] [PubMed] [Google Scholar]
- Maellaro E, Del Bello B, Sugherini L, Santucci A, Comporti M, Casini AF. Purification and characterization of glutathione-dependent dehydroascorbate reductase from rat liver. Biochem. J. 1994;301:471–476. doi: 10.1042/bj3010471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Bell JA. Ascorbic acid protects neurons from injury induced by glutamate and NMDA. Neuroreport. 1990;1:194–196. doi: 10.1097/00001756-199011000-00004. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Bell JA, London ED. Regulation of the NMDA receptor by redox phenomena: inhibitory role of ascorbate. Brain Res. 1990;537:328–332. doi: 10.1016/0006-8993(90)90379-p. [DOI] [PubMed] [Google Scholar]
- May JM. Inhibition of hexose transport and labelling of the hexose carrier in human erythrocytes by an impermeant maleimide derivative of maltose. Biochem. J. 1988;254:329–336. doi: 10.1042/bj2540329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JM, Asard H. Ascorbate Recycling. In: Asard H, May JM, Smirnoff N, editors. Vitamin C. Functions and biochemistry in animals and plants. London: Bios Scientific Publishers; 2004. pp. 139–158. [Google Scholar]
- May JM, Buchs A, Carter-Su C. Localization of a reactive exofacial sulfhydryl on the glucose carrier of human erythrocytes. Biochemistry. 1990;29:10393–10398. doi: 10.1021/bi00497a014. [DOI] [PubMed] [Google Scholar]
- May JM, Cobb CE, Mendiratta S, Hill KE, Burk RF. Reduction of the ascorbyl free radical to ascorbate by thioredoxin reductase. J. Biol. Chem. 1998a;273:23039–23045. doi: 10.1074/jbc.273.36.23039. [DOI] [PubMed] [Google Scholar]
- May JM, Li L, Hayslett K, Qu ZC. Ascorbate Transport and Recycling by SH-SY5Y Neuroblastoma Cells: Response to Glutamate Toxicity. Neurochem. Res. 2006;31:785–794. doi: 10.1007/s11064-006-9077-z. [DOI] [PubMed] [Google Scholar]
- May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J. Biol. Chem. 1997;272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- May JM, Qu ZC. Redox regulation of ascorbic acid transport: Role of transporter and intracellular sulfhydryls. Biofactors. 2004;20:199–211. [Google Scholar]
- May JM, Qu ZC, Cobb CE. Extracellular reduction of the ascorbate free radical by human erythrocytes. Biochem. Biophys. Res. Commun. 2000;267:118–123. doi: 10.1006/bbrc.1999.1906. [DOI] [PubMed] [Google Scholar]
- May JM, Qu Z-C, Mendiratta S. Protection and recycling of a-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch. Biochem. Biophys. 1998b;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- Mefford IN, Oke AF, Adams RN. Regional distribution of ascorbate in human brain. Brain Res. 1981;212:223–226. doi: 10.1016/0006-8993(81)90056-1. [DOI] [PubMed] [Google Scholar]
- Mehlhorn RJ. Ascorbate- and dehydroascorbic acid-mediated reduction of free radicals in the human erythrocyte. J. Biol. Chem. 1991;266:2724–2731. [PubMed] [Google Scholar]
- Miele M, Boutelle MG, Fillenz M. The physiologically induced release of ascorbate in rat brain is dependent on impulse traffic, calcium influx and glutamate uptake. Neuroscience. 1994;62:87–91. doi: 10.1016/0306-4522(94)90316-6. [DOI] [PubMed] [Google Scholar]
- Milby K, Oke A, Adams RN. Detailed mapping of ascorbate distribution in rat brain. Neurosci. Lett. 1982;28:15–20. doi: 10.1016/0304-3940(82)90201-4. [DOI] [PubMed] [Google Scholar]
- Mun GH, Kim MJ, Lee JH, Kim HJ, Chung YH, Chung YB, Kang JS, Hwang YI, Oh SH, Kim JG, Hwang DH, Shin DH, Lee WJ. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J. Neurosci. Res. 2006;83:919–928. doi: 10.1002/jnr.20751. [DOI] [PubMed] [Google Scholar]
- Nazemi M, Staudinger H. Kinetische Untersuchungen zur mitochondrialen NADH-semidehydroascorbate-oxydoreduktase (EC 1.6.5.4) in der Rattenleber. Hoppe Seylers. Z. Physiol Chem. 1968;349:345–348. [PubMed] [Google Scholar]
- Niki E, Noguchi N, Tsuchihashi H, Gotoh N. Interaction among vitamin C, vitamin E, and b-carotene. Am. J. Clin. Nutr. 1995;62(Suppl.):1322S–1326S. doi: 10.1093/ajcn/62.6.1322S. [DOI] [PubMed] [Google Scholar]
- Nualart FJ, Rivas CI, Montecinos VP, Godoy AS, Guaiquil VH, Golde DW, Vera JC. Recycling of vitamin C by a bystander effect. J. Biol. Chem. 2003;278:10128–10133. doi: 10.1074/jbc.M210686200. [DOI] [PubMed] [Google Scholar]
- Pastore P, Rizzetto T, Curcuruto O, Cin MD, Zaramella A, Marton D. Characterization of dehydroascorbic acid solutions by liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2001;15:2051–2057. doi: 10.1002/rcm.476. [DOI] [PubMed] [Google Scholar]
- Patel KB, Stratford MRL, Wardman P, Everett SA. Oxidation of tetrahydrobiopterin by biological radicals and scavenging of the trihydrobiopterin radical by ascorbate. Free Radic. Biol. Med. 2002;32:203–211. doi: 10.1016/s0891-5849(01)00777-8. [DOI] [PubMed] [Google Scholar]
- Qiu S, Li L, weeber ej, May JM. Ascorbate transport `by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2006 doi: 10.1002/jnr.21204. In Press. [DOI] [PubMed] [Google Scholar]
- Ranjan A, Theodore D, Haran RP, Chandy MJ. Ascorbic acid and focal cerebral ischaemia in a primate model. Acta Neurochir. (Wien.) 1993;123:87–91. doi: 10.1007/BF01476291. [DOI] [PubMed] [Google Scholar]
- Rao A, Cha EM, Craig AM. Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J. Neurosci. 2000;20:8344–8353. doi: 10.1523/JNEUROSCI.20-22-08344.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Pierce RC. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994;43:537–565. doi: 10.1016/0301-0082(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Reiber H, Ruff M, Uhr M. Ascorbate concentration in human cerebrospinal fluid (CSF) and serum. Intrathecal accumulation and CSF flow rate. Clin. Chim. Acta. 1993;217:163–173. doi: 10.1016/0009-8981(93)90162-w. [DOI] [PubMed] [Google Scholar]
- Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- Rice ME, Lee EJ, Choy Y. High levels of ascorbic acid, not glutathione, in the CNS of anoxia-tolerant reptiles contrasted with levels in anoxia-intolerant species. J. Neurochem. 1995;64:1790–1799. doi: 10.1046/j.1471-4159.1995.64041790.x. [DOI] [PubMed] [Google Scholar]
- Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1998;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- Rose RC. Transport of ascorbic acid and other water-soluble vitamins. Biochim. Biophys. Acta. 1988;947:335–366. doi: 10.1016/0304-4157(88)90014-7. [DOI] [PubMed] [Google Scholar]
- Seno T, Inoue N, Matsui K, Ejiri J, Hirata K, Kawashima S, Yokoyama M. Functional expression of sodium-dependent vitamin C transporter 2 in human endothelial cells. J. Vasc. Res. 2004;41:345–351. doi: 10.1159/000080525. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Matsunami T, Onishi S. Histochemical demonstration of ascorbic acid in the locus coeruleus of the mammalian brain. Nature. 1960;186:479–480. doi: 10.1038/186479a0. [DOI] [PubMed] [Google Scholar]
- Siushansian R, Wilson JX. Ascorbate transport and intracellular concentration in cerebral astrocytes. J. Neurochem. 1995;65:41–49. doi: 10.1046/j.1471-4159.1995.65010041.x. [DOI] [PubMed] [Google Scholar]
- Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- Spector R. Vitamin homeostasis in the central nervous system. N. Engl. J. Med. 1977;296:1393–1398. doi: 10.1056/NEJM197706162962409. [DOI] [PubMed] [Google Scholar]
- Spector R, Lorenzo AV. Ascorbic acid homeostasis in the central nervous system. Am. J. Physiol. 1973;225:757–763. doi: 10.1152/ajplegacy.1973.225.4.757. [DOI] [PubMed] [Google Scholar]
- Stevenson NR, Brush MK. Existence and characteristics of Na+-dependent active transport of ascorbic acid in guinea pig. Am. J. Clin. Nutr. 1969;22:318–326. doi: 10.1093/ajcn/22.3.318. [DOI] [PubMed] [Google Scholar]
- Takanaga H, Mackenzie B, Hediger MA. Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch. 2004;447:677–682. doi: 10.1007/s00424-003-1104-1. [DOI] [PubMed] [Google Scholar]
- Taylor A, Jacques PF, Nowell T, Perrone G, Blumberg J, Handelman G, Jozwiak B, Nadler D. Vitamin C in human and guinea pig aqueous, lens and plasma in relation to intake. Current Eye Research. 1997;16:857–864. doi: 10.1076/ceyr.16.9.857.5039. [DOI] [PubMed] [Google Scholar]
- Tolbert BM, Ward JB. Dehydroascorbic acid. In: Seib PA, Tolbert BM, editors. Ascorbic Acid: Chemistry, Metabolism, and Uses. Washington, DC: American Chemical Society; 1982. pp. 101–123. [Google Scholar]
- Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen X-Z, Wang YX, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–75. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- VanDuijn MM, Tijssen K, VanSteveninck J, van den Broek PJA, Van der Zee J. Erythrocytes reduce extracellular ascorbate free radicals using intracellular ascorbate as an electron donor. J. Biol. Chem. 2000;275:27720–27725. doi: 10.1074/jbc.M910281199. [DOI] [PubMed] [Google Scholar]
- Vera JC, Rivas CI, Fischbarg J, Golde DW. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature. 1993;364:79–82. doi: 10.1038/364079a0. [DOI] [PubMed] [Google Scholar]
- Villalba JM, Canalejo A, Rodríguez-Aguilera JC, Burón MI, Moore DJ, Navas P. NADH-ascorbate free radical and -ferricyanide reductase activities represent different levels of plasma membrane electron transport. J. Bioenerg. Biomembr. 1993;25:411–417. doi: 10.1007/BF00762467. [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wells WW. Identification of the dehydroascorbic acid reductase and thioltransferase (glutaredoxin) activities of bovine erythrocyte glutathione peroxidase. Biochem. Biophys. Res. Commun. 1999;257:567–571. doi: 10.1006/bbrc.1999.0508. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can. J. Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- Wilson JX. Vitamin C transport in animals and plants. In: Asard H, May JM, Smirnoff N, editors. Vitamin C. Functions and biochemistry in animals and plants. London: Bios Scientific Publishers; 2004. pp. 97–113. [Google Scholar]
- Wilson JX, Jaworski EM, Kulaga A, Dixon SJ. Substrate regulation of ascorbate transport activity in astrocytes. Neurochem. Res. 1990;15:1037–1043. doi: 10.1007/BF00965751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JX, Peters CE, Sitar SM, Daoust P, Gelb AW. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000;858:61–66. doi: 10.1016/s0006-8993(99)02433-6. [DOI] [PubMed] [Google Scholar]
- Winkler BS. In vitro oxidation of ascorbic acid and its prevention by GSH. Biochim. Biophys. Acta. 1987;925:258–264. doi: 10.1016/0304-4165(87)90190-5. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Orselli SM, Rex TS. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic. Biol. Med. 1994;17:333–349. doi: 10.1016/0891-5849(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Wu X, Itoh N, Taniguchi T, Nakanishi T, Tanaka K. Requirement of calcium and phosphate ions in expression of sodium-dependent vitamin C transporter 2 and osteopontin in MC3T3-E1 osteoblastic cells. Biochim. Biophys. Acta. 2003;1641:65–70. doi: 10.1016/s0167-4889(03)00065-x. [DOI] [PubMed] [Google Scholar]
- Yusa T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001;897:104–113. doi: 10.1016/s0006-8993(01)02099-6. [DOI] [PubMed] [Google Scholar]