

Abstract
Aims
Our studies have shown that the association between integrin-associated protein (IAP) and SHPS-1 regulates the response of cells including osteoclasts, osteoblasts, smooth muscle and retinal endothelial cells to Insulin-like growth factor-I (IGF-I). The aims of this study were to determine whether the regulation of IGF-I responsiveness by IAP/SHPS-1 association is a generalized response of endothelial cells, to identify the mechanism by which IAP/SHPS-1 association contributes to changes in endothelial cell responses to IGF-I and to determine whether inhibiting their association alters pathophysiologic changes that occur in vivo.
Methods and Results
Endothelial cells, maintained in 5mmol/l glucose, showed constitutive cleavage of the extracellular domain of IAP (containing the SHPS-1 binding site) and IAP/SHPS-1 association was not detected. In contrast, hyperglycemia inhibited IAP cleavage allowing IAP/SHPS-1 association and IGF-I stimulated SHPS-1 tyrosine phosphorylation. Exposure to an anti-IAP antibody that disrupts IAP/SHPS-1 association inhibited IGF-I stimulated tube formation and increased permeability. Rodent models of endothelial cell dysfunction were used to investigate the role of IAP-SHPS-1 association in endothelial cell function in vivo. Basal IAP/SHPS-1 association was not detected in retinal extracts in normal rats but was fully restored in rats with diabetes. The anti-IAP antibody inhibited IAP/SHPS-1 association and reduced retinal vascular permeability and leukocyte adherence to levels that were similar to non-diabetic rats. The antibody also significantly inhibited aberrant neovascularization that was induced by hypoxia.
Conclusions
Our results demonstrate that the increase in IAP/SHPS-1 association contributes to the pathophysiologic changes in the endothelium that are induced by hyperglycemia and hypoxia.
Keywords: Neovascularization, diabetic retinopathy, hyperglycemia, integrin associated protein, insulin-like growth factor I, vascular permeability, occluding, hypoxia
Introduction
Changes in endothelial cell function in response to hyperglycemia include increased vascular permeability and increased leukostasis, as well as increased cellular proliferation leading to new blood vessel formation. Enhanced responsiveness of vascular cells to IGF-I has been implicated in all these vascular responses to hyperglycemia. Hypophysectomy decreases serum IGF-I concentrations and arrests retinopathy progression [1]. IGF-I levels in vitreous correlate with the presence of proliferative retinopathy [2] and administration of somatostatin analogs which decrease IGF-I levels in vitreous ameliorates retinopathy progression [3]. Mice that over express IGF-I in the retina develop changes associated with diabetic retinopathy, including increased capillary permeability and angiogenesis [4].
The biological responses of smooth muscle and retinal endothelial cells (RECs) to IGF-I are enhanced when cells are cultured in hyperglycemic conditions [5,6]. This response is dependent upon hyperglycemia-facilitated association of two cell surface proteins, integrin associated protein (IAP or CD47) and SHPS-1 [5,7,8]. The aims of this current study were 1) to determine whether the regulation of IGF-I responsiveness by IAP/SHPS-1 association is a generalized response of endothelial cells to hyperglycemia conditions, 2) to identify the mechanisms by which the association between IAP and SHPS-1 contributes to hyperglycemia-mediated changes in endothelial cell responses to IGF-I and 3) to determine if inhibiting their association alters pathophysiologic changes that occur in vivo in rodent models of retinopathy.
Methods
Endothelial cell culture
Primary HUVECs (Lonza) were grown in M-199 (Life Technologies) plus EGM-2 Endothelial Cell Growth Medium supplements (Lonza, Walkersville MD) containing 5 mmol/l glucose. HUVECs were switched to growth medium containing 15 mmol/l glucose for 3 days. 10 mmol/l mannitol was added to the medium containing 5 mmol/l to control for the difference in osmolarity. Cultures were quiesced for 14 hr in serum-free (SF) M-199 containing 5 or 15 mmol/l glucose and exposed to IGF-I (50 ng/ml) (Genentech, San Fransisco, CA), the monoclonal anti IAP antibody, B6H12 (1 μg/ml) as described previously [8]) or the VEGF inhibitor Je-11 (10 μg/ml) (EMD Biosciences, Rockland, MA). RECs were grown as we have described previously [5]. Use of human cells was approved by the University of North Carolina Ethics Committee.
Cell lysis, immunoprecipitation and immunoblotting
Lysates were immunoprecipitated, separated by SDS-PAGE and transferred to Immobilon filters (Millipore, Billerica, MA) prior to immunoblotting to visualize proteins [5]. Antibodies used for immunoblotting were anti-phosphotyrosine (PY99, Santa Cruz, Santa Cruz, CA), anti-SHPS-1 (BD Biosciences, Franklin Lakes, NC), anti-occludin (Invitrogen, Carlsbad, CA) and anti-VEGF R2 (R and D Systems, Minneapolis, MN).
Real-time PCR analysis of VEGF mRNA
Total RNA was harvested using the Qiagen RNAeasy kit. cDNA was made using 1 μg of RNA and High Capacity cDNA Reverse Transcription Kit (Life Technologies, Grand Island, NY). Real-Time PCR reactions were set up using the TaqMan Gene Assay kit and (ABI Catalog number: for VEGFA Hs0090005_ ml and GAPDH Hs 999999905_ml) primer probe sets. Reactions were performed in quadruplicate. ABI SDS 2.2.2 software was used to determine relative amounts of RNA.
In vitro permeability assay
HUVECs were plated in growth medium (15 mmol/l glucose) on collagen coated Transwell inserts (24 well) After 24 hrs medium was changed to SFM-199 (15 mmol/l glucose) containing IGF-I (50 ng/ml) plus B6H12 (1 μg/ml). After 14 hr fluorescently labeled dextran (average MW 70K) was added (Sigma, St Louis, MO) (0.5 mg/ml). After 1 hr medium was collected from the lower chamber and the amount of FITC-dextran was measured in a fluorescence detecting microplate reader (Fluor Imager 595 Molecular Dynamics) (excitation and emission wave lengths 294 and 521 nm).
In vitro tube formation assay
HUVECs maintained in 15 mmol/l glucose were switched to SFM-199 containing IGF-I (50 ng/ml), B6H12 (1 μg/ml) for 14 hr. They were then plated on 24 well plates coated with 500 μl of growth factor reduced matrigel (BD Biosciences) (1.5 × 105 cells/ml/well). After 4 hours the plates were photographed at 10 x and the number of tubes/cm2 area in 6 random areas of each well was determined [9]. One tube is the area between two branch points (shown in figure 2 as the area between two “x” on the image).
Figure 2. The association between IAP and SHPS-1 is required for IGF-I stimulated cell permeability.

a: HUVECs plated on transwell inserts were treated with either IGF-I (50ng/ml) or IGF-I + B6H12 (1 μg/ml) overnight prior to exposure to fluorescently labeled dextran. The graph shows the mean fold increase in dextran permeation through the membrane (mean ± SEM n = 3, **p <0.01 when the amount of dextran that permeated in the presence of IGF-I is compared with either control or IGF-I + B6H12).
b & c: Occludin protein levels were determined by immunoblotting equal amounts of protein from HUVECs (b) and RECs (c) treated as described for fig 2a. To determine that the difference in occludin levels was not due to a different in total protein the lysates were also immunoblotted with an anti-β3 antibody. Bands shown are from discontinuous lanes of the same gel
In vitro measurement of leukocyte adhesion
RECS were plated in 96 well plates (3 × 104 cells/100μl/well) in growth medium (15 mmol/l glucose). After 48–72 hrs SFM-199 (15 mmol/l glucose) was added for 4 hours, +/− TNFα (Life Technologies; 30 ng/ml) for 2 hours. The monolayer was rinsed 3x then 100 μl of labeled leukocytes added. The monolayers were rinsed 3x and then 100 μl/ml of SFM-199 was added prior to visualization with fluorescent microscope (Excitation and emission wave length 485 and 530 nm respectively). Adherent, fluorescent leukocytes were counted.
Preparation of HL-60 leukocytes
1 × 106 leukocytes were incubated with 1 μg/ml Calcein-AM (BD Biosciences) in SFM-199 (15 mm/l glucose) (37°C for 1 hour). The labeled cells were then loaded on to the REC monolayer (100 μl/well). 500 μl of SFM-199 was added to the lower chamber.
Animals for diabetes study
Male Sprague–Dawley rats (Charles River, Wilmington, MA) were housed under 12-h:12-h light–dark conditions and received food and water ad libitum. All protocols were approved by the University of North Carolina at Chapel Hill Animal Care and Use Committee and adhere to the National Institutes of Health guidelines and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research.
Diabetes induction protocol
Control rats received an injection of vehicle. Streptozotocin (STZ) was given by intraperitoneal injection (50 mg kg −1). Hyperglycemia was confirmed 6 days later. At that point insulin injections were commenced (Neutral Protamine Hagedorn Insulin 9U kg−1/day). 20 days post-injection, 10 animals in the STZ group received an injection of control, mouse IgG (50 μg kg−1) and 10 received an injection of B6H12 (50 μg kg−1) every 72 hours for 30 days. Further details are given in Table 1.
Table 1.
Characteristics of the STZ hyperglycemic rat model
| Rat Group | Weight (g)a | Glucose levels mmol/l | Duration of diabetes |
|---|---|---|---|
| Control | 333 ± 23 | 8.14 ± 0.5 | - |
| Diabetic | 306 ± 7 | 25 ± 0.9* | 50 days |
| Diabetic + B6H12 | 276 ± 7 | 23.2 ± 0.95* | 50 days |
the rate of weight gain was attenuated in the diabetic group but no rat lost weight data shown is mean ± SEM n >20
p < 0.05 when compared with control group
In vivo measurement of vascular permeability [10]
Rats were injected with Nembutal (80 mg kg−1) (Southern Anesthesia, West Columbia, SC). Once deep anesthesia had been achieved warmed Evans blue (45 mg kg−1) (Fisher Scientific, Pittsburgh, PA) was injected into the tail vein. Evans Blue dye binds to albumin allowing the measurement of albumin leakage from the vasculature [10]. After 2 hrs a lethal dose of anesthetic (100 mg kg−1) was administered. Blood was collected and centrifuged at 12,000 × g for 5 min. The rats were perfused with 1% paraformaldehyde in citrate then the eyes removed and placed in PBS. The retinas were removed, lyophilized then resuspended in formamide and incubated at 70°C. After 18 the retina/formamide was centrifuged at 13,000 × g for 10 min.
A standard curve was generated using serial dilutions of Evans Blue (30 mg/μl). The absorbance was measured using a Nanodrop spectrophotometer (Thermo-Scientific, Rockford, IL) with excitation and emission wavelengths of 620 and 740 nm. The amount of Evans Blue permeation from each retina was calculated using the following: [Evans Blue (μg)/retina dry weight (g)]/[time − averaged Evans Blue concentration (μg)/plasma (μl) × circulation (h)].
In vivo measurement of leukocyte adhesion
Rats were anesthetized with Nembutal as above. The chest cavity was opened and a 19 gauge needle was inserted into the left ventricle and infused with PBS (300ml/kg) followed with 1% paraformaldehyde (100ml kg−1), 1 % (wt/vol) bovine serum albumin in PBS (20ml kg−1), Concanavalin A FITC (Vector Labs, Burlingame, CA) at a concentration of 40mg/ml in PBS (5 ml kg−1) and finally with PBS (20ml kg−1). Blood was collected and the eyes were removed and placed in paraformaldehyde. After 2 hrs the eyes were rinsed twice in PBS. Retinas were isolated, the hyaloidal vessels and vitreous removed with ora serratas intact. By making 4 incisions 90° apart, the retinas were flattened and then mounted onto microscope slides.
Analysis of retinal homogenates
Retina were removed from the eye and then frozen in modified radioimmunoprecipitation assay buffer with a protease inhibitor cocktail (HALT; Thermoscientific) then analyzed by immunoblotting [5].
Rodent model of retinopathy of prematurity (ROP)
On post natal day (p) 0, litters of 12 to 14 newborn Sprague-Dawley rat pups with their mothers (Charles River) were placed into an incubator (OxyCycler; BioSpherix, New York, New York) and inspired oxygen cycled between 50 % and 10 % every 24 hours. At p12 rat pups were given an intraperitoneal injection of B6H12 (20μg/pup) or control IgG daily for 6 days. After 7 cycles of oxygen fluctuations at p14, pups were placed into room air for 4 days. Oxygen and carbon dioxide levels were monitored daily [11].
Following sacrifice at p18, eyes were fixed in paraformaldehyde, 2 % (wt/vol). Retinas were isolated with ora serratas intact and were placed into PBS. Four incisions 90° apart were made, the retinas were flattened and mounted onto slides [12].
To stain the vasculature, the flattened retinas were permeabilized in 70 % ethanol and then PBS, 1% (vol/vol) Triton X-100. They were incubated with Alexa Fluor 568-conjugated Griffonia simplicifolia (Bandeiraea) isolectin B4 (5 μg/ml) (Invitrogen, Carlsbad, CA) [13]. Images of the retinal blood vessels were captured using a Nikon 80i Research Upright Microscope with Surveyor/TurboScan software (Nikon Inc) and were digitally stored for analysis.
Total retinal area, summed peripheral avascular retinal area, and areas of IVNV were computed in pixels using Image Tool v.3 (The University of Texas, San Antonio) and were converted to square millimeters (using a calibration bar). The IVNV was defined as neovascularization growing into the vitreous at the junction of vascular and avascular retina [14]. For clock hours, flat mounts were divided into 12 clock hours of equal area using Adobe Photoshop (Adobe Systems Inc) and the number of clock hours (0–12) exhibiting IVNV was determined [15,16]. Areas of neovascularization were measured, summed, and expressed as a percentage of total retinal area. Measurements were performed by 2 independent masked reviewers.
Protein estimation
The protein concentration of lysates was determined using a BCA protein assay kit (Thermoscientific).
Statistical Analysis
Chemiluminescent images were obtained from autoradiographs (Thermoscientific) and analyzed as described [5]. The Student’s t test was used to compare differences between treatments. The results that are shown are representative of at least three independent experiments.
Results
Regulation of IAP association with SHPS-1in vitro
To determine whether the hyperglycemia induced increase in IAP/SHPS-1 association was a more generalized response of endothelial cells to glucose we examined IAP/SHPS-1 association in HUVEC cells. Consistent with our previous observations in REC [5] we determined that there was a significant, 5 ± 0.9 fold increase in IAP association with SHPS-1 when HUVECs were cultured in 15 compared with 5 mmol/l glucose [fig 1a (mean ± SEM, n = 3)]. This was associated with a 24 ± 7 fold increase in SHPS-1 phosphorylation in response to IGF-I (Fig 1b mean ± SEM, n = 3) comparable to our previous data in RECs [5]. The lack of IAP/SHPS-1 association in vascular smooth muscle cells maintained in 5 mmol/l glucose is due to cleavage of the extraceullar domain of IAP, the region of IAP that contains the SHPS-1 binding site [7]. Immunoblotting of lysates from HUVEC and REC with the anti-IAP antibody (B6H12), which detects both intact IAP and the residual membrane-associated fragment that is present after cleavage, revealed degradation of IAP in 5 mmol/l glucose (Fig 1c).
Figure 1. Glucose regulation of IAP cleavage and IAP association with SHPS-1.
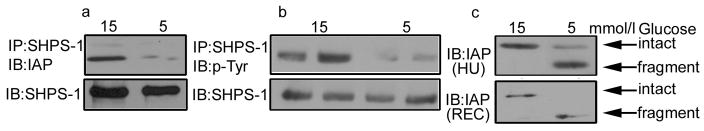
HUVECs and RECs were grown to confluency in either 15 of 5 mmol/l glucose prior to overnight incubation in serum free medium with the appropriate glucose concentration.
a. IAP association with SHPS-1 was determined by immunoprecipitating (IP) HUVEC lysates using an anti-SHPS-1 antibody then immunoblotting (IB) with an IAP antibody (B6H12). Equal amounts of protein were separated by SDS-PAGE and immunoblotted with the anti-SHPS-1 antibody to demonstrate that the different in IAP association with SHPS-1 is not due to difference in SHPS-1 levels.
b. Cell lysates were obtained from HUVECs that had exposed to IGF-I (50 ng/ml) for 5 min. The lysates were immunoprecipitated with an anti-SHPS-1 antibody and immunoblotted for phosphotyrosine (p-Tyr). To control for loading an equal amount of lysate was immunoblotted for SHPS-1. Bands shown are from discontinuous lanes of the same gel.
c. To examine IAP cleavage equal amounts of HUVEC (HU) and REC (REC) lysate were separated by SDS-PAGE and immunoblotted with the anti-IAP antibody B6H12 that recognizes both intact and fragments of IAP.
Disruption in IAP and SHPS-1 association blocks IGF-I stimulated increases in endothelial cell permeability
We next determined if the glucose mediated increase in IAP/SHPS-1 association played a role in the regulation of endothelial permeability in response to hyperglycemia and IGF-I. IGF-I stimulated a significant 2.4 ± 0.14 (mean ± SEM n = 3 p <0.01) increase in the amount of dextran that permeated the HUVEC monolayer in 1 hour. However when cells were incubated with B6H12 which disrupted IAP/SHPS-1 there was no significant increase in dextran permeation compared with control (Fig 2a).
A decrease in the junction protein occludin has been implicated in vascular permeability in diabetes. In both HUVECs and RECs cultured in 15 mmol/l glucose IGF-I stimulated a significant decrease in occludin levels (a 5.3 ± 1.2 and 7.5 ± 1.9 fold decrease respectively; mean ± SEM, n = 3 p <0.01). The decrease was completely blocked by co-incubation with B6H12 (Fig 2b).
IGF-I regulates VEGF production and receptor activation in 15 mM glucose
Previous studies have suggested that the response of cells to VEGF is regulated by IGF-I but whether this response is altered by hyperglycemia is unknown [17,18]. In HUVECs IGF-I stimulated a significant 2 ± 0.5 fold increase (mean ± SEM, n = 3 p<0.01) in VEGF mRNA. This response was dependent upon the association between IAP and SHPS-1 since it was blocked in the presence of B6H12 (Fig 3a).
Figure 3. IGF-I regulates VEGF.
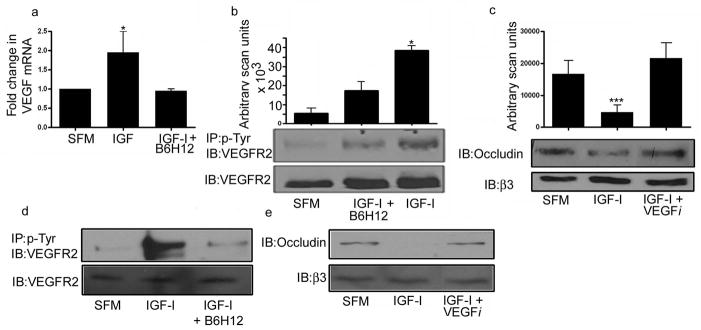
a: RNA was prepared from whole cell lysates. One microgram of total RNA was reverse transcribed. Two ul of cDNA was used in each RT-PCR reaction. A standard curve was generated using the control sample (10 fold dilutions from 1 to 10,000). The graph shows the results of three independent experiments (mean ± SEM, n = 3, *p<0.01 when the amount of VEGF mRNA in the presence of IGF-I is compared with either control or IGF-I + B6H12).
b: HUVECs were grown to confluency in medium containing 15 mmol/l glucose prior to overnight incubation in serum free medium with the same glucose concentration plus IGF-I (50 ng/ml) with or without the addition of B6H12 (1 μg/ml). The extent of VEGFR2 phosphorylation was determined by immunoprecipitating with an anti-phosphotyrosine antibody (p-Tyr) and immunoblotting with an anti-VEGFR2 antibody. To control for differences in protein, cell lysates were also immunoblotted directly with the anti-VEGFR2 antibody. The graph shows the results of 3 independent experiments expressed as arbitrary scanning units (mean ± SEM, n =3 *p<0.05 when the extent of VEGFR2 phosphorylation in the presence of IGF-I is compared with control or IGF-I + B6H12).
c: HUVECs were grown to confluency in medium containing 15 mmol/l glucose prior to overnight incubation in serum free medium with the same glucose concentration plus IGF-I (50ng/ml) with or without the addition of the VEGF inhibitor (VEGFi) Je-11 (10 μg/ml). Equal amounts of cell lysates were separated by SDS-PAGE and the amount of occludin was assessed by immunoblotting. ***p<0.05 when the change in occludin in response to IGF-I was compared to control or IGF-I + VEGFi.
d and e: REC were grown to confluency and treated as for 3b and c. Bands shown are from discontinuous lanes of the same gel. Control and IGF-I treated results are the same as those shown in figure 2b.
To examine the significance of the increase in VEGF mRNA we examined VEGF receptor phosphorylation in HUVECs. IGF-I stimulated a significant 12 ± 9 fold increase (mean ± SEM n = 3 p<0.05) in VEGF R2 phosphorylation and this increase was blocked in the presence of B6H12 (Fig 3b). To determine the significance of this increase we examined the ability of IGF-I to stimulate a decrease in occludin in the presence of the VEGF inhibitor, Je-11. In the presence of this inhibitor the IGF-I-stimulated decrease in occludin levels was attenuated (Fig 3c). We observed a similar response when we examined these changes in retinal endothelial cells grown in 15 mmol/l glucose (Fig 3d and e).
The role of IAP/SHPS-1 in vascular permeability in vivo
STZ induced hyperglycemia in rats was used to determine whether hyperglycemia induced an increase in IAP association with SHPS-1 in vivo and whether this played a role in the hyperglycemia-associated increase in vascular permeability
There was a 2.4 ± 0.6 fold increase in IAP/SHPS-1 association when retinal tissue from the hyperglycemic rats was compared with normoglycemic animals [Fig 4a (mean ± SEM, n = 9 p < 0.05)]. This is somewhat less than the increase in IAP association with SHPS-1 in cultured endothelial cells (Fig 1A and [5]) and this difference is probably due to the fact the retinal lysates contain additional cells types compared with pure cultures of endothelial cells. Treatment of the hyperglycemic rats with B6H12 significantly reduced IAP association with SHPS-1 such that there was no significant difference between the B6H12 treated rats and control treated animals (Fig 4a top panel). Consistent with our in vitro data there was a significant increase in the amount of intact IAP in the retina from hyperglycemic rats compared with normoglycemic, control animals. This increase in intact IAP was inhibited in rats treated with B6H12 (Fig 4a bottom panel).
Figure 4. B6H12 disrupts IAP association with SHPS-1 in vivo thereby inhibiting diabetes increased vascular permeability.

a: Individual retinal lysates from control (CON), diabetic and diabetic rats treated with B6H12 (+B6H12) were immunoprecipitated with an anti-SHPS-1 antibody and immunoblotted with an anti-IAP antibody (upper panel) or the lysates were immunoblotted directly with an anti-IAP antibody, R569, that selectively recognizes only intact IAP (bottom panel). The graph shows the association of IAP with SHPS-1 expressed as arbitrary scanning units (mean ± SEM, n = 9, *p<0.05 when diabetic rats are compared with either control or diabetic rats treated with B6H12.
b: Evans Blue permeation from the retinal vasculature was measured by spectrophotometry and corrected for plasma concentration, retina weight and time. The graph shows the mean amount of Evans Blue harvested from retina from each group of rats (mean ± SEM) (n = 37, con nondiabetic) (n=21, diabetic treated with control IgG) and (n=23, diabetic treated with B6H12). The permeation of Evans Blue from the diabetic retinal vasculature was significantly increased compared with control or diabetic treated with B6H12 (*p<0.05).
c: Individual retinal lysates from nondiabetic, control and diabetic rats treated control IgG, or B6H12 (+B6H12) were immunoblotted directly using an anti-occludin antibody. The graph shows the data expressed as arbitrary scanning units (mean ± SEM, n = 9, *p <0.05 when occluding levels in retina from diabetic animals treated with IgG were compared with either control, nondiabetic or diabetic animals treated with B6H12.
Disruption in IAP association with SHPS-1 impairs the increase in vascular permeability associated with hyperglycemia in vivo
There was a significant, 1.7 ± 0.2 fold, increase in the permeation of Evans Blue from the retinal vasculature of the diabetic rats compared with control (mean ± SEM, n = p<0.05). Treatment of hyperglycemic rats with the anti-IAP antibody restored Evans Blue leakage to a level that was comparable to that of control, normoglycemic rats (Fig 4b). We also examined occludin levels in the normal and diabetic rats. There was a significant (p <0.05) decrease in the amount of occludin detected in the retinal homogenates from the hyperglycemic rats compared with control, however, this was normalized in the hyperglycemic rats treated with B6H12 (Fig 4c).
Disruption of IAP association with SHPS-1 impairs the hyperglycemia-associated increase in leukocyte adhesion
TNFα treatment significantly increased leukocyte adhesion to REC cultured in both 5 and 15 mmol/l glucose. However, significantly more leukocytes adhered to the REC monolayer that had been cultured in 15mmol/l as compared cells cultured in 5 mmol/l. Pretreatment of REC, that had been stimulated with TNFα, with B6H12 significantly decreased leukocyte adhesion to the REC monolayer (Fig 5a).
Figure 5. Disruption in IAP association with SHPS-1 disrupts leukocyte adhesion to endothelial cells in hyperglycemic conditions.

a: The number of fluorescently labeled leukocytes adhering to a confluent monolayer of retinal endothelial cells in 5 mmol/l (NG) or 15mmol/l (HG) glucose, in the presence (+) or absence (−) of TNFα with (+) or without B6H12 (1 μg/ml) was assessed (mean ± SEM n = 3 **p <0.01 when leukocyte adhesion in the presence of TNFα is compared with adhesion in its absence irrespective of the glucose culture conditions, * p<0.05 when leukocyte adhesion in HG + TNFα the presence of B6H12 is compared with its absence).
b: Leukocyte adhesion to the retinal vasculature assessed and expressed as the number of leukocytes/107 pixels. The graph shows the mean ± SEM n = 14 (con) n=22 (diabetic) n=17 (diabetic + B6H12 treatment) *p<0.05 when diabetic rats treated with IgG are compared to nondiabetic or diabetic rats who received B6H12.
The number of leukocytes adhering to the retinal vasculature in the hyperglycemic rats compared with control was increased significantly (1.4 ± 0.2 fold compared with 2.0 ± 0.3, p<0.05). This increase was inhibited in the rats treated with B6H12 (Fig 5b) (1.2 ± 0.2 fold increase, p, NS when compared with control).
The role of IAP/SHPS-1 in regulating tube formation
Neovascularization, which occurs in diabetes in response to hyperglycemia, requires cell proliferation, migration and cellular fusion. We have shown previously that the hyperglycemia induced increase in IAP/SHPS-1 is required for the increased proliferation of retinal endothelial cells to IGF-1 [5]. To extend those observations we analyzed the role of IAP/SHPS-1 formation in vitro tube formation in HUVEC in response to IGF-I. A representative image and quantification of three independent experiments is shown in figure 6a. In the presence of IGF-I there was a significant (32.9 ± 2.61 fold) increase in the number of tubes formed (mean ± SEM, n = 3) (Fig 6a). In contrast in the presence of B6H12 the number of tubes formed was reduced significantly and not different than in the control cultures. The ability of IGF-I to stimulate an increase in tube formation was not altered in the presence of the VEGF inhibitor (Fig 6b).
Figure 6. Disruption of IAP association with SHPS-1 impairs IVNV in a rodent model of retinopathy of prematurity.

a&b: HUVECs were plated on growth factor-reduced matrigel in the presence of IGF-I (50ng/ml) and/or B6H12 (1μg/ml) and/or VEGF inhibitor (10 μg/ml). Tube formation was allowed to proceed for 4 hours at which point photographs were taken at 10x magnification. Tube number was quantified. The graph shows the mean number of tubes formed from 3 independent experiments (mean ± SEM, n = 3 *p<0.05 when the number of tubes in the presence of IGF-I is compared with the number of tubes formed in SFM alone or in the presence of IGF-I + B6H12).
c: Pools of retinal lysates from room air (RA) and ROP rat pups were immunoblotted with an anti-IAP antibody that specifically recognizes intact IAP R569 (upper panel) or with an anti-IAP antibody B6H12 that specifically recognizes both intact and fragmented IAP (middle panel). An actin immunoblot is shown as a loading control (bottom panel).
d, e and f. Lectin-stained retinal flat mounts from pups injected at p12 with (B6H12) or control IgG (con)were analyzed at day 18. The total IVNV area is shown in (d) the number of clock hours of intravitreous neovascularization (IVNV) is shown in (e), and avascular area in (f). *p<0.05 when control (con) is compared to B6H12. Representative images are shown in figure 6g.
Disruption of IAP association with SHPS-1 impairs neoangiogenesis in a rodent model of hypoxic retinopathy
Currently, there are no rodent models of diabetes-accelerated neovascularization comparable to that seen in human subjects with diabetes. However the rodent model of retinopathy of prematurity (ROP) allows the opportunity to test anti-angiogenic strategies in vivo. We first compared the level of IAP in the retinal homogenates from room-air versus ROP rat pups. There was a significant increase in the amount of intact IAP and correspondingly a decrease in IAP fragment in the ROP pups compared with control (Fig 6c). We then tested the ability of B6H12 to impair the intravitreal neovascularization (IVNV) that occurs in the ROP rats. In room air pups, retinal vascularization of the inner capillary plexus extends to the ora serrata and there is no avascular retina or IVNV. In the ROP pups, avascular retina is 30% of the total retinal area at p14 and 25% at p18. Approximately 4 clock hours of IVNV were present at p18 (Fig 6d). Treatment with B6H12 markedly reduced the number of clock hours exhibiting IVNV from (4.1 ± 0.4 to 2.0 ± 1.0 p<0.05) as well as the total IVNV area (0.6 ± 0.1 (% of total area) to 0.39 ± 0.1% p <0.05) (Fig 6d and e). There was no significant difference in the amount of avascular area when the untreated ROP animals were compared with the ROP treated with B6H12 (Fig 6f).
Discussion
The results of this study show that an increase in intact IAP and the increase in its association with SHPS-1 is a generalized endothelial cell response to hypeglycemia and is required for endothelial tube formation and increased endothelial permeability that occur in response to IGF-I during hyperglycemia, as well as for glucose enhanced leukocyte adherence to endothelial cells. Culturing cells in normal glucose resulted in proteolytic cleavage of the region of IAP that contains the SHPS-1 binding site which resulted in loss of IAP/SHPS-1 association and the ability of IGF-I to stimulate SHPS-1 phosphorylation and downstream signaling. In contrast, exposure to 15 mmol/l glucose protected IAP from cleavage which allowed SHPS-1/IAP association and IGF-I stimulated SHPS-1 phosphorylation. The significance of these in vitro findings is highlighted in our in vivo studies in which we demonstrated that not only was there an increase in IAP association with SHPS-1 in retinal homogenates from diabetic rats, but that blocking this increase inhibited pathophysiologic changes that have been identified during the early stages of diabetic retinopathy, for example increased endothelial permeability and leukocyte adherence [19,20]. These results strongly suggest that the increase in SHPS-1/IAP association may be a critical step in the biochemical events that lead to these early changes in the endothelium that occur in response to hyperglycemic stress.
Several studies have linked IGF-I to stimulation of diabetic retinopathy [1–4]. Endothelial cells in culture have been shown to secrete increased amounts of IGF-I in response to hyperglycemia [21,22]. Transgenic overexpression of IGF-I in mice has been associated with several changes in the retina that simulate the changes that occur in diabetic retinopathy including loss of pericytes, thickening of the capillary basement membrane and neovascularization [4]. Administration of IGF-I to patients with diabetes has been associated with the development of retinal edema and some evidence of increased retinopathy progression [23]. Similarly administration of octreotide decreases IGF-I levels in vitreous and retinopathy progression [3]. Our findings suggest inhibition of IGF-I signaling by disrupting IAP/SHPS-1 association represents a potentially effective treatment strategy.
Our data suggest a direct, but complex relationship between IGF-I and VEGF in mediating the changes that occur in the diabetic retina. One prior study clearly indicated that IGF-I stimulates VEGF synthesis during angiogenesis in the retinopathy prematurity model [17]. Our data indicated that during hyperglycemic conditions IGF-I can induce VEGF synthesis and secretion resulting in VEGF receptor activation. Whether the ability of IGF-I to stimulate VEGF actually requires hyperglycemic stress was not addressed in these studies but prior studies have suggested this possibility [24]. VEGF is a stimulant of angiogenesis and inhibiting the VEGF receptor in diabetes has been shown to be antiangiogenic and to inhibit retinal capillary leakage of proteins [25,26]. In our studies we showed that IGF-I can support angiogenesis in a VEGF independent manner suggesting that IGF-I may contribute to the proliferative phase of diabetic retinopathy both by direct stimulation of angiogenesis and indirectly by increasing VEGF production. In contrast to the VEGF independent effects of IGF-I on tube formation, the ability of IGF-I to stimulate a decrease in occludin and thus increase vascular permeability was dependent upon VEGF mediated signaling since it was blocked in the presence of the VEGF inhibitor.
Several junctional proteins have been linked to changes in permeability in retinal endothelium. These include claudin-5, Z0-1, VE cadherin and occludin [27]. Occludin binds within the tight junctions to the SH3-GUK domain of Z01 [28]. Both disassociation from the cell border and lower concentrations of total occludin occur in diabetes [29]. Exposure to VEGF also stimulates these changes and VE cadherin also undergoes phosphorylation and internalization in response to VEGF stimulation [29]. The VE cadherin content is decreased in vessels of diabetic rats and its concentration correlates inversely with vascular permeability [30]. There is also decreased association of VE cadherin and beta catenin in diabetic vessels. These changes are believed to be due primarily to changes in VE cadherin and beta catenin phosphorylation [28]. Both PKC isoforms and c-Src have been shown to phosphorylate these substrates leading to altered vascular permeability. Src phosphorylation of occludin leads to its disassociation from the adherens complex and its degradation [30]. VEGF has been shown to stimulate Src dependent occludin phosphorylation and Src can also phosphorylate Z0-1 leading to changes in its localization [31]. Since IGF-I stimulated SHPS-1 phosphorylation in VSMC enhances Src activation this represents a potential mechanism leading to occludin downregulation in response to hyperglycemia [32,33]. Future studies will be required to determine whether stimulation of IGF-I signaling has direct effects on the localization of VE cadherin, beta catenin or other components of endothelial cell tight junctions and/or an effect mediated through its regulation of VEGF signaling.
The translational impact of these observations is hard to predict. The length of time that the rodents were diabetic is very short compared with the length of time that diabetic retinopathy develops in humans. While the severity of retinopathy in animal models increases with duration of diabetes it remains mild compared with that seen in many patients with diabetes. Additionally, our studies focused on endothelial cells but the development of diabetic retinopathy includes changes in other cell types such as pericytes and retinal pigmented epithelial cells.
Despite these limitations, taken together with our other recent study [5], our data provides evidence that the hyperglycemia induced increase in IAP/SHPS-1 is a generalized response of endothelial cells and it contributes to changes in endothelial cell function, furthermore, our data suggest that this novel biochemical pathway may contribute to the early changes in vascular cell function that occur in diabetes including diabetic retinopathy.
Acknowledgments
This work was supported by a grant from the Juvenile Diabetes Research Foundation (17-2008-1048). The authors wish to thank Ms. Laura Lindsey for her help in preparing the manuscript.
Abbreviations
- IGF-I
Insulin-like growth factor-I
- VEGF
Vascular endothelial growth factor
- IAP
Integrin associated protein
- HUVEC
Human umbilical vein endothelial cells
- ROP
Retinopathy of prematurity
- IVNV
Intravitreal neovascularization
- PKC
Protein kinase C
Footnotes
Contributions
All authors were involved in the conception and design or analysis and interpretation of data. LAM, MEH and DRC contributed to the drafting or revising of the article and all authors gave final approval.
Duality of Interest Statement
The authors declare that there is no duality of interest associated with this manuscript
References
- 1.Merimee TJ, Zapf J, Froesch ER. Insulin-like growth factors. Studies in diabetics with and without retinopathy. N Engl Med. 1983;309:527–530. doi: 10.1056/NEJM198309013090904. [DOI] [PubMed] [Google Scholar]
- 2.Meyer-Schwickerath Vitreous levels of the insulin-like growth factors I and II, and the insulin-like growth factor binding proteins 2 and 3, increase in neovascular eye disease. Studies in nondiabetic and diabetic subjects. J Clin Invest. 1993;92:2620–2625. doi: 10.1172/JCI116877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grant MB, Mames RN, Fitzgerald C, et al. The efficacy of octreotide in the therapy of severe nonproliferative and early proliferative diabetic retinopathy. Diab Care. 2000;23:504–509. doi: 10.2337/diacare.23.4.504. [DOI] [PubMed] [Google Scholar]
- 4.Ruberte J, Ayuso E, Navarro M, et al. Increased in ocular levels of IGF-I in transgenic mice lead to diabetes-like eye disease. J Clin Invest. 2004;113:1149–1157. doi: 10.1172/JCI19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller E, Capps BE, Sanghani R, Clemmons DR, Maile L. Regulation of IGF-I signaling in retinal endothelial cells by hyperglycemia. Invest Opthal & Visual Sci. 2007;8:3878–3887. doi: 10.1167/iovs.07-0014. [DOI] [PubMed] [Google Scholar]
- 6.Maile LA, Capps BE, Ling Y, Xi G, Clemmons DR. Hyperglycemia alters the responsiveness of smooth muscle cells to insulin-like growth factor-I. Endocrinology. 2007;148:2435–43. doi: 10.1210/en.2006-1440. [DOI] [PubMed] [Google Scholar]
- 7.Maile LA, Capps BE, Miller E, et al. Glucose regulation of integrin-associated protein cleavage controls the response of vascular smooth muscle cells to insulin-like growth factor-I. Mol Endocrinol. 2008;22:1226–1237. doi: 10.1210/me.2007-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maile LA, Badley-Clarke J, Clemmons DR. The association between integrin-associated protein and SHPS-1 regulates insulin-like growth factor-I receptor signaling in vascular smooth muscle cells. Mol Biol Cell. 2003;14:3519–28. doi: 10.1091/mbc.E03-04-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5:628–635. doi: 10.1038/nprot.2010.6. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Qaum T, Adamis AP. Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest Ophthalmol Vis Sci. 2001;3:789–94. [PubMed] [Google Scholar]
- 11.Penn JS, Henry MM, Wall PT, Tolman BL. The range of PaO2 variation determines the severity of oxygen induced retinopathy in newborn rats. Invest Ophthalmol Vis Sci. 1995;36:2063–2070. [PubMed] [Google Scholar]
- 12.Chan-Ling T. Glial, vascular and neuronal cytogenesis in whole-mounted cat retina. Microsc Res Tech. 1997;36:1–16. doi: 10.1002/(SICI)1097-0029(19970101)36:1<1::AID-JEMT1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 13.Budd SJ, Thompson H, Harnett ME. Reduction in endothelial tip cell filopedia corresponds to reduced intravitreous but not intraretinal vascularization in a model of ROP. Exp Eye Res. 2009;89:718–727. doi: 10.1016/j.exer.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartnett ME, Mariniuk DJ, Saito Y, Geisen P, Peterson LJ, McColm JR. Triamcinolone reduces neovascularization, capillary density and IGF-I receptor phosphorylation in a model of oxygen-induced retinopathy. Invest Opthalmol Vis Sci. 2006;47:4975–4982. doi: 10.1167/iovs.06-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geisen P, Peterson LJ, Martiniuk D, Uppal A, Saito Y, Hartnett ME. Neutralizing antibody to VEGF reduces intravitreous neovascularization and may not interfere with ongoing intraretinal vascularization in a rat model of retinopathy of prematurity. Mol Vis. 2009;14:345–357. [PMC free article] [PubMed] [Google Scholar]
- 16.Werdich XQ, Penn JS. Specific involvement of Src family kinase activation in the pathogenesis of retinal neovascularization. Invest Opthalmol Vis Sci. 2006;47:5047–5056. doi: 10.1167/iovs.05-1343. [DOI] [PubMed] [Google Scholar]
- 17.Punglia RS, Law M, Hsu J, et al. Regulation of vascular endothelial growth factor expression by insulin-like growth factor-I. Diabetes. 1997;46:1619–1626. doi: 10.2337/diacare.46.10.1619. [DOI] [PubMed] [Google Scholar]
- 18.Smith LE, Shen W, Peruzzi C, et al. Regulation of vascular endothelial growth factor-dependent neovascularization by insulin-like growth factor-1 receptor. Nat Med. 1999;5:1390–1395. doi: 10.1038/70963. [DOI] [PubMed] [Google Scholar]
- 19.Xu X, Zhu Q, Xia S, Zhang S, Gu Q, Luo D. Blood-retinal barrier breakdown induced by activation of protein kinase C via vascular endothelial growth factor in streptozocin-induced diabetic rats. Current Eye Res. 2004;28:251–256. doi: 10.1076/ceyr.28.4.251.27834. [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz BA, Roberts R, Luan H, Peysakhov J, Mao X, Thomas KA. Dynamic contrast-enhanced MRI measurements of passive permeability through blood retinal barrier in diabetic rats. Invest Opthal & Visual Sci. 2004;45:2391–2398. doi: 10.1167/iovs.03-1381. [DOI] [PubMed] [Google Scholar]
- 21.Spoerri P, Ellis E, Tarnuzzer R, Grant M. Insulin-like growth factor: Receptor and binding proteins in human retinal endothelial cell cultures of diabetic and non-diabetic origin. Growth Reg. 1997;7:1–8. doi: 10.1016/s1096-6374(98)80102-0. [DOI] [PubMed] [Google Scholar]
- 22.Spraul CW, Baldysiak-Figiel A, Lang GK, Lang GE. Octreotide inhibits growth factor-induced bovine choriocapillary endothelial cells in vitro. Graefes Arch Clin Exp Ophthalmol. 2002;240:227–231. doi: 10.1007/s00417-002-0441-7. [DOI] [PubMed] [Google Scholar]
- 23.Kolaczynski JW, Caro JF. Insulin-like growth factor-I therapy in diabetes: Physiologic basis, clinical benefits and risks. Ann Intern Med. 1994;120:47–55. doi: 10.7326/0003-4819-120-1-199401010-00009. [DOI] [PubMed] [Google Scholar]
- 24.Treins C, Giorgett-Peraldi S, Murdaca J, Monthouel-Kartmann MN, Van Obberghen E. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor I. Mol Endocrinol. 2005;19:1304–1317. doi: 10.1210/me.2004-0239. [DOI] [PubMed] [Google Scholar]
- 25.Avery RL, Perham J, Piermici DJ, et al. Intravitreal bevacizumab (Avastin) in the treatment of proliferative diabetic retinopathy. Opthal. 2006;113:169Se–169515. doi: 10.1016/j.ophtha.2006.05.064. [DOI] [PubMed] [Google Scholar]
- 26.Ideno J, Mizukami H, Kakehashi A, et al. Prevention of diabetic retinopathy by intraocular soluble ft1 gene transfer in a spontaneously diabetic rat model. In J Mol Med. 2007;19:75–79. [PubMed] [Google Scholar]
- 27.Bucolo C, Ward KW, Mazzon E, Cuzzocrea S, Drago F. Protective effects of a coumarin derivative in diabetic rats. Invest Opthalmol Vis Sci. 2009;50:3846–3852. doi: 10.1167/iovs.08-3328. [DOI] [PubMed] [Google Scholar]
- 28.Steed E, Balda M, Matter K. Dynamics and functions of tight junctions. Trends in Cell Bio. 2009;3:142–149. doi: 10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 29.Leal EC, Martins J, Voabil P, et al. Calcium dobesilate inhibits the alterations in tight junction proteins and leukocyte adhesion to retinal endothelial cells induced by diabetes. Diabetes. 2010;59:2637–2645. doi: 10.2337/db09-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dejana E, Orsenigo F, Lampugnani M. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 31.Steed E, Balda M, Matter K. Dynamics and functions of tight junctions. Trends in Cell Bio. 2009;3:142–149. doi: 10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Elias BC, Suzuki t, Seth A, et al. Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. Trends in Cell Bio. 2009;3:142–149. doi: 10.1074/jbc.M804783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:1559–1569. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen X, Xi G, Radhakrishnan Clemmons DR. Recruitment of Pyk2 to SHPS-1 signaling complex is required for IGF-I-dependent mitogenic signaling in vascular smooth muscle cells. Cell Mol Life Sci. 2010;67:3893–3903. doi: 10.1007/s00018-010-0411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]