

Abstract
Objective
The HIV epidemic has carved contrasting trajectories around the world with sub-Saharan Africa (SSA) being most affected. We hypothesized that mean HIV-1 plasma RNA viral loads (VL) are higher in SSA than other areas, and that these elevated levels may contribute to the scale of epidemics in this region.
Design and Methods
To evaluate this hypothesis, we constructed a database of means of 71,668 VL measurements from 44 cohorts in seven regions of the world. We used linear regression statistical models to estimate differences in VL between regions. We also constructed and analyzed a mathematical model to describe the impact of the regional VL differences on HIV epidemic trajectory.
Results
We found substantial regional VL heterogeneity. The mean VL in SSA was 0.58 log10 copies/mL higher than in North America (95% CI: 0.45 to 0.71); this represents about a 4-fold increase. The highest mean VLs were found in Southern and East Africa, while in Asia, Europe, North America, and South America, mean VLs were comparable. Mathematical modeling indicated that conservatively 14% of HIV infections in a representative population in Kenya could be attributed to the enhanced infectiousness of subjects with heightened VL.
Conclusion
We conclude that community VL appears to be higher in SSA than in other regions and this may be a central driver of the massive HIV epidemics in this region. The elevated VLs in SSA may reflect, among other factors, the high burden of co-infections or the preponderance of HIV-1 subtype C infection.
Keywords: HIV, viral load, co-infection, epidemic, sub-Saharan Africa, mathematical model
INTRODUCTION
Sub-Saharan Africa (SSA) continues to be the region hardest hit by the HIV pandemic. It is home to 70% of the world’s estimated 2.7 million new HIV infections in 2010, as well as 68% of the estimated 34 million persons living with HIV [1]. While in other regions HIV transmission is concentrated in populations with identifiable high-risk sexual or injecting-drug behaviors, in SSA HIV infection is prevalent in the general population with low levels of reported sexual risk behaviors [2]. Multiple behavioral, ecological, and biological host factors and viral characteristics may have contributed to the striking contrast between the HIV epidemic trajectory in SSA and other regions [2], such as differences in concurrency patterns and sexual networks [3], co-infections that drive higher HIV-1 plasma RNA viral load (VL) [4], and sub-type-specific viral dynamics [5]. Yet the interplay of factors contributing to disproportionately high HIV prevalence in the general population in SSA, and not in other regions, remains poorly understood.
HIV-1 VL is a principal determinant of heterosexual HIV transmission [6, 7]. Ample experimental and observational studies have shown that suppression of HIV VL leads to large reduction in HIV transmission [8]. Decreases in community VL have been associated with reductions in HIV incidence [9]. Each one log10 copies/mL increase in VL has been found to more than double the per coital-act probability of HIV transmission [10, 11].
In light of the link between HIV infectiousness and VL level, we hypothesized that a specific biological cofactor contributed to the contrasting HIV epidemic trajectories between SSA and other regions; specifically we hypothesized that community level HIV-1 VL in SSA is higher than in other regions, and is substantially higher to the extent that it can drive divergent epidemic trajectories between SSA and other regions. We examined this hypothesis by addressing two specific research questions: 1) Are VL levels higher in SSA compared to other regions? 2) Can these higher VLs explain, in part, the fulminant HIV transmission dynamics observed in SSA, in contrast to other regions?
METHODS
Subjects and settings
We obtained the mean HIV-1 plasma RNA VL from prospective cohorts of HIV infected, antiretroviral therapy (ART) naïve study participants across seven geographic regions (Table 1 and Supplemental Digital Content (SuppDC) Table S3.1). Overall, the studies included 15 cohorts of men, 10 cohorts of pregnant women, 14 cohorts of non-pregnant women, five cohorts of women with unknown pregnancy status and a total of 71,668 VL measurements (SuppDC Sections 1 and 3). Data from each cohort consisted of the mean VL for subjects in the following CD4 categories: < 200, 200–349, 350–499, and ≥500 cells/μL by sex and pregnancy status. In addition, for each study, we captured the geographic location, viral load assay used, predominant subtype in the region and intervention description. We excluded studies in which the intervention had a potential impact on viral load, such as acyclovir, or used only baseline enrolment data.
Table 1.
Summary of mean HIV-1 plasma RNA viral load by region.
| Region | CD4 count cells/μL
|
||||
|---|---|---|---|---|---|
| <200 | 200–349 | 350–500 | >500 | ||
| North America | Mean HIV RNA (log10 copies/mL) | 4.81 | 4.18 | 4.21 | 3.65 |
| Sample size (N) | 3,758 | 1,966 | 3,199 | 4,700 | |
| Europe | Mean HIV RNA (log10 copies/mL) | 4.81 | 4.27 | 4.04 | 3.71 |
| Sample size (N) | 8,754 | 9,047 | 7,674 | 7,463 | |
| Asia | Mean HIV RNA (log10 copies/mL) | 4.90 | 4.50 | 4.20 | 3.80 |
| Sample size (N) | 129 | 359 | 474 | 908 | |
| South America | Mean HIV RNA (log10 copies/mL) | 4.17 | 4.58 | 4.23 | 3.90 |
| Sample size (N) | 46 | 135 | 114 | 99 | |
| West Africa | Mean HIV RNA (log10 copies/mL) | 5.33 | 4.67 | 4.25 | 3.83 |
| Sample size (N) | 194 | 156 | 136 | 131 | |
| East Africa | Mean HIV RNA (log10 copies/mL) | 5.15 | 4.45 | 4.33 | 4.18 |
| Sample size (N) | 510 | 220 | 510 | 510 | |
| Southern Africa | Mean HIV RNA (log10 copies/mL) | 4.90 | 4.60 | 4.43 | 4.10 |
| Sample size (N) | 377 | 515 | 424 | 452 | |
| South Africa | Mean HIV RNA (log10 copies/mL) | 4.53 | 4.18 | 4.08 | 3.66 |
| Sample size (N) | 5,260 | 2,243 | 4,140 | 1,438 | |
Statistical methods
Mean log10 VL was modeled using a linear regression model with the following predictors: indicators of region (North America, Europe, Asia, South America, West Africa, East Africa, Southern Africa, and South Africa), sex, CD4 category, pregnancy status, and interactions between CD4 category and sex, and between CD4 and pregnancy status (SuppDC Section 3). Pregnancy status was included as a potential confounder because lower VLs have been observed among pregnant women compared to non-pregnant women [30]. Of the 44 cohorts, the four with unknown pregnancy status were treated as not pregnant. The effects of sub-type and assay could not be statistically adjusted due to high co-linearity with region. The model weighted each observation by the sample size. Robust standard errors are reported for the estimated model coefficients (SuppDC Table 3.2). Sensitivity analyses were conducted to assess the impact of model assumptions (SuppDC Section 3.4).
Mathematical modeling methods
We assessed the epidemiological implications of the regional differences in VL by constructing and analyzing a deterministic compartmental mathematical model describing HIV epidemic expansion in a representative SSA population; that of Kisumu, Kenya (SuppDC Section 4). The model calculates the proportion of incident HIV infections that are directly attributable to the VL effect, or population attributable fraction (PAF). It stratifies the population into compartments according to HIV sero-status and stage of HIV infection, sexual-risk activity group, and exposure to a biological cofactor that heightens VL. This cofactor modulates HIV infectiousness according to the empirical relationship between VL and HIV per-coital transmission probability as observed initially by Quinn and colleagues in Uganda [10], and affirmed recently in the Partners in Prevention study [11]. Table 2 summarizes the key assumptions in our model and a detailed description of the model and its parameterization can be found in SuppDC Section 4.
Table 2.
Key assumptions of our mathematical model assessing the epidemiological impact of the heightened HIV-1 plasma RNA viral load in sub-Saharan Africa.*
| Assumption | Parameter value | Source |
|---|---|---|
| Rate ratio increase in HIV-1 coital transmission probability per one-log10 rise in HIV-1 plasma viral load | 2.45 | [10, 11] |
| Logarithmic increase in HIV-1 plasma viral load level in sub-Saharan Africa relative to North America | 0.46 | Statistical analyses of the viral load database with 20% reduction in estimated regional differences to account for potential systematic biases in comparing viral load data across regions |
| Reduction in coital frequency associated with the heightened HIV-1 plasma viral load | 20% | Conservative assumption |
Detailed description of the model and its biological and behavioral parameters can be found in SuppDC Section 4
Recognizing that multiple uncontrolled variables, such as type of VL assay and changes in sexual activity due to co-infection-associated morbidities, may affect VL or its effect on HIV epidemic trajectory, we reported conservative predictions for the impact of the VL effect on HIV epidemic expansion and conducted multiple sensitivity and uncertainty analyses on the model predictions (SuppDC Sections 4.4.3–5 and 4.5). We assumed a 20% smaller increase in mean VL in SSA relative to North America than emerged from our database analyses (0.46 log10 rather than 0.58 log10; the results assuming the full 0.58 log10 increase can be found in SuppDC Section 4.4.2). We also assumed a 20% sexual-activity reduction with the heightened VL to account for potential illness-associated abstinence during co-infections that increase HIV-1 VL. The latter is a conservative assumption because malaria morbidity, a frequent co-infection in SSA, generally lasts for less than 10% of the period of heightened VL due to malaria [4, 31–34]. Moreover, only a very small fraction of the prevalent herpes simplex virus type 2 (HSV-2) sero-positive persons suffer from clinically apparent ulcers, and even among those, HSV-2 reactivations are predominately asymptomatic [35, 36]. Of note, the assumed 20% reduction in sexual activity reflects a reduction throughout HIV infection natural history and not merely a reduction in sexual activity during a transient episode of a specific co-infection, which biases our model results towards a lower impact of the VL effect. Finally, although co-infection-induced VL does not appear to accelerate HIV disease progression to AIDS or death in SSA [14, 37], we accounted for this potential effect in the model to assess whether such mechanism could influence our predictions (SuppDC Sections 4.4.3 and 4.5).
RESULTS
There was striking heterogeneity in mean VL (Table 1). Compared with North America, VL levels were significantly higher in SSA (Figure 1). The estimated mean log10 VL was 0.29 higher (95% CI: 0.11 to 0.47) in West Africa, 0.71 higher (95% CI: 0.48 to 0.93) in East Africa, and 0.74 higher (95% CI: 0.55 to 0.92) in Southern Africa, excluding the South African study sites (SuppDC Table S3.2). The estimated mean log10 VL was modestly, but significantly, higher in Asia than in North America [0.14 higher (95% CI: 0.03 to 0.26)], but no difference was seen in mean log10 VL between Europe or South America and North America (p = 0.66 and 0.49, respectively).
Figure 1.

Differences in mean regional HIV-1 plasma RNA viral load (VL). Estimated differences in mean HIV-1 log10 VL compared with North America by region and for South Africa. The mean HIV-1 log10 VL in North America is 4.2 (95% CI: 4.0 to 4.4). The error bars reflect the 95% confidence intervals. The black marker for South Africa shows results including bDNA assays and the red marker shows results excluding bDNA assays.
Compared to North America, the mean difference in VL across the three resource-poor sub-regions of SSA, East, West, and Southern Africa, with each sub-region weighted by its HIV infected population size, is 0.58 log10 (95% CI: 0.45 to 0.71). South Africa, with its substantially lower burden of tropical co-infections that drive higher HIV-1 VL (see Discussion), particularly malaria [12], and better access to care than much of SSA, was considered separately. Indeed, mean VLs in South Africa were lower than those in North America. However, this anomaly may be largely due to the use of the bDNA viral load assay for mainly subtype C infections which, in this laboratory, has been associated with consistently lower VL results (SuppDC Figure S3.2) [13]. When data from this laboratory are excluded, mean VL borders on being significantly higher in South Africa than in North America [0.19 log10 (95% CI: −0.02 to 0.39) versus −0.33 log10 (95% CI: −0.49 to −0.18) with inclusion of the bDNA assay results]. SuppDC Section 3.5 includes a discussion and further analysis of the data from South Africa.
Applying our mathematical model to assess the epidemiological implications of such differences in VL in a setting representative of a hyper-endemic HIV epidemic in SSA (Kisumu, Kenya), the VL effect substantially augmented HIV epidemic expansion (Figure 2A). The VL effect contributed an excess HIV prevalence (prevalence with no VL effect subtracted from prevalence with the VL effect) of 2.9% (95% CI: 1.4% to 3.6%) at the epidemic peak, and 2.7% (95% CI: 1.5% to 3.2%) at the endemic equilibrium (CI estimated using the CI of the mean VL in resource-poor SSA). The VL effect fuelled an excess HIV incidence rate (incidence rate with no VL effect subtracted from incidence rate with the VL effect) of 0.69 (95% CI: 0.39 to 1.04) per 100 person-year at the epidemic peak, and 0.48 (95% CI: 0.27 to 0.56) per 100 person-year at endemic equilibrium. The proportion of incident HIV infections that is directly attributable to the VL effect (PAF), increased with time, reaching 14.4% (95% CI: 7.7% to 20.5%) at the epidemic peak in the mid to late 1990s (Figure 2B). The proportion of cumulative incident HIV infections that is directly attributable to the VL effect (PAFCum) is 13.9% by 2010.
Figure 2.

The impact of the heightened HIV-1 plasma RNA viral load (VL) on HIV epidemic trajectory. Simulated HIV epidemic trajectory in Kisumu, Kenya. (A) HIV prevalence in presence of the HIV-1 VL effect compared to HIV prevalence in absence of this effect. (B) Fraction of incident HIV-1 infections (population attributable fraction) at each point in time ( PAF ), and cumulatively since 1980 ( PAFCum), due to the VL effect. The empirically measured prevalences are extracted from several studies (SuppDC Section 4.2).
Our calculations show that the relative impact of the VL effect is largest in the majority of the population exhibiting the lowest sexual risk behavior, the general population. HIV prevalence in the low-risk group increased by 22.5%, while that in the higher risk groups increased by 4.4–11.1% (SuppDC Table S4.3). We estimate that in Kisumu, with an adult population of about 200,000, the VL effect has contributed, directly or indirectly through onward transmission, over 30,000 excess HIV infections from 1980 to 2010 out of a total of approximately 135,000 HIV infections during this period. Additional results of the epidemiological impact of the VL effect can be found in SuppDC Section 4.
The substantial impact of the VL effect was robust to multiple sensitivity and uncertainty analyses (SuppDC Sections 3.4, 4.4.3–5, and 4.5). We performed a broad range of sensitivity analyses to the magnitude of the heightened VL, reduction in sexual activity associated with the heightened VL (the heightened VL could be due to co-infections that lead to co-morbidities as discussed in Methods and Discussion), and enhancement of the rate of disease progression associated with the heightened VL. We found that a reduction in coital frequency with the elevated VL greater than 34% is needed to balance the increase in infectiousness due to the VL effect, while any increase in VL greater than 0.25 log10 would result in considerable population-level impact for this effect. The strong effect was robust to variations in the model parameters, including both the behavioral and biological inputs used to parameterize the model. As expected, our model predictions are primarily sensitive to the exact magnitudes of both the heightened VL and the assumed level of sexual activity reduction associated with it. The lower is the reduction in sexual activity, the higher is the impact of the VL effect.
DISCUSSION
We found substantial regional differences in VL with SSA having about four times the VL levels found elsewhere. The highest VLs were seen in Southern and East Africa. Modeling the impact of the elevated VL in a representative population, Kisumu, Kenya, we demonstrated that the elevated VL in SSA could be a central driver of the massive HIV epidemics that erupted in this part of the world. The VL effect is particularly important driver of HIV transmission among the general population and may explain, in part, the general population HIV epidemics that uniquely characterize this region. The robustness of our results to multiple sensitivity and uncertainty analyses points to a potentially substantial role for this biological cofactor in shaping HIV epidemic trajectories in SSA and warrants further investigation.
The higher regional VL in SSA poses a question about the causes of such elevated VL levels. We suspect that the high burden of infectious diseases beyond HIV in SSA, particularly malaria, tuberculosis, HSV-2, helminthes and other tropical diseases [14], may have caused to a large extent the higher VL in SSA. Mounting epidemiological and laboratory evidence suggests that these co-infections induce transient, but substantial increases in HIV-1 VL, due to enhanced HIV replication associated with the immune response (see SuppDC Section 2 for description of evidence). A recent systematic review and meta-analysis found that acute malaria increases HIV-1 VL by 0·67 log10 (95% CI: 0.15, 1.19), active tuberculosis by 0.40 log10 (95% CI: 0.13–0.67), and HSV-2 infection by 0·18 log10 (95% CI: 0.01, 0.34 ) [15]. Furthermore, high levels of serum immune activation markers have been found in African populations compared to those in industrialized countries [16–18], possibly reflecting extended exposure to a range of pathogens. Together, the comparatively high burden of infectious diseases in SSA, and the links among co-infections, immune activation and increased HIV replication, suggest that VL may be higher in this region than elsewhere, and that immune activation due to co-infections may be an important causal mechanism.
The first work to explore the impact on HIV epidemic trajectory of a co-infection that increases HIV-1 VL, for the case of malaria, suggested a small, but tangible effect in fuelling HIV spread [4]. But the impact of a single co-infection on HIV transmission may be relatively limited, while the combined effects of multiple recurrent or persistent co-infections could potentially result in considerable population-level impact. The transient effect of one co-infection increasing VL may not be discernible at the population level, but the cumulative effect of multiple potentially overlapping co-infections on raising the VL, throughout the course of HIV infection, may be substantial as schematized in Figure 3. Such effect would alter the natural history of HIV infection for the individual and consequently the epidemic trajectory for the community.
Figure 3.
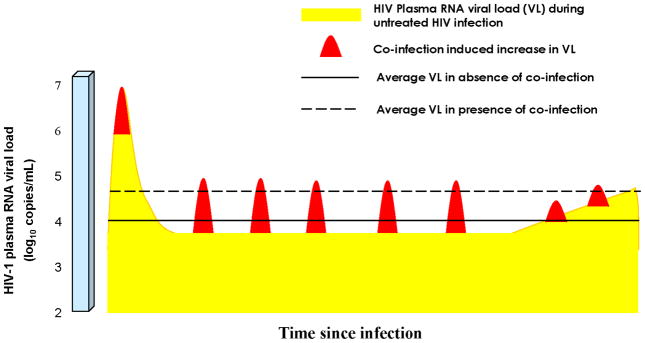
Potential role of co-infections in causing higher community HIV-1 plasma RNA viral load (VL). Schematic diagram of the potential role of the high co-infection disease burden in sub-Saharan Africa (SSA) in increasing mean HIV-1 VL (diagram not to scale). At low co-infection prevalence, such as in resource-rich settings, the natural course of HIV-1 VL resembles that in yellow. In SSA, repeated co-infections resulting in bursts of viremia (in red), may lead, at the population level, to a higher mean HIV-1 VL over the course of HIV-1 infection, and greater aggregate person-time above the threshold HIV-1 VL for HIV-1 transmission. This figure is an adaptation from that in Cohen et al [38].
Another reason, in addition to co-infections, that may contribute to explaining the elevated mean VLs in SSA is virus subtype. Emerging data suggest that persons infected with HIV-1 subtype C infection, the dominant type in SSA particularly in Southern Africa, may maintain high VLs even after acute infection [19].
Our approach assumes a log-linear relationship between HIV-1 VL and HIV per coital-act transmission probability, based primarily on empirical evidence from cohort studies of sero-discordant couples [10, 11]. These studies may under- or over-estimate the relationship between VL and risk of transmission due to selection bias such as the selection of more “resistant” couples in the recruitment of discordant partnerships with high-VL infected individuals. A linear relationship between VL and risk of transmission, assuming implicitly, for example, an intuitive concept for the establishment of the infection in terms of clonal expansion of a single infecting virion, would imply a stronger effect for the higher VL on transmission, and more differential epidemiologic impact as a consequence of the differences in community VL.
To our knowledge, the database we assembled in this study of summary measures of more than 70,000 VL measurements from cohorts representing every major region of the globe, is the most extensive ever analyzed to assess the regional VL differences and their impact on HIV transmission. However, there are limitations to these exploratory analyses. We used heterogeneous datasets from studies not designed to investigate this effect. Our hypothesis of higher community VL in SSA was examined using regional VL data measured on subjects recruited for different reasons (Table S3.1). The recruitment strategies could, in principle, be a source of selection bias where the VL data were measured on subjects who may not necessarily represent the community of HIV-infected persons, and this may confound the assessment of the regional VL differences. Of note, however, this vast amount of VL data was collected from subjects who were recruited using diverse recruitment strategies. The diversity of the modes of recruitment suggests a minimal bias in recruitment of persons who are more likely to have higher (or lower) VL in one region as opposed to another.
The widest difference in VL between SSA and developed settings was observed in the CD4 ≥ 500 category (Table 1), a category that potentially could encompass samples including persons within acute infection. The observed differences in VL could accordingly reflect differences in the distribution of persons across HIV natural history stages. However, all except one of the VL studies in this analysis have targeted recruitment of HIV sero-prevalent subjects, suggesting minimal contribution of acute infection. Beyond acute infection, we stratified our analysis in four CD4 categories and this should implicitly, at least in part, correct for potential differences between the regions in the distribution of persons across HIV natural history stages.
Our ecological analysis did not control for VL assay type which could potentially affect the observed regional VL differences. While we were not able to control for VL assay, PCR-based assays were used in both North America and SSA. VL assays would have to provide consistently lower absolute VL readings in North America and Europe, and higher readings in SSA, for this difference in VL to be seen. Historically, the opposite trend has been observed; commercially available RNA tests are generally optimized to detect subtype B, which predominates in North America and Europe, and are frequently suboptimal in detecting HIV-1 subtypes found in other parts of the world [20]. Furthermore, despite the heterogeneity of assays used within North America and Europe, mean VLs were similar. Another limitation of our database is the small number of VL cohorts from Asia and South America, where only a single country is represented from each region. Finally, pregnancy data were incomplete in five cohorts.
Nevertheless, our study provides a tantalizing “smoking gun”, suggesting both substantially higher VLs in SSA compared to other regions, and a potentially profound VL effect on HIV infectiousness and epidemic trajectory. This is despite conservative assumptions in the mathematical model for the magnitude of the heightened VL and the potential impact of co-infection-associated morbidities on sexual activity.
Several lines of evidence support our findings. The VL differences we found (~ 0.3 to 0.7 log10) are similar to those observed earlier (0.7 log10) in comparing a cohort of 49 Malawians to a cohort of 61 US and Swiss HIV-positive patients matched by CD4 count [21]. Yet, existing evidence suggests that African descent among those residing in North America and Europe is associated with lower, not higher, VL [22, 23]. Therefore, it is likely that ecological factors, such as frequently recurrent or persistent co-infections and HIV-1 sub-type, rather than host biology factors, explain these regional VL differences. Recent data further indicate that individuals living in areas of high malaria prevalence have more than twice the odds of being HIV-positive than individuals living in areas with low malaria prevalence (odds ratio of 2.24, 95% CI 1.62–3.12 for men and 2.44, 95% CI 1.85–3.21 for women) [24].
Additional lines of evidence support our thesis of higher HIV infectiousness per coital act in SSA compared to other regions. A recent systematic review and meta-analysis found HIV transmission probability per coital act in low-income countries (predominantly SSA countries) to be six times that in high-income settings [25]. The VL regional differences identified here of 0.58 log10 implies that the per-coital transmission probability in SSA is only 1.68 times that in North America and Europe (SuppDC Table S4.2); smaller than that observed in this meta-analysis [25]. This suggests that other factors may also contribute to the higher HIV infectiousness in SSA. The recent landmark HPTN 52 clinical trial found four-fold higher HIV sero-conversion rate among the sero-discordant partnerships in the African sites compared to the non-African sites [8], further supporting higher HIV infectiousness in SSA.
One of the truly remarkable research advances of the past two years was the demonstration of the very high efficacy of ART in reducing HIV transmission [8, 26]. This outcome attests to the critical role of VL in driving HIV epidemics. Observational studies and intervention trials designed specifically to elucidate the role of co-infections in HIV infectiousness and epidemic trajectory – such as the on-going trial evaluating the impact of helminthes treatment on HIV-1 VL in Kenya [27] – are essential. They will determine whether aggressive prevention and treatment of selected, persistent or frequently recurrent, co-infections should be included in randomized controlled trials of combination HIV prevention packages, and in HIV prevention programs and policy recommendations. As we are learning from trials evaluating HSV-2 treatment for HIV prevention [28, 29], this may require development of new regimens that target the biological mechanisms underpinning these interactions.
A potential role for co-infections in the exceptionally fulminant spread of HIV in Africa is of great interest because these infections offer feasible intervention points with co-benefits that lie at the intersection of HIV prevention and other major health programs. Until we can assure immediate and sustained access to ART for all people diagnosed with HIV infection, complementary strategies will be essential. Addressing co-infections potentially may offer such a complementary strategy for the control of HIV in SSA.
Supplementary Material
Acknowledgments
The authors are grateful to the investigators who generously shared their data with us or assisted in obtaining different datasets:
Burkina Fasos: Nicolas Nagot, Philippe Mayaud and Philippe Van de Perre for the ANRS 1285 Study Group; Denmark: Nicolai Lohse and Niels Obel for the Danish HIV Cohort Study; India: Amita Gupta, Ramesh S. Paranjape, Madhuri Thakar, Nikhil Gupte and Robert Bollinger; Kenya: Jared Baeten, Julie Overbaugh, Kishorchandra Mandaliya, Grace John Stewart, Phelgona Otieno, Julie Overbaugh and Dorothy Mbori-Ngacha; Malawi: James Kublin, Padmaja Patnaik, Irving Hoffman, Bill Miller, Charles Jere, Richard Pendame, Terrie Taylor and Malcolm Molyneux; Malawi; Tanzania; Zambia: Taha Taha, Lynda Emel, Tom Fleming, Elizabeth Brown, Anthony Mwatha, Lei Wang, Moses Sinkala, George Kafulafula, Gernard Msamanga, Megan Valentine and Robert Goldenberg for HPTN 024 Team; Peru: Aldo Lucchetti, Rosario Zuñiga, Jorge Sanchez, Connie Celum, Jared Baeten, Richard Zuckerman, Wil Whittington, Jesus Peinado and Juan Guanira; South Africa: Gavin Churchyard, Katherine Fielding, Sinead Delany, Nkuli Mlaba, Godspower Akpomiemie, Tim Clayton, Wendy Stevens, Helen Rees, Philippe Mayaud, Gabriela Paz Bailey and David Lewis; Spain: Jordi Casabona for the PISCIS Study Group; Tanzania: Wafaie Fawzi, Donna Spiegelman, Ellen Hertzmark, Ferdinand Mugusi, and Gernard Msamanga, for the Tanzania Vitamin and HIV Infection Trial Team; The Netherlands: Frank de Wolf and Colette Smit for the Dutch HIV Monitoring Foundation; Uganda: Neil French; Canada: D. William Cameron and Curtis L. Cooper; USA: John T. Brooks and Rose Baker for HOPS/HIV Insight, Alvaro Munoz and Lisa Jacobson for the MACS cohort and Ronald Bosch for the ACTG trials group; various datasets: Christl Donnelly, Steve Self, Thomas Skillman, and Daniel Meade
Footnotes
Conflicts of interest: None.
AUTHORS AND CONTRIBUTORS
The concept of this study was conceived by LJA. All authors contributed to the study design. RVB and LJA collected the HIV-1 plasma RNA viral load data. RVB constructed and managed the database and was responsible for collating the data. HJ conducted the statistical analyses. LJA conducted the mathematical modeling analyses and wrote the first draft of the paper. All authors contributed to the analysis, discussion of the results, and writing of the manuscript.
Disclose funding received for this work: Primarily, the Qatar National Research Fund (QNRF), a Qatar Foundation funded program (NPRP 08-068-3-024). RVB acknowledges also NCRR/NIH (5 KL2 RR025015) and CFAR/NIH (P30 AI027757) funding. Additional support was provided by the Fred Hutchinson Cancer Research Center and the HIV Vaccine Trials Network. Some of the data provided to this study were supported in part by the AIDS Clinical Trials Group funded by the National Institute of Allergy and Infectious Diseases (AI 68636, AI 38858, AI 68634, and AI 38855).
References
- 1.UNAIDS. [accessed 3 January 2012];UNAIDS 2011 World AIDS Day Report. 2011 (available at http://www.unaids.org/en/media/unaids/contentassets/documents/unaidspublication/2011/JC2216_WorldAIDSday_report_2011_en.pdf.
- 2.Pettifor AE, Levandowski BA, Macphail C, Miller WC, Tabor J, Ford C, et al. A tale of two countries: rethinking sexual risk for HIV among young people in South Africa and the United States. J Adolesc Health. 2011;49:237–243. e231. doi: 10.1016/j.jadohealth.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris M. Sexual networks and HIV. Aids. 1997;11:S209–S216. [PubMed] [Google Scholar]
- 4.Abu-Raddad LJ, Patnaik P, Kublin JG. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science. 2006;314:1603–1606. doi: 10.1126/science.1132338. [DOI] [PubMed] [Google Scholar]
- 5.Kaul R, Cohen CR, Chege D, Yi TJ, Tharao W, McKinnon LR, et al. Biological factors that may contribute to regional and racial disparities in HIV prevalence. Am J Reprod Immunol. 2011;65:317–324. doi: 10.1111/j.1600-0897.2010.00962.x. [DOI] [PubMed] [Google Scholar]
- 6.Fideli US, Allen SA, Musonda R, Trask S, Hahn BH, Weiss H, et al. Virologic and immunologic determinants of heterosexual transmission of human immunodeficiency virus type 1 in Africa. AIDS Res Hum Retroviruses. 2001;17:901–910. doi: 10.1089/088922201750290023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray RH, Wawer MJ, Brookmeyer R, Sewankambo NK, Serwadda D, Wabwire-Mangen F, et al. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet. 2001;357:1149–1153. doi: 10.1016/S0140-6736(00)04331-2. [DOI] [PubMed] [Google Scholar]
- 8.Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011;365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das M, Chu PL, Santos GM, Scheer S, Vittinghoff E, McFarland W, et al. Decreases in community viral load are accompanied by reductions in new HIV infections in San Francisco. PLoS One. 2010;5:e11068. doi: 10.1371/journal.pone.0011068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinn TC, Wawer MJ, Sewankambo N, Serwadda D, Li CJ, Wabwire-Mangen F, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. New England Journal of Medicine. 2000;342:921–929. doi: 10.1056/NEJM200003303421303. [DOI] [PubMed] [Google Scholar]
- 11.Lingappa JR, Hughes JP, Wang RS, Baeten JM, Celum C, Gray GE, et al. Estimating the impact of plasma HIV-1 RNA reductions on heterosexual HIV-1 transmission risk. PLoS One. 2010;5:e12598. doi: 10.1371/journal.pone.0012598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sim J, et al. AIDS 2000. Durban; 2000. Log10 differences between viral load by assays (bDNA vs. PCR) [Google Scholar]
- 14.Morgan D, Mahe C, Mayanja B, Whitworth JA. Progression to symptomatic disease in people infected with HIV-1 in rural Uganda: prospective cohort study. BMJ. 2002;324:193–196. doi: 10.1136/bmj.324.7331.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnabas RV, Webb EL, Weiss HA, Wasserheit JN. The role of coinfections in HIV epidemic trajectory and positive prevention: a systematic review and meta-analysis. Aids. 2011;25:1559–1573. doi: 10.1097/QAD.0b013e3283491e3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizzardini G, Piconi S, Ruzzante S, Fusi ML, Lukwiya M, Declich S, et al. Immunological activation markers in the serum of African and European HIV-seropositive and seronegative individuals. Aids. 1996;10:1535–1542. doi: 10.1097/00002030-199611000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Kalinkovich A, Weisman Z, Leng Q, Borkow G, Stein M, Greenberg Z, et al. Increased CCR5 expression with decreased beta chemokine secretion in Ethiopians: relevance to AIDS in Africa. J Hum Virol. 1999;2:283–289. [PubMed] [Google Scholar]
- 18.Cohen CR, Moscicki AB, Scott ME, Ma Y, Shiboski S, Bukusi E, et al. Increased levels of immune activation in the genital tract of healthy young women from sub-Saharan Africa. AIDS. 2010;24:2069–2074. doi: 10.1097/QAD.0b013e32833c323b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novitsky V, Ndung’u T, Wang R, Bussmann H, Chonco F, Makhema J, et al. Extended high viremics: a substantial fraction of individuals maintain high plasma viral RNA levels after acute HIV-1 subtype C infection. AIDS. 2011;25:1515–1522. doi: 10.1097/QAD.0b013e3283471eb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emery S, Bodrug S, Richardson BA, Giachetti C, Bott MA, Panteleeff D, et al. Evaluation of performance of the Gen-Probe human immunodeficiency virus type 1 viral load assay using primary subtype A, C, and D isolates from Kenya. J Clin Microbiol. 2000;38:2688–2695. doi: 10.1128/jcm.38.7.2688-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dyer JR, Kazembe P, Vernazza PL, Gilliam BL, Maida M, Zimba D, et al. High levels of human immunodeficiency virus type 1 in blood and semen of seropositive men in sub-Saharan Africa. J Infect Dis. 1998;177:1742–1746. doi: 10.1086/517436. [DOI] [PubMed] [Google Scholar]
- 22.Gras L, Jurriaans S, Bakker M, van Sighem A, Bezemer D, Fraser C, et al. Viral load levels measured at set-point have risen over the last decade of the HIV epidemic in the Netherlands. PLoS One. 2009;4:e7365. doi: 10.1371/journal.pone.0007365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller V, von Wyl V, Yerly S, Boni J, Klimkait T, Burgisser P, et al. African descent is associated with slower CD4 cell count decline in treatment-naive patients of the Swiss HIV Cohort Study. Aids. 2009;23:1269–1276. doi: 10.1097/QAD.0b013e32832d4096. [DOI] [PubMed] [Google Scholar]
- 24.Cuadros DF, Branscum AJ, Crowley PH. HIV-malaria co-infection: effects of malaria on the prevalence of HIV in East sub-Saharan Africa. Int J Epidemiol. 2011 doi: 10.1093/ije/dyq256. [DOI] [PubMed] [Google Scholar]
- 25.Boily MC, Baggaley RF, Wang L, Masse B, White RG, Hayes RJ, et al. Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies. Lancet Infect Dis. 2009;9:118–129. doi: 10.1016/S1473-3099(09)70021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donnell D, Baeten JM, Kiarie J, Thomas KK, Stevens W, Cohen CR, et al. Heterosexual HIV-1 transmission after initiation of antiretroviral therapy: a prospective cohort analysis. Lancet. 2010;375:2092–2098. doi: 10.1016/S0140-6736(10)60705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walson JL. Empiric Therapy of Helminth Co-infection to Reduce HIV-1 Disease Progression (THE or PHE) ClinicalTrial.gov. 2008 [Google Scholar]
- 28.Hayes R, Watson-Jones D, Celum C, van de Wijgert J, Wasserheit J. Treatment of sexually transmitted infections for HIV prevention: end of the road or new beginning? Aids. 2010;24 (Suppl 4):S15–26. doi: 10.1097/01.aids.0000390704.35642.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanton C, Abu-Raddad LJ, Weiss HA. Time to Refocus on HSV Interventions for HIV Prevention? J Infect Dis. 2011;204:1822–1826. doi: 10.1093/infdis/jir653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen RHN, Gange SJ, Serwadda D, Wabwire-Mangen F, Sewankambo KN, Wawer M, et al. Reduced odds of livebirth associated with HIV-RNA viral load: Rakai, Uganda. XIV International AIDS Conference (); Stockholm. Barcelona: International AIDS Society; 2002. p. 395. [Google Scholar]
- 31.Bruce-Chwatt LJ. A Longitudinal Survey of Natural Malaria Infection in a Group of West African Adults. West Afr Med J. 1963;12:199–217. [PubMed] [Google Scholar]
- 32.Rogier C, Ly AB, Tall A, Cisse B, Trape JF. Plasmodium falciparum clinical malaria in Dielmo, a holoendemic area in Senegal: no influence of acquired immunity on initial symptomatology and severity of malaria attacks. Am J Trop Med Hyg. 1999;60:410–420. doi: 10.4269/ajtmh.1999.60.410. [DOI] [PubMed] [Google Scholar]
- 33.Snow RW, Craig MH, Newton CR, Steketee RW. Working Paper No 11, Disease Control Priorities Project. Bethesda, Maryland: Fogarty International Center, National Institutes of Health; 2003. The public health burden of Plasmodium falciparum malaria in Africa: Deriving the numbers. http://mednet3.who.int/prioritymeds/report/append/610snow_wp11.pdf. [Google Scholar]
- 34.Kublin JG, Patnaik P, Jere CS, Miller WC, Hoffman IF, Chimbiya N, et al. Effect of Plasmodium falciparum malaria on concentration of HIV-1-RNA in the blood of adults in rural Malawi: a prospective cohort study. Lancet. 2005;365:233–240. doi: 10.1016/S0140-6736(05)17743-5. [DOI] [PubMed] [Google Scholar]
- 35.Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, et al. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abu-Raddad LJ, Magaret AS, Celum C, Wald A, Longini IM, Jr, Self SG, et al. Genital herpes has played a more important role than any other sexually transmitted infection in driving HIV prevalence in Africa. PLoS ONE. 2008;3:e2230. doi: 10.1371/journal.pone.0002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaffar S, Grant AD, Whitworth J, Smith PG, Whittle H. The natural history of HIV-1 and HIV-2 infections in adults in Africa: a literature review. Bull World Health Organ. 2004;82:462–469. [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen MS, Pilcher CD. Amplified HIV transmission and new approaches to HIV prevention. J Infect Dis. 2005;191:1391–1393. doi: 10.1086/429414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.