

Abstract
The observation that the T-bet transcription factor allows tissue-specific upregulation of intracellular osteopontin (Opn-i) in plasmacytoid dendritic cells (pDCs) suggests that Opn might contribute to the expression of interferon-α (IFN-α) in those cells. Here we show that Opn deficiency substantially reduced Toll-like receptor 9 (TLR9)–dependent IFN-α responses but spared expression of transcription factor NF-κB–dependent proinflammatory cytokines. Shortly after TLR9 engagement, colocalization of Opn-i and the adaptor molecule MyD88 was associated with induction of transcription factor IRF7–dependent IFN-α gene expression, whereas deficient expression of Opn-i was associated with defective nuclear translocation of IRF7 in pDCs. The importance of the Opn–IFN-α pathway was emphasized by its essential involvement in cross-presentation in vitro and in anti–herpes simplex virus 1 IFN-α response in vivo. The finding that Opn-i selectively coupled TLR9 signaling to expression of IFN-α but not to that of other proinflammatory cytokines provides new molecular insight into the biology of pDCs.
Increasing evidence that innate immune responses can determine both the type and intensity of adaptive immune responses has stimulated great interest in the underlying regulatory mechanisms. The recognition phase of innate responses depends on a series of pattern-recognition receptors, including Toll-like receptors (TLRs) expressed by dendritic cells (DCs), which detect a wide range of pathogen-associated molecules carried by bacteria and viruses1. Engagement of the TLR9 family of endosomal receptors by microbe-derived DNA triggers the production of large amounts of interferons α and β (IFN-αβ)2, key mediators that regulate the development of both innate and adaptive immunity3–6. However, the pathway initiated by TLR9 engagement that culminates in IFN-αβ production is not fully understood.
The main cell type responsible for robust production of IFN-αβ is the plasmacytoid DC (pDC), a specialized subset of DCs that can be distinguished from conventional DCs (cDCs) according to surface markers, including B220 (ref. 2). The engagement of TLR9 on pDCs by CpG oligonucleotides, which mimic microbial DNA, leads to production of IFN-αβ that in turn activates other cells, including cDCs, to elaborate cytokines, present antigen and promote the development of T helper type 1 (TH1) effector and memory cells3–6. Although production of IFN-αβ by pDCs augments cross-presentation by cDC7–9, the ability of pDCs themselves to cross-present proteins to CD8+ T cells has been not established.
Studies have indicated that robust IFN-αβ production by ligation of TLR9 on pDCs depends on interaction between the adaptor protein MyD88 and transcription factor IRF7, which is activated by the kinases IRAK4 and IRAK1 and the adaptor molecule TRAF6 in the signaling complex10,11. However, the unique ability of pDCs but not other cell types, including cDCs, to successfully use this IRF7-dependent pathway to express IFN-αβ after TLR9 ligation is not fully understood. Although pDCs can retain CpG ligands in TLR9-containing endosomal vesicles for longer periods of time than can cDCs12, that characteristic might not fully account for robust IFN-αβ expression by pDCs. Additional pDC-specific factors may be needed to fully endow this cell type with the capacity to produce IFN-α and IFN-β after TLR9-dependent signaling.
The phosphoprotein osteopontin (Opn) is essential for efficient development of TH1 immune responses13–15. Opn is expressed in activated T cells, DCs and macrophages within 24 h of viral or bacterial infection and may account for resistance to microbial infection13,16. Dysregulated expression of Opn, in contrast, has been associated with autoimmune disease in mouse models14 and in several types of human autoimmune disorders, including lupus nephritis, multiple sclerosis and rheumatoid arthritis17–22. Although most studies have focused on the activity of secreted Opn as a cytokine or chemokine14,15,23, a portion of newly synthesized Opn is retained as intracellular Opn (Opn-i) in the cytoplasm and distinct from secretory vesicles24–26. Given the early and decisive effect of Opn on type 1 immunity and evidence of its expression in activated DCs27,28, we investigated its possible involvement in mediating the specialized activation of DC subsets.
We found that engagement of TLR9 resulted in transcription factor T-bet–dependent induction of expression of the gene encoding Opn in pDCs but not cDCs. Moreover, intracellular expression of Opn in pDCs was required for TLR9-dependent expression of IFN-α but not that of other proinflammatory cytokines, as shown by studies of Opn-deficient and genetically reconstituted pDCs. Additional analyses indicated that localization of Opn-i together with the MyD88-containing signal transduction complex after TLR9 engagement was essential for efficient nuclear translocation of IRF7 and associated IFN-α gene expression. The biological importance of Opn in IFN-α responses was emphasized by the finding that Opn-deficient mice developed impaired IFN-α-dependent natural killer (NK) cell responses to tumors and reduced IFN-α responses after infection with herpes simplex virus 1 (HSV-1).
RESULTS
T-bet-dependent Opn expression in pDCs
The function of Opn expression in immunity has been studied mainly in macrophages, NK cells and T cells29–31. The findings that activated human DCs and mouse Langerhans cells express Opn28,32 prompted us to examine its expression in pDCs (B220+CD11clo) and cDCs (B220−CD11chi). Although both pDCs and cDCs express TLR9, CpG-dependent ligation elicited considerable expression of the gene encoding Opn in pDCs but not cDCs within 24 h, whereas ligation of CD40 or TLR4 did not enhance expression of the gene encoding Opn by either DC subset (Fig. 1a). A dose-response analysis of the effect of CpG (oligodeoxynucleotide 1668 (ODN-1668); CpG-B) on Opn RNA and protein expression confirmed that low concentrations of CpG (about 0.1 μg/ml) were sufficient to induce expression of the gene encoding Opn in pDCs (Fig. 1b).
Figure 1.
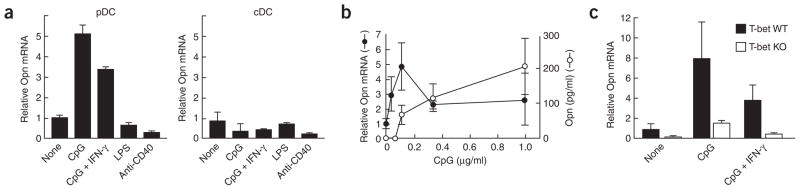
Opn expression in pDCs and cDCs 24 h after stimulation. (a) Opn mRNA in splenic pDCs (left) or cDCs (right) stimulated with 1 μg/ml of CpG-B (ODN-1668); 1 μg/ml of CpG-B plus 10 ng/ml of recombinant mouse IFN-γ protein; 1 μg/ml of lipopolysaccharide (LPS); or 5 μg/ml of anti-CD40. Relative Opn mRNA is based on Opn mRNA in pDCs without stimulation. (b) Opn mRNA and secreted protein in pDCs and culture supernatants, respectively. (c) Opn mRNA concentrations in T-bet wild-type (T-bet WT) and T-bet-deficient (T-bet KO) pDCs. Concentrations of CpG-B and recombinant mouse IFN-γ were 1 μg/ml and 10 ng/ml, respectively. Relative Opn mRNA is based on Opn mRNA in T-bet wild-type pDCs without stimulation. Opn mRNA and protein were measured by quantitative real-time PCR of cDNA and ELISA, respectively, with triplicate wells, and are presented with error bars. Data are representative of at least three independent experiments.
Opn expression in activated T cells depends on transcriptional regulation by T-bet14, an essential factor in TH1 lineage commitment33. Because T-bet is also expressed in pDCs after CpG stimulation34, we sought to determine whether this transcription factor might regulate Opn expression in pDCs. We found that T-bet was required for expression of the gene encoding Opn in pDCs after CpG-dependent activation (Fig. 1c). Reduced Opn expression in T-bet-deficient pDCs did not reflect the absence of a putative IFN-γ intermediary35, as provision of recombinant IFN-γ along with CpG did not reconstitute Opn expression by T-bet-deficient pDCs (Fig. 1c).
Opn and cytokine expression by CpG-activated pDCs
We next sought to determine whether defective expression of the gene encoding Opn might alter cytokine expression by pDCs after TLR9 engagement. A functional ‘signature’ of TLR9 engagement of pDCs is IFN-α expression. The IFN-α response of T-bet-deficient (Fig. 2a) and Opn-deficient pDCs (Fig. 2b) was impaired substantially. In contrast, neither Opn deficiency nor T-bet deficiency impaired production of the transcription factor NF-κB–dependent proinflammatory cytokines interleukin 6 (IL-6) and tumor necrosis factor (TNF; Fig. 2a, b).
Figure 2.

Cytokine production by pDCs. (a) IFN-α (left) and IL-6 (right) in 24-hour pDC culture supernatants of T-bet wild-type (T-bet WT) versus T-bet-deficient (T-bet KO) cells stimulated with various concentrations of CpG-B (ODN-1668). The pDCs were plated at a density of 1.0 × 106 cells/ml. (b) Comparison of secreted IFN-α protein, cellular IFN-α mRNA, secreted IL-6 and TNF (left to right) for Opn wild-type (Opn WT) and Opn-deficient (Opn KO) pDCs plated at a density of 1.0 × 106 cells/ml with CpG-B (1 μg/ml for mRNA samples; otherwise, concentrations along horizontal axes) and collected at 24 h. (c) Comparison of secreted IFN-α, IL-6 and TNF (left to right) for Opn wild-type (Opn WT) and Opn-deficient (Opn KO) pDCs after CpG-A stimulation. The pDCs (0.2 × 106 cells/ml) were plated with CpG-A (ODN-D19; concentration, horizontal axis) and were collected at 24 h for analysis. (d) IFN-α from 24-hour pDC culture supernatants (1.0 × 106 cells/ml) from Opn WT and Opn KO mice stimulated with imiquimod (R837). Protein and mRNA were measured by ELISA and real-time PCR in triplicate wells, except for c, for which the IFN-α ELISA was done in duplicate wells. Data are representative of two to three experiments.
We initially examined the response to ODN-1668 (CpG-B) rather than to ODN-D19 (CpG-A) because TLR9 expression in heterologous cells confers responsiveness to CpG-B but not to CpG-A, which may use additional interactions with non-TLR9 surface structures36,37. We then examined the more potent CpG-A (ODN-D19). Concentrations of CpG-A up to 10 μg/ml, which stimulated robust IFN-α responses by wild-type pDCs, failed to induce substantial responses by Opn-deficient pDCs, although IL-6 and TNF responses were relatively unimpaired (Fig. 2c). Engagement of the TLR9 family member TLR7 by imiquimod also induced efficient IFN-α responses by pDCs (Fig. 2d), whereas the response of Opn-deficient pDCs was reduced by approximately 75% (Fig. 2d). Impaired IFN-α production by Opn-deficient pDCs did not reflect a generalized resistance to activation after TLR9 ligation, as inhibition of cytokine expression was selective and Opn-deficient and wild-type pDCs had similar increases in expression of major histocompatibility complex classes I and II, CD80, CD86 and CD40 after CpG activation both in vitro and in vivo (Supplementary Fig. 1 online).
Intracellular Opn and the induction of IFN-α
To gain insight into the mechanism underlying Opn-dependent IFN-α cytokine expression production, we sought to determine whether this effect was mediated by secreted Opn (extracellular) or Opn-i. Involvement of secreted Opn was unlikely, because the addition of recombinant Opn to cultures of Opn-deficient pDCs did not enhance IFN-α production and the addition of Opn-neutralizing antibodies to cultures of pDCs with wild-type Opn did not inhibit IFN-α production (Supplementary Fig. 2 online). Opn-i can regulate the cellular migration and motility of macrophages and osteoblasts and is distinct from secreted Opn, as demonstrated by pulse-chase experiments and confocal microscopy24–26. To evaluate the possible function of Opn-i in pDCs, we reconstituted Opn-deficient pDCs by infecting them with a lentivirus expressing either full-length Opn or a mutant lacking the signal sequence to prevent targeting to secretory vesicles. Supernatants of Opn-deficient pDCs reconstituted with full-length Opn contained concentrations of Opn that increased in direct proportion to the full-length Opn lentivirus titer used to infect the cells, whereas supernatants of cells infected with lentivirus expressing mutant Opn had much less Opn at 6 h (Fig. 3a) and 24 h (data not shown). However, infection with mutant Opn lentivirus reconstituted IFN-α responses more efficiently than did infection with full-length Opn lentivirus, as indicated by dose-response comparison of the two vectors (Fig. 3b). Additional analysis showed that increasing intracellular concentrations of Opn up to 1 fg/cell resulted in a roughly proportional increase in IFN-α secretion (Fig. 3c). Notably, however, expression of Opn in cDCs was not sufficient to allow robust IFN-α responses to CpG-A (data not shown), suggesting that this DC subset lacks an additional element required for robust IFN-α responses. Robust IFN-α responses of pDCs may reflect, in part, the ability of these cells to retain TLR9 ligands in endosomal vesicles for more time than do cDCs12. Although inclusion of CpG-B (ODN-1668) in cationic lipids such as DOTAP (N-[1-(2,3-dioleoyloxy)propyl]-N, N, N-trimethylammonium methyl-sulfate) can enhance endosomal retention time12, DOTAP in complex with CpG-B or CpG-A failed to restore the IFN-α responses of Opn-deficient pDCs after they were pulsed with DOTAP–CpG-A or DOTAP–CpG-B (Fig. 3d).
Figure 3.

Intracellular Opn is required for IFN-α production. (a–c) Lentivirus infection of pDCs stimulated with CpG-B (0.2 μg/ml). (a) Extracellular Opn protein measured by ELISA in 6-hour culture supernatants of sorted splenic Opn-deficient pDCs infected with lentivirus to express full-length Opn (fOpn), mutant Opn (ΔOpn) or GFP control (GFP). HIV capsid protein p24, determined by ELISA, indicates the concentration of lentivirus used for infection (horizontal axis). (b) IFN-α in 24-hour culture supernatants of splenic Opn-deficient pDCs transfected with lentiviral reagents (GFP, ΔOpn and fOpn). (c) IFN-α (left vertical axis) or intracellular Opn (right vertical axis) in culture supernatants and cells. ‘Titration’ of mutant Opn lentivirus (ΔOpn-lenti) into bone marrow–derived Opn-deficient pDCs (1 × 106 cells/ml) was followed by CpG-B stimulation (ODN-1668; 0.2 μg/ml), then analysis 24 h later. (d) IFN-α production in 24-hour culture supernatants of Opn wild-type (WT) and Opn-deficient (KO) bone marrow–derived pDCs after a 20-minute pulse with DOTAP in complex with CpG-B (ODN-1668; left) or CpG-A (ODN-D19) with (+) or without (−) DOTAP (right). Data are representative of at least three independent experiments.
Analyses of Opn-dependent responses to CpG
MyD88-IRF7–mediated signaling is responsible for the induction of IFN-α after TLR9 engagement. As the intracellular localization of Opn is unrelated to secretory granules and Opn-i is distinct from the secreted form, according to pulse-chase experiments, Opn may associate with components of the TLR9-MyD88–associated signaling complex, leading to IRF7 nuclear translocation and IFN-α gene expression12. We first investigated this issue using a biochemical approach (Fig. 4) and then confirmed it microscopically (Fig. 5). We found that CpG-dependent activation of Opn wild-type but not Opn-deficient bone marrow–derived DCs was associated with a substantial increase in nuclear IRF7 (Fig. 4a), whereas IRF7 protein concentrations in total cell lysates were similar for Opn wild-type and Opn-deficient cells. IRF7 in wild-type nuclear extracts appeared as a doublet including an upper band that has been associated with phosphorylation after CpG treatment. Defective Opn-i expression quenched IFN-α production and IRF7 nuclear translocation but had little or no effect on the expression of NF-κB-dependent proinflammatory cytokines, including IL-6 and TNF (Fig. 2b). We therefore analyzed the activation status of NF-κB in nuclear lysates from bone marrow–derived DCs according to chemiluminescence detection of the binding of NF-κB to DNA oligonucleotides containing the relevant consensus binding sequence. There were no differences in active NF-κB in lysates from Opn wild-type versus Opn-deficient (Fig. 4b), confirming the selective action of Opn-i on IRF7-dependent IFN-α expression but not NF-κB-dependent proinflammatory cytokine responses.
Figure 4.

Biochemical analysis of Opn-i. (a) Nuclear translocation of IRF7 in total bone marrow–derived Opn wild-type (WT) and Opn-deficient (KO) DCs treated with CpG-B (0.4 μM) for 3 h before extraction of nuclear and total lysates. (b) Activation of NF-κB in total bone marrow–derived DCs stimulated for 1 h with 0.3 μM CpG-B. Control competitor and noncompetitor oligonucleotides were added to some samples (+, below graph) to confirm sequence-specific binding of NF-κB in lysate samples. (c) Immunoblot (IB) of protein complexes immunoprecipitated (IP) from HEK293 cells cotransfected for 48 h with Opn, Flag-MyD88 and TLR9 (lane 1) or Opn, Flag-Mink (serine kinase) and TLR2 (lane 2; negative control) followed by stimulation for 3 h with 0.3 μM CpG-B. (d) Opn expression and Ifna4 activation in HEK293 cells transiently transfected with mutant Opn–expressing lentivirus (concentrations, below graph) along with IRF7 and/or MyD88 cDNA. (e) MyD88-dependent Ifna4 activation after transfection of MyD88 expression vector (concentrations, below graph) into HEK293 cells along with mutant Opn and/or IRF7. Data are representative of at least three independent experiments.
Figure 5.

Localization of Opn together with MyD88 and TLR9. (a, b) Immuno-fluorescence and confocal microscopy of Opn (green) and MyD88 (red; a) or TLR9 (red; b) in pDCs. After treatment of pDCs with 0.5 μM of CpG-B for 10 min, cells were fixed and Opn, MyD88 and TLR9 were detected with mouse anti-Opn (2A1), rabbit anti-MyD88 and rabbit anti-TLR9, followed by secondary antibodies. Original magnification, ×400 (first three columns) and ×800 (single-cell images, far right).
To further define the relationship of Opn to TLR9 and MyD88, we expressed Opn together with Flag-tagged MyD88 and TLR9 in HEK293T cells. We found that MyD88 precipitated together with Opn. This result was unlikely to reflect a nonspecific concentration-dependent effect, because Opn was similar in experimental and control lanes in immunoblots, as were Flag-MyD88 and the control ligand Flag-Mink (Fig. 4c). To extend that biochemical analysis, we determined localization of Opn together with MyD88 and TLR9 in pDC by immunofluorescence and confocal microscopy. At 10 min after activation by 0.5 μM CpG ODN-1668, Opn localized together with both MyD88 (Fig. 5a) and TLR9 (Fig. 5b).
Those data suggested that Opn was more likely to interact with the TLR9-MyD88 signaling complex, leading to IRF7-dependent activation of IFN-α, rather than enhancing the efficiency of endosomal trafficking of TLR9 ligands. To further evaluate that hypothesis, we determined the effect of Opn expression on IRF7-dependent activity of the Ifna4 promoter in HEK293T cells. Expression of Opn resulted in a dose-dependent increase in Ifna4 promoter activity that depended fully on coexpression of IRF7 (Fig. 4d). Additional analysis indicated that coexpression of MyD88 was required for optimal Opn-dependent induction of Ifna4 promoter activity (Fig. 4e).
pDCs require Opn for efficient cross-presentation
The uptake and processing of exogenous antigen in the major histocompatibility complex class I pathway may contribute to protective immunity against viruses and tumors. Although IFN-α production by pDCs may enhance cross-presentation by cDCs or mixed DC populations7,38,39, whether pDCs themselves can cross-present antigen has remained an open issue. A pDC population of more than 99% purity (B220+CD11cloCD3ε−CD19−NK1.1−) obtained after repeated sorting by flow cytometry (Fig. 6a) showed efficient Opn-dependent cross-presentation of ovalbumin (OVA; Fig. 6b). In contrast, Opn deficiency did not affect peptide presentation to CD8+ T cells (Fig. 6c). Cross-presentation by pDCs was dependent on TAP1 (transporter associated with antigen processing 1; Fig. 6d, left and middle), whereas TAP1 deficiency did not affect peptide presentation to CD8+ T cells (Fig. 6d, right). Although bone marrow–derived pDCs may acquire myeloid DC–like features after virus stimulation, as judged by upregulated surface CD11b expression40, this was not the case for CpG-activated pDCs (Supplementary Fig. 1). Finally, defective cross-presentation by Opn-deficient pDCs was associated with a defective IFN-α response during antigen uptake (Fig. 6e).
Figure 6.

In vitro OVA cross-presentation by pDCs. (a) Purity of splenic CD11c+ cells (B220+CD11c+CD3ε−CD19−NK1.1−) after repeated sorting by flow cytometry. (b) OVA cross-presentation by Opn wild-type (WT) or Opn Opn-deficient (KO) pDCs. Purified bone marrow–derived pDCs were first cultured for 24 h with (+ CpG) or without (− CpG) 0.2 μg/ml of CpG-B in the presence of soluble OVA (50 μg/ml), then OT-I T cell proliferation was measured. (c) OT-I peptide presentation by Opn WT or Opn KO pDCs. Purified splenic pDCs were cultured for 18 h with (+) or without (−) 0.2 μg/ml of CpG-B in presence of OT-I peptide (1 μg/ml). (d) OVA cross-presentation (left and middle) and OT-I peptide direct presentation (right) by purified splenic pDCs from B6 wild-type mice (WT) and B6 mice deficient in transporter associated with antigen processing (TAP1-deficient). The pDCs were treated for 20 h with 50 μg/ml of OVA or 1 μg/ml of OT-I peptide with 0.2 μg/ml of CpG-B, then OT-I T cell proliferation and IFN-γ production were assessed. (e) IFN-α production by pDCs incubated with OVA and CpG (0.2 μg/ml of CpG-B) before culture together with OT-I T cells. Data are representative of at least four independent experiments.
Opn-dependent cross-presentation by pDCs is mediated by IFN-α
We noted that providing IFN-α to CpG-stimulated Opn-deficient pDCs during antigen uptake (that is, before washing pDCs to set up culture together with OT-I CD8+ T cells) remedied the Opn-deficient phenotype and allowed efficient cross-presentation (Fig. 7a). Moreover, adding IFN-α to resting pDCs, which are unable to cross-present antigen, replaced the need for CpG-dependent activation and allowed these cells to cross-present antigen to CD8+ T cells, suggesting that the TLR9 signals are dispensable for ‘licensing’ of pDCs (Fig. 7b) and cDCs (Fig. 7c) in the presence of IFN-α. Consistent with that hypothesis, recombinant IFN-α (1,000 U/ml) failed to ‘license’ IFN-αβ receptor–deficient pDCs to cross-present OVA antigen (Supplementary Fig. 3 online).
Figure 7.

IFN-α-dependent cross-presentation. (a, b) Purified bone marrow–derived Opn-deficient (Opn KO) and Opn wild-type (Opn WT) pDCs were treated with or without CpG-B (0.2 μg/ml) and/or recombinant IFN-α (rIFN-α; 1 × 108 to 5 × 108 units = 1 mg; key) in the presence of OVA (50 μg/ml). OT-1 T cell proliferation was measured. (a) Supplementary effect of recombinant IFN-α in antigen cross-presentation by CpG-treated pDCs. (b) Treatment with recombinant IFN-α without CpG is sufficient to ‘license’ pDCs for antigen cross-presentation. (c) ‘Licensing’ of cDCs from Opn KO and Opn WT mice after treatment of cells with (100 U/ml; key) or without (0 U/ml; key) recombinant IFN-α. Data are representative of at least three independent experiments.
As anticipated from those data, the addition of recombinant Opn to culture supernatants or the neutralization of extracellular Opn (Supplementary Fig. 4 online) did not alter the ability of pDCs to cross-prime CD8+ T cells. In contrast, lentiviral transfection of Opn enhanced the capacity of the pDCs to cross-prime and was associated with increased IFN-α after infection with Opn-expressing lentivirus (Supplementary Fig. 4). These findings indicate that cross-presentation by pDC requires Opn-i-dependent expression of IFN-α after TLR9-dependent activation.
Development of anti-HSV-1 responses in Opn-deficient mice
We tested the physiological importance of those findings in relation to mouse anti-HSV-1 responses. The IFN-α response of Opn-deficient pDCs exposed to ultraviolet irradiation–treated HSV-1 was impaired substantially (Fig. 8a). Moreover, in contrast to the response of Opn wild-type mice, the IFN-α response of Opn-deficient mice 48 h after HSV-1 infection was nearly abolished and the response of TLR9-deficient mice was substantially reduced (Fig. 8b). Defective IFN-α responses were associated with a failure of Opn-deficient mice to develop significant anti-HSV-1 delayed-type hypersensitivity responses after challenge with ultraviolet irradiation–treated HSV-1 (Fig. 8c).
Figure 8.
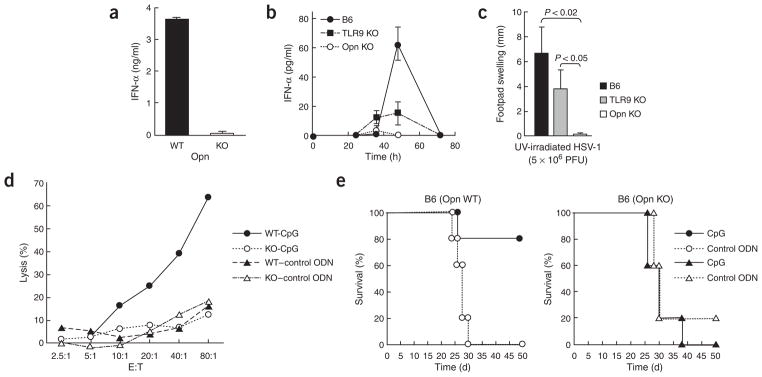
Opn-dependent in vivo response to HSV-1 infection. (a) IFN-α concentrations from Opn wild-type (WT) and Opn-deficient (KO) pDC culture supernatants after incubation in vitro with ultraviolet irradiation–treated (UV-irradiated) HSV-1 for 24 h. (b) Serum IFN-α in B6, B6 TLR9-deficient (TLR9 KO) and B6 Opn-deficient (Opn KO) mice injected intraperitoneally with HSV-1 (1 × 106 PFU/mouse). Data represent three mice per group. (c) HSV-1-specific delayed-type hypersensitivity responses in B6, TLR9 KO and Opn KO mice injected intraperitoneally with HSV-1 (5 × 106 PFU/mouse) and challenged 6 d later in the left footpad with ultraviolet irradiation–treated HSV-1 (1 × 105 PFU/mouse). Error bars indicate mean ± s.d. of three mice per group. (d) 51Cr-release assay of NK cells from B6 Opn wild-type (WT) and Opn-deficient (KO) mice, evaluated against YAC-1 cells. Draining lymph nodes of mice injected with CpG (ODN-1585; -CpG) or control GpC (–control ODN) were pooled. Data are representative of at least three independent experiments. (e) Survival of Opn WT and Opn KO mice injected intraperitoneally with 5 × 103 B16 cells plus 100 μg CpG (ODN-1585) or control GpC (control ODN). There were five mice in each group.
IFN-α-dependent natural killer cell responses require Opn
NK cells are known to be activated by IFN-α produced by pDCs2,41. We also tested the in vivo importance of the observations outlined above based on published findings that inoculation of mice with CpG elicits an IFN-α-dependent NK cell cytotoxic response that protects against tumor growth42. Inoculation of Opn-deficient mice with CpG failed to elicit either enhanced NK cell activity (Fig. 8d) or protection from the lethal growth and dissemination of B16 melanoma cells (Fig. 8e), in contrast to the vigorous NK cell responses and associated antitumor protection found in Opn wild-type mice after CpG inoculation (Figs. 8d, e).
DISCUSSION
Although there is good evidence that engagement of TLR9 by unmethylated CpG oligonucleotides elicits robust expression of the canonical pDC cytokine IFN-α, the biochemical events underlying that process are not fully understood. The observation that the T-bet transcription factor allowed upregulation of Opn in pDCs indicated the possibility that Opn might contribute to the IFN-α expression pathway. Although Opn deficiency did not affect TLR9-dependent expression of NF-κB-dependent proinflammatory cytokines (including IL-6 and TNF), IFN-α responses were almost completely abolished. Defective IFN-α responses reflected an acute absence of Opn rather than an Opn-dependent developmental defect of pDCs, as lentivirus-mediated expression of Opn in Opn-deficient pDCs restored TLR9-dependent IFN-α responses.
An intracellular form of Opn (mutant Opn lacking a signal sequence) was at least as effective as full-length Opn in reconstituting IFN-α production by pDCs, consistent with the findings that provision of extracellular Opn failed to reconstitute Opn-deficient DCs and neutralization of extracellular Opn did not impair IFN-α production by pDCs. Although much attention has been given to the biological activity of Opn secreted by activated T cells and other cell types, the findings reported here have shown that the intracellular form of Opn found in cytoplasm24–26 is biologically active when expressed in pDCs. Although Opn-i has been linked to the motility and cell fusion of macrophages, fibroblasts and osteoclasts24,25, the data provided here indicate that lineage-specific expression of Opn-i in pDCs is important in the differentiated function of these cells. The basis for the localization of Opn to endosomal membranes of pDCs shortly after CpG-dependent engagement of TLR9 has not been fully determined. It may be relevant that Opn contains several dileucine motifs, including one (SQIKRLLSE) similar to a dileucine motif found in mouse CD4, that targets the latter to lysosomal and endosomal membranes and is regulated by phosphorylation of the upstream serine43. As Opn-i can be highly phosphorylated26 by several serine-threonine kinases, phosphorylation-dependent or functional changes in Opn after TLR9 engagement may regulate both the localization of Opn-i and its potential interactions with other members of the MyD88-IRF7 signaling complex.
Biochemical analysis of heterologous HEK293T cells transfected with relevant expression vectors indicated that shortly after TLR9 engagement, Opn localized together with a complex that included MyD88, whereas confocal microscopy indicated that Opn localized together with MyD88 and TLR9. Those interactions are functionally meaningful, as TLR9 engagement of Opn-deficient pDCs failed to induce nuclear translocation of IRF7, and Ifna4 promoter activity in heterologous cells was enhanced by Opn in a dose-dependent way when that IRF7 was coexpressed. The synergistic interaction between Opn and IRF7 was enhanced by overexpression of MyD88 (we did not directly evaluate the effect of endogenous concentrations of (human) MyD88 on the response).
There is good evidence that a TLR9-linked MyD88 complex containing TRAF6, IRAK1 and IRAK4 (also called the ‘cytoplasmic transductional–transcriptional processor’44) is required for the phosphorylation and nuclear translocation of IRF7 and robust upregulation of IFN-α in pDCs but not cDCs10. The capacity of pDCs to mediate robust IFN-α responses after activation reflects several cell-specific features, including efficient endosomal connections to TLR9-containing ligands and Opn-i-dependent enhancement of IRF7-dependent IFN-α gene expression. Additional pathways leading to IFN-α expression may allow both cDCs and Opn-deficient pDCs to express type I interferons after cell-cell interactions between non-TLR9 surface molecules engaged at high concentrations of activated DCs.
These findings suggest a molecular mechanism for skewing TLR9 signals toward IRF7 rather than IRF5–NF-κB45, which would allow for robust expression of IFN-α but not proinflammatory cytokines such as TNF. Titration of lentiviral-based expression of Opn-i indicated that an increase in intracellular concentration of Opn-i was needed to support robust IFN-α responses. That finding opens the possibility that the intensity and duration of Opn-i induction by microbial or mammalian DNA complexes in the environment regulate the amount of IFN-α gene expression by pDCs.
The contribution of an Opn–IFN-α axis to the antigen-presenting activity of pDCs was supported by data showing that Opn-dependent expression of IFN-α ‘licensed’ pDCs for cross-presentation after TLR9 ligation. IFN-α and or CpG can induce cross-presentation by heterogeneous DCs in vitro or heterogeneous antigen-presenting cells in vivo7,46–48. The finding that IFN-α was sufficient to ‘license’ both pDC and cDC cross-presentation suggests that experimental examples of TLR9-independent induction of cross-presentation by pDCs may exploit a cytokine milieu rich in IFN-α. Those findings also account for the ability of recombinant IFN-α to stimulate cross-priming in vivo without TLR9 engagement, provided that DCs express IFN-αβ receptors7,46,48. Published studies have indicated that Opn expression is an essential component of type 1 immune responses after HSV-1 infection13. Opn-deficient mice failed to develop substantial IFN-α responses shortly after HSV-1 infection and did not mount delayed-type hypersensitivity responses after challenge with HSV-1; thus, impaired expression of IFN-α after HSV-1 infection of Opn-deficient mice may be responsible for those abnormal phenotypes. The route and type of viral infection may determine the contribution of TLR9-Opn–dependent IFN-α expression, as RNA viruses such as influenza can induce MyD88-independent IFN-α responses49 that may bypass Opn-dependent immune mechansisms50.
There is considerable evidence linking dysregulated IFN-αβ expression to the pathogenesis of systemic lupus erythematosus6, although this connection is not straightforward5,51,52. Increased serum IFN-α and expression of a characteristic set of IFN-α-inducible genes in blood cells have been associated with systemic lupus erythematosus, and a subset of these IFN-α-induced genes correlates with disease progression53. Treatment with IFN-α results in the production of antinuclear and anti–double-stranded DNA antibodies in a subgroup of patients and, in an animal model, genetic deficiency in IFN-αβ receptors attenuates the development of mouse lupus54. However, the contribution of type I interferon (IFN-αβ) to systemic lupus erythematosus is complex and may be affected by interactions with other cytokines5. For example, subsets of disease characterized by pathogenic amounts of type II interferon (IFN-γ) responses may worsen after experimental manipulations that decrease reception of type I interferon–mediated signals, possibly reflecting compensatory increases in type II interferon51. The studies reported here suggest that Opn may exert a ‘double-barreled’ effect on expression of IFN-α and IFN-γ: T cell–dependent elaboration of secreted Opn can promote increased TH1–dependent IFN-g, whereas pDC expression of Opn-i can promote increased IFN-αβ. ‘Dissection’ of the separate and combined effects of these two Opn-dependent pathways may provide new insight into the involvement of interferons in immunity and autoimmune disease.
METHODS
Animals and reagents
Opn-deficient (Spp1−/−) mice55, a gift from D. Denhardt and S. Rittling (Rutgers University, New Brunswick, New Jersey), were back-crossed to C57BL/6 (B6) mice for 15 generations. T-bet-deficient (Tbx21−/−) mice33 were backcrossed to B6 mice for seven generations. B6, BALB/c, B6 Rag2−/−, B6 Tap1−/− and B6 OT-I TCR-transgenic mice were obtained from The Jackson Laboratory. B6 TLR9-deficient mice were provided by S. Akira (Osaka University, Osaka, Japan) and D. Golenbock (University of Massachusetts Medical Center, Worcester, Massachusetts). All work involving animals was done in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care. CpG DNA (ODN-1668 and ODN-D19) and control ODN (GpC) were synthesized with phosphorothioated modifications, as described12,56. CpG DNA (ODN 1585) and control GpC DNA (ODN 2118) had phosphorothioation at various positions as described42. The B16-Flt3L melanoma cell line, engineered to generate ligand for the receptor tyrosine kinase Flt3 (Flt3L), was a gift from G. Dranoff (Dana-Farber Cancer Institute, Boston, Massachusetts). Recombinant mouse Opn, IFN-α and IFN-γ proteins were obtained from R&D Systems, Cell Sciences and BD Pharmingen, respectively. Antibodies to Opn were a gift from D.T. Denhardt (2A1 and 2C5; Rutgers University, New Brunswick, New Jersey) or were purchased from R&D Systems (AF808). DNA constructs for TLR4, TLR9 and Flag-MyD88 were a gift from R. Medzihtov (Yale University, New Haven, Connecticut), and the Flag-MINK construct has been described57. Imiquimod (R837) was purchased from InvivoGen.
DC subset purification
Splenic DC samples obtained from mice 2 weeks after subcutaneous injection on the back with 2 × 106 to 5 × 106 B16-Flt3L cells were enriched for the CD11c+ population using CD11c MACS beads (Miltenyi). Selected cells were stained with phycoerythrin-conjugated anti-B220, fluorescein isothiocyanate–conjugated anti-CD11c, biotin-conjugated anti-CD3ε, anti-CD19 and anti-NK1.1, followed by CyChrome-conjugated streptavidin (all from Phamingen). For ‘gating out’ of dead cells, 7-amino-actinomycin D was added. The pDCs (B220+CD11c+CD3ε−CD19−NK1.1−7-AAD−) and cDCs (B220−CD11c+CD3ε−CD19−NK1.1−7-AAD−) were purified by cell sorting (FACSAria; BD Biosciences). Bone marrow–derived DCs were prepared as described58. A total of 2 × 106 bone marrow cells/ml were cultured in RPMI complete medium with 100 ng/ml of recombinant Flt3L (R&D Systems). Half of the culture medium was replaced on day 5 and cells were collected on day 9 or day 10, were stained with antibodies and then were sorted by a cell sorter with markers described above.
OT-I TCR-transgenic CD8+ T cell purification
Cells from spleens and lymph nodes were collected from B6 OT-I TCR-transgenic mice. Samples were enriched for OT-I cells by depletion of other cells using rat antibodies to CD19, B220, Mac-1, Gr-1, DX5 and CD4 (Pharmingen) and sheep anti-rat beads (Dynal). Purity was checked by flow cytometry and the Vβ5+CD8+ double-positive population was more than 90% pure.
Enzyme-linked immunosorbent assay (ELISA)
Except for Opn analysis, ELISA kits were used according to the manufacturers’ protocols (IFN-α, Endogen; IFN-γ, TNF and IL-6, BD Pharmingen; and HIV p24, Perkin Elmer). The Opn ELISA was done as described14. Assays were done in triplicate unless stated otherwise. Error bars in ELISA figures indicate mean ± s.e.m.
RNA and cDNA preparation and real-time PCR
Total RNA was extracted from cells with an RNeasy kit (Qiagen). Synthesis of cDNAs was primed with oligo(dT) followed by synthesis by Moloney murine leukemia virus reverse transcriptase (Ambion). The cDNA was analyzed by real-time PCR with an ABI 7700 (Applied Biosystems). QuantiTect SYBR Green PCR (Qiagen) was used to detect expression of Spp1 (encoding Opn) and Ifna, with Actb (encoding β-actin) as an internal control. Primers were as follows: Opn forward, 5′-GCCTGTTTGGCATTGCCTCCTC-3′, and reverse, 5′-CACAGCATTCTGTGGCGCAAGG-3′; β-actin forward, 5′-TGTTACCAACTGGGACGACA-3′, and reverse, 5′-CTGGGTCATCTTTTCACGGT-3′; primer sequences for detecting subsets of IFN-α were obtained from Clontech. Error bars indicate the maximum and minimum values calculated from s.d. and negative change in cycling threshold (−ΔΔCT) values from triplicate PCR as described in the Applied Biosystems manuals.
Construction of lentiviral Opn expression vectors and infection of pDCs
IRES2-GFP was inserted into the pLenti6/V5 plasmid (Invitrogen) as described59 and was used as a control green fluorescent protein (GFP) vector. Full-length (wild-type, secreted) Spp1 (Opn) cDNA was generated by RT-PCR from B6 splenic DC mRNA with Moloney murine leukemia virus reverse transcriptase (Ambion) and the Expand High Fidelity PCR System (Roche). XhoI and BamHI sites were introduced into the PCR primers for cloning into the pLenti-IRES-GFP vector. For intracellular accumulation of Opn, the truncated Spp1 (Opn) vector without a signal sequence was generated by PCR-mediated mutagenesis, deleting codons 1–15 with the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). The accuracy of both full-length Spp1 (Opn) and deleted Spp1 (Opn-i) was confirmed by DNA sequencing. Lentiviral stocks were generated by transfection of HEK293 cells with lentivirus packaging constructs (Invitrogen) using LipofectAmine 2000 (Invitrogen). Viral supernatants were collected 72 h later. Cells were infected with lentivirus in the presence of 5 μg/ml of polybrene by 30 min centrifugation at 1,300g followed by 3 h at 37 °C followed by replacement of three quarters of fresh medium with or without CpG. ELISA was used for the titration of lentiviral particles for the HIV p24 capsid protein (1 ng/ml p24 is about 1 × 103 to 1 × 105 titer units; Perkin-Elmer).
DOTAP treatment of CpG
CpG (3 μg) was mixed with DOTAP (15 μg; Roche) in 12 μl of PBS for a total ‘cocktail’ volume of 30 μl. The DOTAP-CpG ‘cocktail’ was incubated for 15 min at 25 °C and then was added to pDC culture supernatants at various concentrations of CpG. For the DOTAP-CpG ‘pulse’, CpG in complex with DOTAP was added at various concentrations to pDC culture supernatants and cells were incubated for 20 min at 37 °C. After three washes, cells were plated in complete medium.
Immunoprecipitation and immunoblot
HEK293 cells were transfected with various vectors using Lipofectamine 2000 (Invitrogen). Then, 48 h later, cells were activated for 3 h with 0.3 μM CpG (ODN-1668) and were collected in lysis buffer including 1% Triton-X. Immunoprecipitation used 2 μg/ml of monoclonal anti-Flag (Sigma) or 2 μg/ml of monoclonal anti-Opn (2A1) and anti-mouse IgG Sepharose beads (eBioscience). Precipitated complexes were separated by SDS-PAGE and were blotted onto nitrocellulose membrane, followed by immunoblot with rabbit anti-TLR9 (eBioscience), mouse mono-clonal anti-Flag (Sigma) or mouse monoclonal anti-Opn and horseradish peroxidase–conjugated secondary antibodies and chemiluminescence (ECL; Amersham). IRF7 nuclear translocation was detected in nuclear lysates from total bone marrow–derived DCs prepared as described60. Lysate samples were separated by SDS-PAGE, were transferred to nitrocellulose and were probed with 0.2 μg/ml of rabbit anti-IRF7 (Zymed) followed by with horse-radish peroxidase–conjugated anti-rabbit IgG (secondary antibody) and enhanced chemiluminescence.
Ifna4 promoter reporter assay
HEK293 cells were seeded on 24-well plates at a density of 1.2 × 105 cells/well before being transiently transfected with 250 ng of reporter plasmid pNiFty2-IFA-SEAP (InvivoGen) together with various expression plasmids. Total amounts of DNA were kept constant by supplementation with empty control vector lacking Spp1 (Opn) cDNA sequence. After 21 h, cells were collected and ‘SEAP’ (secreted form of embryonic alkaline phosphatase) activity was measured with the QUANTI-Blue system (Invivo-Gen). The internal control was β-galactosidase activity expressed by the pCMVβ vector (Clontech).
NF-κB detection assay
Active NF-κB in total bone marrow–derived DC nuclear lysates was measured by chemiluminescence ELISA-based assay with the NF-κB p65 Transcription Kit (Pierce). Nuclear lysates were obtained from bone marrow–derived DCs activated for 1 h with 0.3 μM CpG (ODN-1668). Lysates were added to assay plate wells bound to NF-κB target DNA fragments. Active NF-κB bound to the consensus sequence was incubated with primary antibody to NF-κB p65, then with a secondary horseradish peroxidase–conjugated antibody. Chemiluminescence detection was used to quantify the NF-κB in lysates. Results are presented as relative light units. The competitor duplex included the GGGACTTTCC motif for NF-κB binding; the noncompetitor duplex had the following nucleotide substitutions (underlined): GCCACTTTCC.
Immunofluorescence
After pDCs were obtained from total Flt3L bone marrow–derived DCs by bead purification (negative selection to deplete samples of CD3+CD19+NK1.1+ cells and then positive selection to obtain B220+ pDCs), purity was confirmed by flow cytometry. The pDCs were treated for 10 min with 0.5 μM CpG (ODN-1668) and were fixed with 4% paraformaldehyde. Opn, MyD88 and TLR9 were detected with mouse anti-Opn (2A1), rabbit anti-MyD88 (Sigma) and rabbit anti-TLR9 (eBioscience), respectively, followed by secondary Alexa Fluor 488–conjugated anti-mouse IgG (Opn) or Alexa Fluor 546–conjugated anti-rabbit IgG (MyD88 and TLR9; Molecular Probes). Nuclei were stained with Hoechst 33258 (Molecular Probes). Stained cells were viewed with a confocal microscope (Zeiss LSM 510) with a 63× objective lens.
In vitro cross-presentation assay
Purified DC subsets at a density of 5 × 105 cells/well in a 96-well U-bottom plate were treated for 16–24 h with 50 μg/ml of soluble OVA (purity, 99%; Sigma) for cross-presentation and with or without 0.2 μg/ml of CpG (ODN-1668). For peptide antigen presentation, 1 μg/ml of SIINFEKL peptide was used instead of OVA. After being washed three times, various numbers of DCs were cultured together with 1 × 105 OT-I CD8+ T cells/well in U-bottomed 96-well plates. Cells were pulsed with 5 μCi/well of [3H]thymidine on day 3 (1 μCi/well for peptide presentation) and then were collected 17–24 h later. T cell proliferation was evaluated by incorporation of [3H]thymidine from triplicate wells. IFN-γ secretion at day 3 was evaluated with an IFN-γ ELISA kit (BD Pharmingen).
HSV-1 infection
B6 mice were infected intraperitoneally with ultraviolet irradiation–treated HSV-1 (KOS; 5 × 106 plaque-forming units (PFU)/mouse) before mouse sera from peripheral blood was collected by retro-orbital bleeding and was assessed by ELISA. Delayed-type hypersensitivity was assessed by measurement of footpad swelling 24 h after injection of ultraviolet irradiation–treated HSV (1 × 105 PFU/mouse) into the right footpads of mice 6 d after HSV-1 infection.
In vivo NK cell activity assay and B16 melanoma cell injection
These methods were adopted from a published procedure42. B6 Opn wild-type and Opn-deficient mice were injected in the footpads with 100 μg CpG ODN-1585 or control ODN-2118. On day 2, draining lymph nodes from two mice were pooled and total lymph node cells were used in a standard 51Cr-release assay against 5 × 103 YAC-1 cells/well. The proportion of the NK cell population (DX5+CD3ε−) in the total lymph node cells was determined by flow cytometry. B6 Opn wild-type and Opn-deficient mice were injected intraperitoneally with B16F10 melanoma cells (5 × 103) and 100 μg of CpG ODN-1585 or control ODN-2118.
Supplementary Material
Acknowledgments
We thank S. Turley for critical reading; D. Laznik for technical assistance; and A. Angel for assistance with the manuscript and figures. Supported by the National Institutes of Health (AI48125 and AI12184 to H.C.; T32 CA70083 to M.L.S; and CA48126 and AI56296 to L.H.G.), the Ellison Medical Foundation (L.H.G.) and the Juvenile Diabetes Research Foundation (M.L.S.).
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 2.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 3.Krug A, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 5.Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20:539–550. doi: 10.1016/s1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- 6.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (α/β) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 7.Le Bon A, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 8.Boonstra A, et al. Flexibility of mouse classical and plasmacytoid-derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll-like receptor ligation. J Exp Med. 2003;197:101–109. doi: 10.1084/jem.20021908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salio M, Palmowski MJ, Atzberger A, Hermans IF, Cerundolo V. CpG-matured murine plasmacytoid dendritic cells are capable of in vivo priming of functional CD8 T cell responses to endogenous but not exogenous antigens. J Exp Med. 2004;199:567–579. doi: 10.1084/jem.20031059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int Immunol. 2005;17:1367–1378. doi: 10.1093/intimm/dxh318. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Honda K, et al. Spatiotemporal regulation of MyD88–IRF7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 13.Ashkar S, et al. Eta-1 (osteopontin): an early component of Type 1 (cell-mediated) immunity. Science. 2000;287:860–864. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- 14.Shinohara ML, et al. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci USA. 2005;102:17101–17106. doi: 10.1073/pnas.0508666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyazaki T, et al. Implication of allelic polymorphism of osteopontin in the development of lupus nephritis in MRL/lpr mice. Eur J Immunol. 2005;35:1510–1520. doi: 10.1002/eji.200425672. [DOI] [PubMed] [Google Scholar]
- 16.Nau GJ, et al. Attenuated host resistance against Mycobacterium bovis BCG infection in mice lacking osteopontin. Infect Immun. 1999;67:4223–4230. doi: 10.1128/iai.67.8.4223-4230.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sibalic V, Fan X, Loffing J, Wuthrich RP. Upregulated renal tubular CD44, hyaluronan, and osteopontin in kdkd mice with interstitial nephritis. Nephrol Dial Transplant. 1997;12:1344–1353. doi: 10.1093/ndt/12.7.1344. [DOI] [PubMed] [Google Scholar]
- 18.Yu XQ, et al. A functional role for osteopontin in experimental crescentic glomer-ulonephritis in the rat. Proc Assoc Am Phys. 1998;110:50–64. [PubMed] [Google Scholar]
- 19.Hudkins KL, et al. Osteopontin expression in human crescentic glomerulonephritis. Kidney Int. 2000;57:105–116. doi: 10.1046/j.1523-1755.2000.00813.x. [DOI] [PubMed] [Google Scholar]
- 20.Steinman L, Martin R, Bernard C, Conlon P, Oksenberg JR. Multiple sclerosis: deeper understanding of its pathogenesis reveals new targets for therapy. Annu Rev Neurosci. 2002;25:491–505. doi: 10.1146/annurev.neuro.25.112701.142913. [DOI] [PubMed] [Google Scholar]
- 21.Xu G, et al. Role of osteopontin in amplification and perpetuation of rheumatoid synovitis. J Clin Invest. 2005;115:1060–1067. doi: 10.1172/JCI23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comabella M, et al. Plasma osteopontin levels in multiple sclerosis. J Neuroimmunol. 2005;158:231–239. doi: 10.1016/j.jneuroim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Gravallese EM. Osteopontin: a bridge between bone and the immune system. J Clin Invest. 2003;112:147–149. doi: 10.1172/JCI19190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zohar R, et al. Intracellular osteopontin is an integral component of the CD44-ERM complex involved in cell migration. J Cell Physiol. 2000;184:118–130. doi: 10.1002/(SICI)1097-4652(200007)184:1<118::AID-JCP13>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki K, et al. Colocalization of intracellular osteopontin with CD44 is associated with migration, cell fusion, and resorption in osteoclasts. J Bone Miner Res. 2002;17:1486–1497. doi: 10.1359/jbmr.2002.17.8.1486. [DOI] [PubMed] [Google Scholar]
- 26.Zhu B, et al. Osteopontin modulates CD44-dependent chemotaxis of peritoneal macrophages through G-protein-coupled receptors: evidence of a role for an intracellular form of osteopontin. J Cell Physiol. 2004;198:155–167. doi: 10.1002/jcp.10394. [DOI] [PubMed] [Google Scholar]
- 27.Wilson HL, O’Neill HC. Identification of differentially expressed genes representing dendritic cell precursors and their progeny. Blood. 2003;102:1661–1669. doi: 10.1182/blood-2002-08-2426. [DOI] [PubMed] [Google Scholar]
- 28.Renkl AC, et al. Osteopontin functionally activates dendritic cells and induces their differentiation toward a Th1-polarizing phenotype. Blood. 2005;106:946–955. doi: 10.1182/blood-2004-08-3228. [DOI] [PubMed] [Google Scholar]
- 29.Cantor H. T-cell receptor crossreactivity and autoimmune disease. Adv Immunol. 2000;75:209–233. doi: 10.1016/s0065-2776(00)75005-x. [DOI] [PubMed] [Google Scholar]
- 30.Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–1061. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diao H, et al. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity. 2004;21:539–550. doi: 10.1016/j.immuni.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Weiss JM, et al. Osteopontin is involved in the initiation of cutaneous contact hypersensitivity by inducing Langerhans and dendritic cell migration to lymph nodes. J Exp Med. 2001;194:1219–1230. doi: 10.1084/jem.194.9.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabo SJ, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 34.Lugo-Villarino G, Ito S, Klinman DM, Glimcher LH. The adjuvant activity of CpG DNA requires T-bet expression in dendritic cells. Proc Natl Acad Sci USA. 2005;102:13248–13253. doi: 10.1073/pnas.0506638102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, Glimcher LH. T-bet is required for optimal production of IFN-γ and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci USA. 2003;100:7749–7754. doi: 10.1073/pnas.1332767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hemmi H, Kaisho T, Takeda K, Akira S. The roles of Toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J Immunol. 2003;170:3059–3064. doi: 10.4049/jimmunol.170.6.3059. [DOI] [PubMed] [Google Scholar]
- 37.Lee SW, et al. Effects of a hexameric deoxyriboguanosine run conjugation into CpG oligodeoxynucleotides on their immunostimulatory potentials. J Immunol. 2000;165:3631–3639. doi: 10.4049/jimmunol.165.7.3631. [DOI] [PubMed] [Google Scholar]
- 38.Durand V, Wong SY, Tough DF, Le Bon A. Shaping of adaptive immune responses to soluble proteins by TLR agonists: a role for IFN-α/β. Immunol Cell Biol. 2004;82:596–602. doi: 10.1111/j.0818-9641.2004.01285.x. [DOI] [PubMed] [Google Scholar]
- 39.Haeryfar SM. The importance of being a pDC in antiviral immunity: the IFN mission versus Ag presentation? Trends Immunol. 2005;26:311–317. doi: 10.1016/j.it.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. 2004;5:1227–1234. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baccala R, Kono DH, Theofilopoulos AN. Interferons as pathogenic effectors in autoimmunity. Immunol Rev. 2005;204:9–26. doi: 10.1111/j.0105-2896.2005.00252.x. [DOI] [PubMed] [Google Scholar]
- 42.Ballas ZK, et al. Divergent therapeutic and immunologic effects of oligodeoxynucleotides with distinct CpG motifs. J Immunol. 2001;167:4878–4886. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]
- 43.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 44.Honda K, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF7 in Toll-like receptor signaling. Proc Natl Acad Sci USA. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takaoka A, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 46.Cho HJ, et al. IFN-αβ promote priming of antigen-specific CD8+ and CD4+ T lymphocytes by immunostimulatory DNA-based vaccines. J Immunol. 2002;168:4907–4913. doi: 10.4049/jimmunol.168.10.4907. [DOI] [PubMed] [Google Scholar]
- 47.Datta SK, et al. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol. 2003;170:4102–4110. doi: 10.4049/jimmunol.170.8.4102. [DOI] [PubMed] [Google Scholar]
- 48.Heit A, et al. Cutting edge: Toll-like receptor 9 expression is not required for CpG DNA-aided cross-presentation of DNA-conjugated antigens but essential for cross-priming of CD8 T cells. J Immunol. 2003;170:2802–2805. doi: 10.4049/jimmunol.170.6.2802. [DOI] [PubMed] [Google Scholar]
- 49.Barchet W, et al. Dendritic cells respond to influenza virus through TLR7- and PKR-independent pathways. Eur J Immunol. 2005;35:236–242. doi: 10.1002/eji.200425583. [DOI] [PubMed] [Google Scholar]
- 50.Abel B, Freigang S, Bachmann MF, Boschert U, Kopf M. Osteopontin is not required for the development of Th1 responses and viral immunity. J Immunol. 2005;175:6006–6013. doi: 10.4049/jimmunol.175.9.6006. [DOI] [PubMed] [Google Scholar]
- 51.Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol. 2004;173:2134–2142. doi: 10.4049/jimmunol.173.3.2134. [DOI] [PubMed] [Google Scholar]
- 52.Li J, et al. Deficiency of type I interferon contributes to Sle2-associated component lupus phenotypes. Arthritis Rheum. 2005;52:3063–3072. doi: 10.1002/art.21307. [DOI] [PubMed] [Google Scholar]
- 53.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kono DH, Baccala R, Theofilopoulos AN. Inhibition of lupus by genetic alteration of the interferon-α/β receptor. Autoimmunity. 2003;36:503–510. doi: 10.1080/08916930310001624665. [DOI] [PubMed] [Google Scholar]
- 55.Rittling SR, et al. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13:1101–1111. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- 56.Nakano H, Yanagita M, Gunn MD. CD11c+B220+Gr-1+ cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med. 2001;194:1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCarty N, et al. Signaling by the kinase MINK is essential in the negative selection of autoreactive thymocytes. Nat Immunol. 2005;6:65–72. doi: 10.1038/ni1145. [DOI] [PubMed] [Google Scholar]
- 58.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med. 2002;195:695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu D, et al. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol. 2004;5:516–523. doi: 10.1038/ni1063. [DOI] [PubMed] [Google Scholar]
- 60.Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.