

Abstract
Objective
To determine the efficacy of osteopontin (OPN) targeting in hepatocellular cancer (HCC).
Summary/Background
OPN is associated with HCC growth and metastasis and represents a unique therapeutic target.
Methods
OPN and EMT markers, α-smooth muscle actin (SMA), vimentin and tenascin-c, were measured in archived human HCC tissues from metastatic (n=4) and non-metastatic (n=4) settings. Additional studies utilized human Sk-Hep-1 (high OPN expression) and Hep3b (low OPN expression) HCC cells. An RNA aptamer (APT) that avidly binds (Kd=18 nM; t 1/2=7 hrs) and ablates OPN binding was developed. Adhesion, migration/invasion, and EMT markers were determined with APT or a mutant control aptamer (Mu-APT). RFP-Luc-Sk-Hep-1 were implanted into NOD-scid mice livers and followed using bioluminescence imaging. After verification of tumor growth, at week 3, APT (0.5 mg/kg; n=4) or Mu-APT (0.5 mg/kg; n=4) was injected Q48h. When mice were sacrificed at week 8, tumor cells were re-isolated and assayed for EMT markers.
Results
OPN and EMT markers were significantly increased in the metastatic cohort. APT inhibited Sk-Hep-1 adhesion and migration/invasion by 5- and 4-fold, respectively. APT significantly decreased EMT protein markers, SMA, vimentin and tenascin-c. In contrast, APT did not alter Hep3B adhesion, or migration/invasion. EMT markers were slightly decreased. In the in vivo model, at weeks 6-8, APT inhibited HCC growth by >10-fold. SMA, vimentin and tenascin-c mRNAs were decreased by 60%, 40% and 49% in RFP+ Sk-Hep-1 recovered by FACS. (p<0.04 vs Mu-APT for all)
Conclusions
APT targeting of OPN significantly decreases EMT and tumor growth of HCC.
Introduction
Hepatocellular cancer (HCC) is the fifth most common solid organ tumor and the third leading cause of cancer-related death in the world. In the United States, the incidence of HCC has tripled during the period from 1975 to 2005.[1] The extremely poor prognosis of patients with HCC is largely due to the high rate of tumor recurrence after locoregional therapy or intrahepatic metastasis at presentation.[1] With the exception of the recently approved drug, sorafenib, systemic therapies are otherwise nonspecific and ineffective.[1] As a result, the search for alternative molecular targets has focused, in part, on osteopontin (OPN) as a key mediator of metastatic behavior in HCC.[2] Initially discovered as an inducible tumor promoter, OPN is overexpressed in tumors, is the major phosphoprotein secreted by malignant cells in advanced metastatic cancer, is a key mediator of tumor cell migration and metastasis, is a lead marker of HCC progression and metastasis, and induces epithelial-mesenchymal transition (EMT).[3, 4] Originally, described in embryogenesis and organ development, EMT is a process by which a polarized cell undergoes multiple biochemical changes as it transforms into a mesenchymal cell phenotype.[5] New properties include enhanced migratory capacity, invasiveness, elevated resistance to apoptosis, and greatly increased production of ECM components. Numerous investigators have implicated OPN as an important regulator of growth and demonstrated that blockade of OPN cell surface binding significantly decreases HCC growth and metastasis. [3, 6, 7]
RNA aptamers are a unique emerging class of therapeutic agents.[8, 9] They are relatively short (12-30 nt) ss RNA oligonucleotides that assume a stable three-dimensional shape to tightly and specifically bind selected protein targets to elicit a biological response. As a member of the family of emergent RNA therapeutic agents, aptamers are unique in that they disrupt function at the protein level. RNA aptamers effectively target extracellular targets, such as OPN. As a secreted, extracellular protein, OPN is an ideal molecular target for RNA aptamer-based therapy.
In this study, we examine the role of RNA aptamer mediated blockade of extracellular OPN in HCC. Our results demonstrate that: 1) expression of OPN and EMT markers are significantly increased in metastatic human HCC tissue samples, 2) RNA aptamer (APT) binding of OPN decreases in vitro correlates of growth and metastasis in high OPN expressing Sk-Hep-1 cells, and 3) APT inhibits SK-Hep-1 tumor growth and expression of EMT markers in an in vivo model of experimental tumor growth. Our results suggest that OPN may be a viable clinically relevant target for HCC therapy. Phase 1 clinical trials are planned.
Methods
Cell Culture
SK-Hep-1 and Hep3B cells were obtained from American Tissue Culture Collection, Manassas, VA and maintained as monolayer cultures in Minimum Essential Medium (MEM eagle) with 2 mM L-glutamine and Earle's BSS adjusted to contain 1.5 g/L sodium bicarbonate, 0.1 mM non-essential amino acids, and 1.0 mM sodium pyruvate, and 10% fetal bovine serum at 37°C and 5% CO2. Following 5 passages, cells were utilized for in vitro or in vivo studies. For protein lysates preparation, cells were plated on 100 mm dishes overnight to obtain 70-80% confluence.
OPN-R3 Aptamer
The sequences for the OPN aptamers are as follows: OPN-R3: 5’-CGGCCACAGAAUGAAAAACCUCAUCGAUGUUGCAUAGUUG-3’ Mutant OPN-R3: 5'-CGGCCACAGAAUGAAUCAUCGAUGUUGCAUAGUUG-3'. Commercially synthesized OPN-R3 aptamers contain 2'-OMe C, 2'-OMe U, A, and G and were used for in vivo and in vitro studies.
HCC Specimens
Archival HCC specimens with de-identified clinical history are a gift from Prof. K.J. Kao, Koo Foundation Sun Yat Sen Cancer Center (KFSYSCC), Taipei, Taiwan. The KFSYSCC Tumor Bank was established in 1990 with subsequent collection of all borderline and malignant tumors removed during surgery. When the surgical specimen is removed from the patient, it is placed in a sterile container and transported immediately to surgical pathology, where tissues were processed by a pathologist. Tumor tissue that is not necessary for diagnosis is selected and provided by a pathologist to the tissue procurement service personnel. Specimens were snap frozen in liquid nitrogen within one hour of surgery. Informed consent is obtained from every patient.
A total of 8 specimens were randomly selected from the HCC tissue bank. The specimens fall within two categories, Non-Metastatic: primary resection specimens associated with >5 year patient survival in the absence of metastases and Metastatic: primary resection specimens associated with distant metastases within 5 years of resection. All HCC samples were obtained with informed consent from patients who underwent curative resection. (Table 1) The 8 samples were derived from 6 male and 2 female patients, predominantly hepatitis B−positive (5/8), Chinese patients with a mean age of 44.3 ± 2.1 years. All samples were histopathologically diagnosed as HCC according to Edmonson's classification.[10] Primary HCC samples ranged from 1.3 cm to 7.2 cm in diameter with a median diameter of 3.6 cm. Total RNA was extracted from each sample using TRIzol (Invitrogen, Carlsbad, California) according to the manufacturer's instructions. For protein analysis, tissues were ground in liquid-N2-cooled mortars and transferred into an homogenization tube with PMSF lysis buffer (0.8% NaCl, 0.02% KCl, 1% SDS, 10% Triton X-100, 0.5% sodium deoxycholic acid, 0.144% Na2HPO4, and 0.024% KH2PO4, 2 mM phenylmethylsulfonyl fluoride, pH 7.4). After homogenizing, samples were sonicated for 30 s and clarified by centrifugation at 12,000 × g for 10 min at 4 °C. The protein concentration was determined by the Bio-Rad DC protein assay kit (Hercules, CA).
Table 1.
Description of Human HCC Tissue Samples
| Sample Number | Metastatic | Positive Margins | Vascular Invasion | Size (cm) | Multifocal | Age (Yrs) |
|---|---|---|---|---|---|---|
| 1 | Yes | No | No | 1.3 | No | 55 |
| 2 | Yes | No | No | 3.2 | No | 47 |
| 3 | Yes | No | No | 3.9 | No | 46 |
| 4 | Yes | No | No | 2.6 | No | 44 |
| 5 | No | No | No | 7.2 | No | 58 |
| 6 | No | No | No | 3.7 | No | 43 |
| 7 | No | No | No | 4.6 | No | 55 |
| 8 | No | No | No | 2.7 | No | 47 |
Western Blot Analysis
Samples were lysed in buffer (0.8% NaCl, 0.02 KCl, 1% SDS, 10% Triton X-100, 0.5% sodium deoxycholic acid, 0.144 % Na2HPO4 and 0.024% KH2PO4, pH 7.4) and centrifuged at 12000 × g for 10 min at 4 °C. Protein concentration was determined by absorbance at 650 nm using protein assay reagent. Cell lysate (50 μg/lane) were separated by SDS-12% PAGE, and the products were electrotransferred to PVDF membrane (Amersham Pharmacia, Piscataway, NJ). The membrane was blocked with 5% skim milk PBS-0.05% Tween for 1 h at room temperature. After being washed three times, blocked membranes were incubated with primary antibody for 1 hr at room temperature, washed three times in PBS-0.05% Tween, and incubated with horseradish peroxidase conjugated secondary antibody for 1 hr at room temperature. After an additional three times washing, bound peroxidase activity were detected by the ECL detection system (Amersham Pharmacia, Piscataway, NJ). For quantification, the bands were scanned into an AlphaImager 3400 (Alpha Innotech, San Leandro, CA) and normalized by dividing the measured density of protein bands by the density of the loading control.
Real Time-PCR (RT-PCR) Analysis
Real-time PCR was performed with the two-step reaction protocol using iQ SYBR Green detection kit (Bio-Rad Laboratories, Hercules, CA). First-strand cDNA were synthesized from 1-μg total RNA using the iScript Select cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA) at 48°C for 30 min. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the endogenous control. The primer sets were used for the quantitative PCR analysis are listed above. Real-time PCR parameters used were as follows: 95°C for 3 min; 95°C for 30 s, 55°C for 35 s for 40 cycles; 95°C for 1 min, and 55°C for 10 min. PCR was performed with iQ SYBR Green super mix, using the iCycler iQ Real-time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). The 2-delta-delta Ct value was calculated following GAPDH normalization.
RT-PCR Primer Sequences
α-SMA (Length: 166bp): 5’-TAGCACCCAGCACCATGAAGAT-3’; 5’-GAAGCATTTGCGGTGGACAATG-3’ Tenascin-C (Length: 119bp): 5’-AGCATCACCCTGGAATGGAGGA-3’; 5’-TGTGGCTTGTTGGCTCTTTGGA-3’ Vimentin (Length: 200bp): 5'-AGAACGTGCAGGAGGCAGAAGAAT-3'; 5'-TTCCATTTCACGCATCTGGCGTTC-3' OPN (Length: 160bp): 5’-AGTTTCGCAGACCTGACATCCAGT-3’; 5’-TTCATAACTGTCCTTCCCACGGCT-3’
Adhesion And Migration/Invasion Assays
Adhesion assays was performed on 96-well microtiter plates coated with 10 μg/ml Matrigel. Cells (105) were exposed to 100 nM aptamer or 2 μg Ab for 30 min. Cells were trypsinized and resuspended in DMEM with 1% BSA, 1 mM MgCl2, 0.5 mM CaCl2 at a concentration of 1× 106 cells/ml. 1×105 cells (100 μl) were added into each well and placed for 30 min at 37°C in 5% CO2 humidified air incubation. Non-adherent cells were removed by gently washing the wells three times with phosphate-buffered saline (PBS) with 1 mM MgCl2 and 0.5 mM CaCl2. Adherent cells were fixed with 3.7% paraformaldehyde for 10 min at room temperature, followed by rinsing with PBS, and stained with 0.4% crystal violet for 10 min. After extensive rinsing, the dye was released from the cells by addition of 30% acetic acid, and the microtiter plates were read in a microplate reader (Molecular Devices, Berkeley, California) at 590 nm. The invasion assay was carried out in a Boyden Chamber system (Corning, NY). Cells were seeded at a density of 105 cells per well in triplicate in the upper chamber of 12 well transwells (8 μm pore ). After being incubated and treated with 100 nM aptamer or 2 ug Ab at 37°C with 5% CO2 for 24 hours, the cells were fixed in 3.7% paraformaldehyde in phosphate-buffered saline for 10 min. The cells on the top surface of the filters were wiped off with cotton swabs. Following three wash with PBS, the filters were stained with 0.4% crystal violet for 10 min, and the dye was detected as described for the in vitro adhesion assay.
Mouse Xenograft Model
Animal handling and procedures were approved by the Duke University Animal Care and Use Committee. 6-week-old NOD scid mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). Double labeled luciferase-red fluorescent (RFP) Sk-Hep-1 cells (106) were implanted under the liver capsule. The mice were anesthetized with intraperitoneal ketamine (75 mg/kg) and xylazine (10 mg/kg). Bioluminescence is reported as the sum of detected photons per second from a constant region of interest (photons/second/region of interest). Ten minutes after administration of luciferase substrates (D-luciferin, 150 mg/kg), anesthetized mice were imaged with the IVIS 100 Imaging System (Xenogen, Alameda,CA) following the company's manual. Initial in vivo images at day 2 were obtained to establish baseline tumor volume. Following confirmation of stable tumor growth, OPN-R3 (10 mg/kg) or OPN-R3-mutant (10 mg/kg) were injected into the mouse tail vein every 2 days beginning at week 3. Images were analyzed with the use of software AxioVision LE (Carl Zeiss Micro Imaging, Thornwood, NY) After imaging, tissues we restored at -80°C for RNA and protein analysis, in 1% formalin solution for immunohistochemical analysis, or immediately prepared for flow cytometry cell sorting. Tissue stored at -80°C was processed in a manner identical to that listed for the human HCC samples.
Fluorescence Activated Cell Sorting (FACS)
Fresh primary tumor was obtained. Single cell suspension was prepared as reported by Siemann. (Keng et al 1990) The tissues were finely minced with surgical scissors and transferred to 10ml collagenase-PBS solution (1×PBS, PH7.4; 0.025% collagenase, 0.05%pronase, 0.04% Dnase). After 1 hour incubation at 37°C, the tissue pellets were centrifuged at 1000rpm for 10 min at 4°C, and washed 3 times with 5 ml 1×PBS. The tissue homogenate was gently passed through 70-um-pore nylon mesh filter at 4°C. RFP labeled cell sorting was performed using BD FACStar using an air-cooled argon laser operated at 100mW RFP fluorescence with a 620/10 band pass filter. The RFP positive cells were collected in 1 × PBS and kept in -80°C freezer.
Statistical Analysis
Data are expressed as mean ± SEM. Analysis was performed using Student's t test. Values of p<0.05 were considered significant.
Results
Human HCC expression of OPN and EMT markers
Human HCC specimens were examined for protein expression of OPN and the EMT markers, SMA, vimentin and tenascin-c. These were classified according to the development of metastatic disease within three years following “curative” resection. The specimens fall within two categories, Non-Metastatic: primary resection specimens associated with >5 year patient survival in the absence of metastases and Metastatic: primary resection specimens associated with distant metastases within three years of resection. (Figure 1) Alpha-fetoprotein (AFP) expression was uniformly present among all of the samples, with exception of a single sample in the metastatic group with minimal AFP. OPN, SMA, vimentin and tenascin-c protein were found at significantly higher levels in cell lysates from tumors which exhibited subsequent metastases. OPN was approximately 25 -fold greater in the metastatic group in comparison to those of the nonmetastatic controls. Similarly, SMA, vimentin and tenascin-c were 11-, 15- and 24 -fold greater, respectively, in the metastatic group. These results suggest that OPN and EMT marker expression is significantly greater in HCC with a pro-metastatic phenotype.
Figure 1.

Expression of alpha-fetoprotein, osteopontin and epithelial mesenchymal transition (EMT) marker proteins in resected HCC samples. The specimens fall within two categories, Non-Metastatic: primary resection specimens associated with >5 year patient survival in the absence of metastases (n=4) and Metastatic: primary resection specimens associated with distant metastases within three years of resection. EMT markers are tenascin-c, vimentin, and SMA (α-smooth muscle actin). AFP, alpha-fetoprotein; OPN, osteopontin.
Sk-Hep-1 and Hep3B expression of OPN and EMT markers
To block the potential effects of secreted circulating extracellular OPN, we have developed an RNA aptamer (APT) targeting OPN.[11] The in vitro Kd was 18 nM; the in vivo Kd is 32 nM. Half life is approx. 8 hours. (unpublished data) The efficacy of APT to block cell surface binding of OPN was assessed in SK-Hep-1 and Hep3B cells using immunofluorescence.(Figure 2a) SK-Hep-1 and Hep3B were transfected with a plasmid construct encoding for OPN with an N-terminal YFP tag. Cells were exposed to APT (5 nM) or Mu-APT for 6 hours and extent of OPN cell surface binding was determined. In both Sk-Hep-1 and Hep3B, there was near total APT-mediated ablation of OPN binding to the cell surface. To determine whether APT localizes to the extracellular space or may also be taken up into the cell, APT was modified to express a Cy3 fluorophore tag. After incubation with Sk-Hep-1 cells in the presence and absence of the transfection agent, lipofectamine, only those cells incubated with APT and lipofectamine showed fluorescence after 72 hours. (Figure 2b) Cells treated with only APT showed no intracellular fluorescence. This indicates that APT is not taken up by Sk-Hep-1 cells and exerts its effects in the extracellular environment.
Figure 2a. APT blocks cell surface OPN binding.
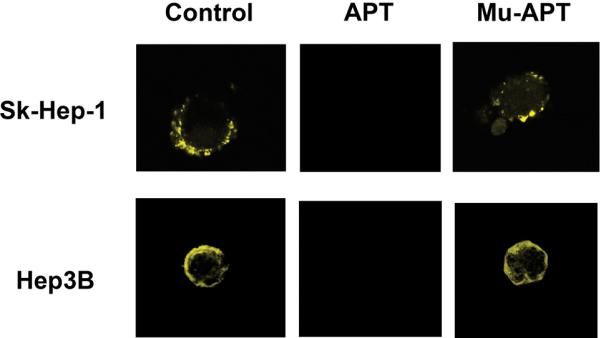
SK-Hep-1 or Hep3B cells were transfected with YFP-OPN (OPN cDNA fused in frame into pE-Yellow Fluorescent Protein). Cells were cultured in the presence of APT (5 nM × 6 hrs) targeting OPN or mutant APT. Untreated cells served as Controls. Fluorescent microscopy was performed. Images are representative of three experiments.
Figure 2b. APT remains in the extracellular space.

APT was modified to express a Cy3 fluorophore tag. APT (5 nM) was incubated with Sk-Hep-1 cells. In selected instances, cells were also incubated in the presence of the transfection agent, lipofectamine, to serve as a positive control. Fluorescent microscopy was performed at 0, 24, 48 and 72 hours. Images are representative of three experiments.
To examine the role of differential OPN and EMT marker expression on in vitro correlates of HCC growth and metastasis, Sk-Hep-1 and Hep3B cells in culture were selected. Sk-Hep-1 secrete OPN and exhibit aggressive OPN-dependent in vitro indices of adhesion, migration and invasion. In contrast, Hep3B served as negative controls which do not secrete OPN and exhibit the opposite phenotype. EMT marker protein expression was examined. (Figure 2c) Sk-Hep-1 expressed significantly higher levels of OPN, SMA, vimentin and tenascin-c in cell lysates when compared to Hep3B. (p<0.01 for all EMT markers) In the presence of APT (5 nM × 6 hrs), OPN expression in Sk-Hep-1 and Hep3B lysates was unchanged. However, EMT markers were significantly decreased in Sk-Hep-1 by APT treatment; vimentin expression was 7-fold lower when compared to mutant APT (Mu-APT) controls. (p<0.01 APT vs Mu-APT) SMA and tenascin-c protein expression was ablated in Sk-Hep-1 lysates exposed to APT. Hep3B expression of OPN was quite minimal and in this setting, EMT marker proteins were also decreased by APT, although to a lesser extent than Sk-Hep-1. These data suggest that APT-mediated inactivation of extracellular OPN decreases expression of EMT markers.
Figure 2c.

Expression of osteopontin and epithelial mesenchymal transition (EMT) marker proteins, tenascin-c, vimentin, and SMA (α-smooth muscle actin), in Sk-Hep-1 and Hep3B cells. Cells were cultured in the presence of APT (5 nM × 6 hrs) targeting OPN or mutant APT. Untreated cells served as Controls. Blot is representative of three experiments.
Sk-Hep-1 and Hep 3B adhesion and migration/invasion studies were then performed in the presence and absence of APT and Mu-APT. (Figure 3) At baseline, the relative extent of adhesion and migration/invasion was significantly less in Hep3B vs Sk-Hep-1. This observation is consistent with the decreased levels of OPN and EMT marker expression noted in Hep3B. In the presence of APT, Sk-Hep-1 cell adhesion and migration/invasion were decreased by 5– and 4–fold, respectively, when compared to Mu-APT and untreated controls. (p<0.05 vs Mu-APT and control for both adhesion and migration/invasion) While there was slightly decreased OPN seen in APT-treated Hep3B (Figure 2c), this did not translate into any significant differences in adhesion, migration and invasion. We speculate that the relatively greater extent of OPN expressed in Sk-Hep-1 versus Hep3B overshadows the small change resulting from APT mediated decrease in Hep3B OPN. These result indicate that blockade of extracellular OPN by APT significantly decreases in vitro correlates of growth and metastasis.
Figure 3.

Osteopontin dependent adhesion and migration/invasion in Sk-Hep-1 and Hep3B cells. Cells were cultured in the presence of APT (5 nM × 6 hrs) targeting OPN or mutant APT. Untreated cells served as Controls. Data are presented as mean ± SEM of three experiments.
(*, p<0.01 vs. Sk-Hep-1 + Mu-APT; #, p<0.01 vs Sk-Hep-1 + Mu-APT)
OPN aptamer and murine model of Sk-Hep-1 HCC
A murine NOD-scid xenograft model of HCC was utilized to assess the role of OPN blockade using the APT. Double labeled luciferase-red fluorescent protein (RFP) Sk-Hep-1 cells (106 cells) were implanted under the liver capsule. Initial in vivo bioluminescence images were obtained on day 2 to establish baseline tumor volume. Following confirmation of stable tumor growth, APT (0.5 mg/kg; n=4) or Mu-APT (0.5 mg/kg; n=4) were injected via tail vein every 2 days beginning at week 3 until the animals were sacrificed at week 8. Untreated animals served as Controls (n=4). Bioluminescence was measured weekly.(Figure 4a and b) In both the Mu-APT and Control group, there was aggressive and unabated growth over the course of the 8 weeks. In contrast, the APT bioluminescence signature was 10-fold less by 7-8 weeks post implantation.(p<0.01 vs Control and Mu-APT at weeks 6, 7 and 8) At necropsy, bulky tumor was present in both Controls and Mu-APT. In comparison, APT animals had visibly less tumor burden. These results indicate that ablation of extracellular OPN in HCC is associated with significantly decreased tumor growth.
Figure 4a.

In vivo bioluminescence of RFP-Luc-Sk-Hep-1 cells implanted orthotopically in NOD-scid mice. Double labeled luciferase-red fluorescent (RFP) Sk-Hep-1 cells (106 cells) were implanted under the liver capsule. Initial in vivo bioluminescence images were obtained on day 2 to establish baseline tumor volume. Following confirmation of stable tumor growth, APT (10 mg/kg; n=4) or Mu-APT (10 mg/kg; n=4) were injected via tail vein every 2 days beginning at week 3 until the animals were sacrificed at week 8. Untreated animals served as Controls (n=4). Bioluminescence was measured weekly.
Figure 4b.

Representative images of in vivo bioluminescence of RFP-Luc-Sk-Hep-1
OPN and EMT Marker Expression
RFP-Sk-Hep-1 tumors and liver were dissociated and tumor cells were recovered using FACS. RT-PCR was utilized to measure the expression of mRNA for the EMT markers, SMA, vimentin and tenascin-c. (Data not shown) In this setting, SMA, vimentin and tenascin-c mRNAs were significantly decreased by 60%, 40% and 49% in RFP+ Sk-Hep-1 recovered by FACS. (p<0.04 vs Mu-APT for all) These data indicate that inactivation of extracellular OPN is associated with decreased expression of EMT markers.
To determine the OPN cell surface receptor which regulates OPN-dependent EMT marker expression, in vitro studies were performed with Sk-Hep-1 cells. OPN is known to bind to CD44 and αvβ3 integrin receptors. SK-Hep-1 cells were exposed to APT, MU-APT, blocking Ab to CD44, and RGD (a competitive ligand inhibitor of integrin binding) in the presence or absence of exogenous OPN (50 nM). Polyclonal IgG and RGE served as negative controls. (Figure 5) RT-PCR was performed for EMT markers following 6 hours of incubation. In the presence of APT, Sk-Hep-1 expression of SMA, vimentin and tenascin-c mRNA was decreased by 4-, 3- and 3-fold in comparison to controls. (p<0.01 vs Controls) This profile was similar in the setting of RGD blockade. The presence of CD44 Ab did not alter the EMT marker profile. The addition of 10-fold excess OPN to compete with APT or RGD resulted in restoration of the EMT profile similar to that seen with Controls. These results suggest that OPN acts via integrin cell surface receptors to mediate expression of EMT markers.
Figure 5.

qRT-PCR of tenascin-c, vimentin, and SMA mRNA in RFP-Luc-Sk-Hep-1 cells in vitro. SK-Hep-1 cells were exposed to APT, MU-APT, blocking Ab to CD44, and RGD (a competitive ligand inhibitor of integrin binding) in the presence or absence of exogenous OPN (50 nM). Polyclonal IgG and RGE served as negative controls. qRT-PCR was performed for EMT markers, tenascin-c, vimentin, and SMA, following 6 hours of incubation. Data are presented as mean ± SEM of three experiments.
(*, p<0.01 vs Control, Mu-APT, RGE, CD44 Ab, IgG, OPN+APT and OPN+RGD)
Discussion
In this paper, we have demonstrated that OPN and EMT marker expression is associated with aggressive growth and metastasis in a sample of resection specimens. An RNA aptamer was developed which binds secreted OPN in the extracellular space. Autocrine and paracrine binding of OPN to the cell surface is ablated by the aptamer. In an in vivo xenotransplant model of Sk-Hep-1 HCC, this aptamer results in significant diminution of growth. Analysis of tumor cells in in vitro and ex vivo settings demonstrates a significant decrease in EMT marker expression by this aptamer. OPN binding to cell surface integrin receptors is required for EMT marker expression. These data suggest that APT targeting of OPN significantly decreases EMT and tumor growth of HCC.
HCC is the most common primary malignancy of the liver.[1] In the United States, liver cancer incidence and death rates exhibit the highest annual percent increase of the top 15 cancers. While resection and transplant technologies have advanced greatly, greater than 70% of HCC patients present with advanced disease. Historically, HCC has been characterized as a tumor which is resistant to chemo and radiation therapies. Presently, sorafenib is the only chemotherapy agent approved for advanced HCC. There are ongoing trials using brivinib ( an inhibitor of FGF1 and VEGFR1), bevacizumab and erlotinib, doxorubicin and ABT-869 (a receptor tyrosine kinase inhibitor of VEGF).
With regard to OPN, numerous investigators have implicated OPN as an important regulator of metastatic behavior. [3] Medico and coworkers used cDNA microarrays to identify OPN as a major target for the transcription factor, hepatocyte growth factor, and demonstrated that OPN mediated cell adhesion in MLP-29 murine liver cancer cells.[12] Subsequently, using hepatitis B-positive human HCC samples, Ye and coworkers used microarray gene expression profiling to examine changes associated with HCC metastasis.[3] These authors found that OPN correlated with the metastatic potential of primary HCC. Additional in vitro studies showed that OPN neutralizing antibody significantly blocked invasion of SK-Hep-1 cells. Using archived HCC resection specimens, OPN mRNA expression correlated closely with intrahepatic metastasis, early recurrence and late-stage/higher grades of HCC. Additional immunohistochemistry studies found that OPN is expressed primarily on cancerous cells, especially in HCC with capsular invasion and in areas adjacent to stromal cells.
Additional studies have examined in vivo consequences of OPN ablation. In a nude mouse model of HCC pulmonary metastasis, OPN specific antibody significantly decreased lung metastasis of HCCLM3 cells.[3] Zhao and colleagues used polyethylenimine nanoparticles to deliver a short-hairpin RNA for depletion of OPN expression in HCC cells. This resulted in inhibition of HCC cell growth, anchorage-independent growth, adhesion with fibronectin and invasion through extracellular matrix in vitro, and suppressed tumorigenicity and lung metastasis in nude mice.[6] In an alternative approach, Sun and coworkers used lentiviral delivery of micro-RNA against OPN and suppressed in vitro proliferation and in vivo tumor growth of HCCLM3.[7]
HCC remains one of the most common and aggressive human malignancies worldwide. The extremely poor prognosis of patients with HCC is largely due to the high rate of tumor recurrence or intrahepatic metastasis at presentation or following locoregional therapy.[13] Current systemic therapies are largely nonspecific and ineffective. In this regard, experimental data indicate that OPN plays a central regulatory role in the metastatic behavior and local growth of HCC. As a secreted, extracellular protein, OPN is a promising molecular target for an RNA aptamer-based therapy.
RNA aptamers represent a unique emerging class of therapeutic agents.[8, 9] They are relatively short (12-30 nt) ss RNA oligonucleotides that assume a stable three-dimensional shape to tightly and specifically bind selected protein targets to elicit a biological response. As a member of the family of emergent RNA therapeutic agents, aptamers are unique in that they disrupt function at the protein level. RNA aptamers can effectively target extracellular targets, such as OPN. Aptamers possess a number of useful properties as potential therapeutic molecules. They are relatively small (8 kD to 15 kD) synthetic compounds that possess high affinity and specificity for their target proteins (Kd ~ 0.05-10 nM). Animal studies have shown that aptamers are well tolerated, exhibit low or no immunogenicity, and are thus likely suitable for repeated administration as therapeutic compounds.[8, 9] As synthetic compounds, site-specific modifications can be made to aptamers to rationally alter their bioavailability and mode of clearance. For example, 2’fluoro pyrimidine-modified aptamers in the 10 kD to 12 kD size range have a short circulating half-life (~10 minutes) following bolus intravenous administration, but simple conjugation of the aptamer to a high molecular weight inert carrier molecule (eg. PEG) increases its circulating half-life substantially (6-12 hours). Like antibodies, aptamers possess binding affinities in the low nanomolar to picomolar range, but are also heat stable, lack immunogenicity, and possess minimal interbatch variability. Aptamers are generated by in vitro screening of complex nucleic-acid based combinatorial shape libraries (>1014 shapes per library) employing a process termed SELEX by Tuerk and Gold (for Systematic Evolution of Ligands by EXponential Enrichment).[14] The SELEX process consists of iterative rounds of affinity purification and amplification of ligands from combinatorial oligonucleotide libraries to yield high affinity and high specificity antagonists to proteins. This technology has enabled the generation of high affinity and high specificity antagonists to a myriad of proteins including reverse transcriptases, proteases, cell adhesion molecules, infectious viral particles and growth factors.[8]
Our APT was previously described.[11] Briefly, the in vitro Kd is 18 ± 0.2 nM. It was modified to increase its biologic half-life, incorporating 2’-O-methyl substituted nucleotides, 5’-cholesterol modification and 3’-inverted deoxythymidine. Additional in vivo PK data were obtained following tail vein injection: t1/2 = 7.76 hr; Tmax = 3 hr; Cmax = 13.2 mM; MRT(area) = 9 hr; and AUC = 161.9mmol-hr/L. The measured in vivo APT Kd = 57.2 nM; for reference, median plasma OPN level in HCC (500 nM) is approx. 10-fold greater.[15, 16] The predicted secondary structure of OPN-R3 contains the usual stem-loop structure of RNA aptamers. Based upon these parameters, the dosing in this study is excessive but was so chosen based upon our previous studies and as proof of principle. As APT is developed for potential clinical studies, more rational dosing will be required.[11]
Recently, EMT has been demonstrated to play an important role in the development of the invasive and metastatic cancer phenotypes.[17, 18] A number of clinical studies have demonstrated a positive correlation between inducible factors associated with EMT and poor clinical outcomes in breast, ovary, colorectal, and lung cancer, melanoma and HCC.[19] Originally, described in embryogenesis and organ development, EMT is a process by which a polarized cell undergoes multiple biochemical changes as it transforms into a mesenchymal cell phenotype.[5] New properties include enhanced migratory capacity, invasiveness, elevated resistance to apoptosis, and greatly increased production of ECM components. Completion of EMT is associated with degradation of underlying basement membrane and the formation of a mesenchymal cell that migrates from the original epithelial. A number of distinct molecular processes, such as activation of transcription factors, expression of specific cell-surface proteins, reorganization and expression of cytoskeletal proteins, production of ECM-degrading enzymes, and expression of specific microRNAs are required to initiate EMT. Some of these serve as biomarkers for EMT, including N-cadherin, vimentin, fibronectin, β-catenin, α-smooth muscle actin, tenascin-c, Snail, Slug and Twist, among others. A recent publication by Nagai and colleagues found that sorafenib treatment of HepG2 and Huh7 HCC cell lines inhibited expression of the EMT markers, Snail and vimentin, and EMT-associated functions in wound healing and migration assays.[17] These findings support our observation that inhibition of EMT is associated with significantly decreased in vitro and in vivo correlates of growth and metastasis.
The relationship between OPN and EMT in HCC has not been previously characterized. In other settings, Saika et al. found that OPN is upregulated in the injured mouse lens prior to initiation of EMT. Using OPN-null mice, these authors found that absence of OPN was associated with inhibition of EMT as measured by SMA, transforming growth factor-β, and collagen type 1.[20] In non-small cell lung cancers, Goparaju and coauthors found that OPN expression was associated with increased expression of the EMT markers, matrix metalloproteinase (MMP)-2, Snail-1, Snail-2, TGFβ1-R, MMP-9, N-cadherin, vimentin, SOX-8, and SOX-9.[21] Certainly, in our current study, OPN expression in HCC was also associated with integrin-dependent expression of EMT markers and enhanced in vitro measures of growth and metastasis. Aptamer blockade of OPN also decreased local growth of HCC. Given the relative lack of agents with efficacy in HCC, aptamer targeting of OPN may prove to be a therapeutic option for the future. We hope to proceed with clinical trials in the near future.
Acknowledgments
Supported by NIH R01 GM065113 (PCK), T32 GM069331 (LJT), and T32 CA093245 (SDB)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomas MB, Jaffe D, Choti MM, et al. Hepatocellular carcinoma: consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2010;28:3994–4005. doi: 10.1200/JCO.2010.28.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wai PY, Kuo PC. Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev. 2008;27:103–118. doi: 10.1007/s10555-007-9104-9. [DOI] [PubMed] [Google Scholar]
- 3.Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat.Med. 2003;9:416–423. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 4.Mi Z, Bhattacharya SD, Kim VM, et al. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis. 2011;32:477–487. doi: 10.1093/carcin/bgr009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J, Dong L, Lu B, et al. Down-regulation of osteopontin suppresses growth and metastasis of hepatocellular carcinoma via induction of apoptosis. Gastroenterology. 2008;135:956–968. doi: 10.1053/j.gastro.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 7.Sun BS, Dong QZ, Ye QH, et al. Lentiviral-mediated miRNA against osteopontin suppresses tumor growth and metastasis of human hepatocellular carcinoma. Hepatology. 2008;48:1834–1842. doi: 10.1002/hep.22531. [DOI] [PubMed] [Google Scholar]
- 8.Que-Gewirth NS, Sullenger BA. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 2007;14:283–291. doi: 10.1038/sj.gt.3302900. [DOI] [PubMed] [Google Scholar]
- 9.Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Mol.Cancer Ther. 2006;5:2957–2962. doi: 10.1158/1535-7163.MCT-06-0172. [DOI] [PubMed] [Google Scholar]
- 10.Edmondson HA, Steiner PE. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer. 1954;7:462–503. doi: 10.1002/1097-0142(195405)7:3<462::aid-cncr2820070308>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 11.Mi Z, Guo H, Russell MB, et al. RNA Aptamer Blockade of Osteopontin Inhibits Growth and Metastasis of MDA-MB231 Breast Cancer Cells. Mol Ther. 2009;17:153–161. doi: 10.1038/mt.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medico E, Gentile A, Lo CC, et al. Osteopontin is an autocrine mediator of hepatocyte growth factor-induced invasive growth. Cancer Res. 2001;61:5861–5868. [PubMed] [Google Scholar]
- 13.Greten TF, Korangy F, Manns MP, Malek NP. Molecular therapy for the treatment of hepatocellular carcinoma. Br J Cancer. 2009;100:19–23. doi: 10.1038/sj.bjc.6604784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuerk C, MacDougal S, Gold L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc Natl Acad Sci U S A. 1992;89:6988–6992. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Ki SS, Lee SD, et al. Elevated plasma osteopontin levels in patients with hepatocellular carcinoma. Am J Gastroenterol. 2006;101:2051–2059. doi: 10.1111/j.1572-0241.2006.00679.x. [DOI] [PubMed] [Google Scholar]
- 16.Xie H, Song J, Du R, et al. Prognostic significance of osteopontin in hepatitis B virus-related hepatocellular carcinoma. Dig Liver Dis. 2007;39:167–172. doi: 10.1016/j.dld.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Nagai T, Arao T, Furuta K, et al. Sorafenib inhibits the hepatocyte growth factor-mediated epithelial mesenchymal transition in hepatocellular carcinoma. Mol Cancer Ther. 2011;10:169–177. doi: 10.1158/1535-7163.MCT-10-0544. [DOI] [PubMed] [Google Scholar]
- 18.van Zijl F, Zulehner G, Petz M, et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169–1179. doi: 10.2217/fon.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 20.Saika S, Shirai K, Yamanaka O, et al. Loss of osteopontin perturbs the epithelialmesenchymal transition in an injured mouse lens epithelium. Lab Invest. 2007;87:130–138. doi: 10.1038/labinvest.3700508. [DOI] [PubMed] [Google Scholar]
- 21.Goparaju CM, Pass HI, Blasberg JD, et al. Functional heterogeneity of osteopontin isoforms in non-small cell lung cancer. J Thorac Oncol. 2010;5:1516–1523. doi: 10.1097/JTO.0b013e3181eba6bd. [DOI] [PMC free article] [PubMed] [Google Scholar]