

Abstract
Recent genome-wide association studies have linked polymorphisms in two atophagy genes, Atg16L1 and IRGM, with Crohn’s Disease. Now, experiments with Atg16L1 transgenic mice indicate multiple roles for autophagy in inflammatory bowel disease via effects on Paneth cells, a runaway inflammasome, and the proinflammatory cytokine IL-1b.
Autophagy is a cytoplasmic homeostasis process that cleanses the interior of all eukaryotic cells. It turns over stable or aggregated proteins, removes surplus or damaged organelles, and eliminates intracellular microbes (Levine and Deretic, 2007). A hallmark feature of autophagy is the formation within the cytosol of membrane crescents corralling cytoplasmic targets into the double-membrane-bound autophagosome, an emblematic feature of autophagy. Autophagosomes fuse with lysosomes, thus degrading the captured cargo. Autophagy plays a role in aging, degenerative diseases, cancer, and immunity. In its immunological manifestations (Levine and Deretic, 2007), autophagy promotes MHC II antigen presentation of endogenous antigens, acts as an effector of Th1/Th2 polarization, governs T cell repertoire and homeostasis, and acts as an antimicrobial mechanism that can be activated by Toll-like receptors (TLR) (Delgado et al., 2008).
Autophagy is best understood in yeast, which was the origin of the Atg nomenclature used for many components of the pathway. Autophagosome formation in eukaryotes is driven by two key Atg conjugation systems: (1) a covalent protein conjugate, Atg5-Atg12, noncovalently complexed with Atg16 (or Atg16L1 in mammals); and (2) a protein-lipid conjugate of Atg8 (LC3 in mammals) with phosphatidylethanolamine at its C terminus. The Atg5-Atg12/Atg16 complex stimulates LC3 lipidation. In this process, Atg16L1 “marks the spot” where the conjugation systems converge to generate nascent autophagosomes (Fujita et al., 2008). Mammalian Atg16L1 contains three distinct regions (Figure 1A): the N-terminal portion interacting with Atg5, the coiled-coil domain (CCD) necessary for Atg16L1 oligomerization and Atg5-Atg12 association, and the WD repeat domain, which is absent in yeast.
Figure 1. Atg16L Roles in Crohn’s Disease.
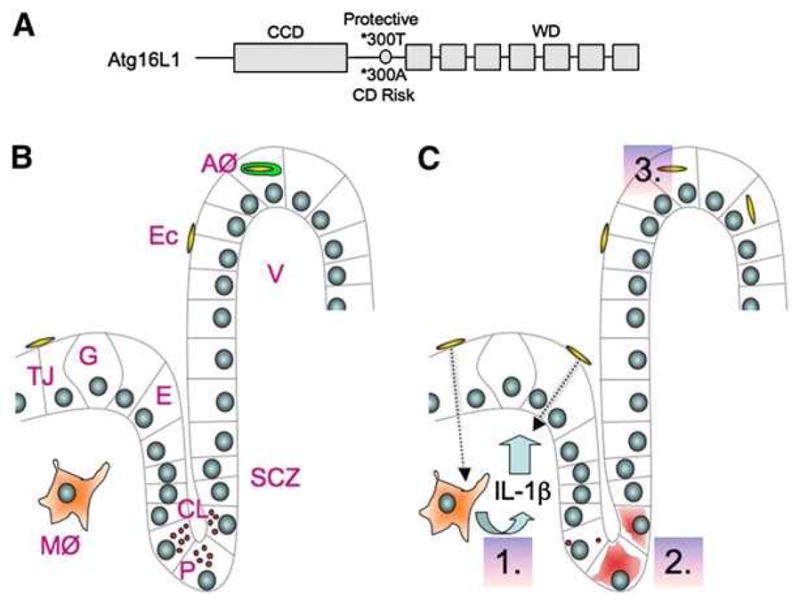
(A) Schematic of Atg16L1 features.
(B) Normal ileal crypt of Lieberkühn (CL) and villus (V). AØ, autophagosome (detected in cell culture); E, enterocyte; E.c., adherent-invasive E. coli; G, Goblet cell; MØ, macrophage; P, Paneth cell; SCZ, stem cell zone; TJ, tight junction.
(C) Dotted arrow, microbial translocation (proposed). 1.–3., effects of ATG16L1 mutations. 1. Increased IL-1b activation (in macrophages from Atg16L1 transgenic mice) accompanied by experimentally induced intestinal inflammation and mortality in vivo (not shown). IL-1b can dilate tight junctions (demonstrated in vitro) and may enhance microbial translocation. 2. Fewer granules or diffuse granule contents in the cytoplasm of Paneth cells (in ileal sections from Atg16L1 HM hypomorphic mice and uninvolved portions of ileocolic resection specimens from CD patients). 3. Reduced autophagy of invasive bacteria (in cultured epithelial cells rendered Atg16L1*300A by siRNA knockdown of endogenous Atg16L1 and complemented with Atg16L1*300A).
Recent genome-wide association (GWA) studies have linked autophagy with Crohn’s Disease (CD), a major form of chronic inflammatory bowel disease (Xavier and Podolsky, 2007). CD develops predominantly at anatomical sites (terminal ileum and colon) where commensal bacteria dramatically increase in mass (Xavier and Podolsky, 2007). It is believed that CD results from a “perfect storm” of ongoing challenge by normal gut flora and an aberrant innate immunity response. The latest GWA breakthroughs have expanded the role of innate immunity components beyond the already implicated Nod2 (Kanneganti et al., 2007) to include autophagy based on association with Atg16L1 (Cadwell et al., 2008; Saitoh et al., 2008) and an autophagy-linked factor, IRGM, involved in clearing bacteria (Singh et al., 2006).
Until the two new reports from the groups of Shizuo Akira (Saitoh et al., 2008) and Herbert Virgin (Cadwell et al., 2008), little was known (but much was being guessed) about the role of Atg16L1 and autophagy in CD. The two teams generated different Atg16L1 transgenic mice and came to diverse but not mutually exclusive conclusions. Saitoh et al. (2008) generated Atg16L1 DCCD mice, with the Atg16L1 gene deleted for the CCD domain. The Atg16L1 DCCD mice die within 1 day of birth, a phenomenon previously seen with the Atg5−/− knockout mice. Atg16L1-deficient MEFs were null for autophagy. Saitoh et al. tested Atg16L DCCD fetal liver-derived macrophages for proinflammatory cytokine production in response to LPS and found elevated IL-1b production (Figures 1B and 1C). Exposure of Atg16L1 DCCD macrophages to commensal bacteria such as Escherichia coli elicited abnormally high IL-1b processing. Next, lethally irradiated mouse chimeras reconstituted with Atg16L1 DCCD embryonic liver cells were subjected to experimentally induced colitis with dextran sulfate. The result was a 100% 10-day mortality of the Atg16L1 DCCD mouse chimeras, elevated IL-1b and IL-18 in the sera, and distal colon inflammation. Death was avoided and pathology was reversed by IL-1b- and IL-18-neutralizing antibodies (Saitoh et al., 2008).
Can elevated IL-1b help understand CD pathogenesis? The Atg16L1 DCCD effects on IL-1b are reminiscent of CD Nod2 variants, which also elicit elevated IL-1b in murine models (Kanneganti et al., 2007). The components of IL-1b signaling are often abnormal in intestinal tissue specimens from inflammatory bowel disease patients, and increased IL-1b may, among other effects, increase epithelial barrier permeability (Al-Sadi et al., 2008), possibly enhancing microbial product translocation. Complementing these findings, the elegant studies by Saitoh et al. have also uncovered a previously unappreciated role for autophagy in inflammasome function. The inflammasome occupies a special place in the innate immunity hall of fame. Upon proper stimulation, the inflammasome proteolytically converts inactive substrates into biologically highly active inflammation mediators, which includes the processing of pro-IL-1b into IL-1b. It now turns out that autophagy downregulates this system by a posttranslational mechanism requiring reactive oxygen species and a TLR binding adaptor molecule known as TRIF. Although the exact details are not available yet, it would be interesting to know whether autophagy targets the inflammasome or its substrates for degradation.
Anticipating that an outright Atg16L1 knockout would be lethal, Cadwell et al. (2008) took a different approach and generated hypomorphic Atg16L1HM mice expressing Atg16L1 at 30% of the wild-type levels. The strategy resulted in viable Atg16L1HM progeny. Autophagy was reduced (but not completely eliminated) as shown in Atg16L1HM MEFs and ileal specimens. Although Atg16L1 is expressed in both the crypt and the villus of the ileum (Figure 1), changes were observed only in Paneth cells, which have a previously suspected role in CD (Xavier and Podolsky, 2007). Paneth cells are found at bottom of ileal crypts (Figure 1B), and exocytose granules containing antimicrobial products—a-defensins, lysozyme, and secretory phospholipase A2. These antimicrobials are believed to protect the neighboring stem cells from microbial insults and keep gut microbial flora under check. Paneth cells in the Atg16L1HM ileum contained fewer granules and showed diffuse cytoplasmic lysozyme staining (Figure 1C), leading to the conclusion that autophagy affects Paneth cell function. As autophagy intersects with the lysosomal pathway, and secretory granules often correspond to specialized lysosomes, autophagy could affect the biosynthesis or quality control of Paneth cell granules. Alternatively, the cells with fewer granules might be a result of Paneth cell exhaustion, compensating for changes elsewhere in the epithelium. As with the studies by Saitoh et al., the Atg16L1HM findings by Cadwell et al. showed parallels with Nod2 effects: expression in CD-like mutant Nod2 mouse alleles affects Paneth cells by reducing these cells’ Nod2-dependent production of a-defensins (Kanneganti et al., 2007).
How do the elegant studies with Atg16L1 DCCD and Atg16L1HM mice relate to the actual Atg16L1 alleles in CD patients? The CD risk allele Atg16L1*300A is a single amino acid polymorphism (an Ala residue at the position 300 between CCD and WD; Figure 1A). In cultured epithelial cells, the Atg16L1*300A allele had no effect on housekeeping autophagy but caused a defective autophagic response to invasive bacteria (Figures 1B and 1C) (Kuballa et al., 2008). In addition, the Atg16L1*300A protein was unstable in infected epithelial cells, a feature not seen with the protective Atg16L1*300T allele. Thus, Atg16L1*300A has a hypomorphic phenotype (Kuballa et al., 2008), meaning that Atg16L1HM mice may recapitulate some aspects of this Atg16L1 CD risk allele. Collectively, these studies affirm several long-term or recent notions about CD and provide insight into the role of autophagy in this disease, an aspect that will become more complete once the effects of other autophagy factors, including IRGM, are revealed.
References
- Al-Sadi R, Ye D, Dokladny K, Ma TY. J Immunol. 2008;180:5653–5661. doi: 10.4049/jimmunol.180.8.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. Nature. 2008 doi: 10.1038/nature07416. in press. Published online October 5, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. Mol Biol Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Lamkanfi M, Nunez G. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. PLoS ONE. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Deretic V. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Nature. 2008 doi: 10.1038/nature07383. in press. Published online October 5, 2008. [DOI] [PubMed] [Google Scholar]
- Singh SB, Davis AS, Taylor GA, Deretic V. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]