

Abstract
The current classification of human sporadic prion diseases recognizes six major phenotypic subtypes with distinctive clinicopathological features, which largely correlate at the molecular level with the genotype at the polymorphic codon 129 (methionine, M, or valine, V) in the prion protein gene and with the size of the protease-resistant core of the abnormal prion protein, PrPSc (i.e. type 1 migrating at 21 kDa and type 2 at 19 kDa). We previously demonstrated that PrPSc typing by Western blotting is a reliable means of strain typing and disease classification. Limitations of this approach, however, particularly in the interlaboratory setting, are the association of PrPSc types 1 or 2 with more than one clinicopathological phenotype, which precludes definitive case classification if not supported by further analysis, and the difficulty of fully recognizing cases with mixed phenotypic features. In this study, we tested the inter-rater reliability of disease classification based only on histopathological criteria. Slides from 21 cases covering the whole phenotypic spectrum of human sporadic prion diseases, and also including two cases of variant Creutzfeldt–Jakob disease (CJD), were distributed blindly to 13 assessors for classification according to given instructions. The results showed good-to-excellent agreement between assessors in the classification of cases. In particular, there was full agreement (100 %) for the two most common sporadic CJD subtypes and variant CJD, and very high concordance in general for all pure phenotypes and the most common subtype with mixed phenotypic features. The present data fully support the basis for the current classification of sporadic human prion diseases and indicate that, besides molecular PrPSc typing, histopathological analysis permits reliable disease classification with high interlaboratory accuracy.
Introduction
Transmissible spongiform encephalopathies (TSEs), or prion diseases, are rare and fatal neurodegenerative disorders characterized by tissue deposition of a misfolded isoform of the cellular prion protein (PrPC), commonly referred to as PrPSc [34].
Prion diseases in humans can be sporadic, familial, or acquired. The sporadic form, by far the most common, comprises a broad spectrum of clinicopathological variants that show heterogeneity in disease duration, symptomatology, and type or regional distribution of brain lesions [30]. Prion strains, defined by their distinct phenotypes upon serial transmission to syngenic animals, are believed to be the main cause of this phenotypic diversity [1]. In addition, the host variability in the gene encoding PrPC (PRNP), as determined in humans by polymorphisms or mutations, has also been recognized to be a source of disease heterogeneity [6, 8, 9].
Although uncertainties remain regarding the molecular basis of prion strains, circumstantial evidence suggests that the strain phenotypes are enciphered in distinct tertiary or even quaternary structures of PrPSc (reviewed in [1]). In human prion disease, different PrPSc profiles or types characterized by distinctive physicochemical properties such as size after protease treatment, degree of protease resistance, and glycoform ratio have been shown to correlate with the phenotypic heterogeneity of the disease and, to some extent, with the strain properties after transmission [5, 7, 18, 20–22, 25–27, 32, 33, 36, 39].
In sporadic human TSEs, for example, the combination of two main PrPSc types commonly referred to as type 1 and type 2, showing distinct sizes of the unglycosylated protease-resistant C-terminal core (21 vs. 19 kDa), and the genotype at the PRNP polymorphic codon 129, encoding for either methionine (M) or valine (V), is significantly related to the phenotypic variability of the disease, and has provided a molecular basis for its classification [25, 28]. The latter currently includes six major phenotypic variants, each resulting from a specific codon 129 genotype/PrPSc type (1, 2, or 1 + 2) combination, with only three exceptions: MM1 and MV1 cases appear phenotypically indistinguishable and have been merged into one subtype (MM/MV 1), whereas the MM2 and MV2 combinations are both associated with two subtypes with distinctive histopathological features. The MM2 subtype showing confluent vacuoles and a predominant cortical pathology has been designated MM2-cortical (MM 2C), whereas that characterized by prominent atrophy of thalamic and inferior olivary nuclei has been defined as either the sporadic form of fatal insomnia (sFI) or as sCJD MM2-thalamic (MM 2T) [24, 28]. Similarly, the most common MV2 subtype with cerebellar amyloid plaques of the kuru type has been designated MV 2-kuru (MV 2K), whereas the rare MV2 cases with cortical confluent vacuoles have been denoted MV2-cortical (MV 2C).
The six sporadic prion disease subtypes differ from each other in several clinicopathological and biochemical features, and five of them (MM/MV1, VV2, VV1, MM/MV 2C, and MM 2T) appear to propagate in animal models as different prion strains [2, 14–17, 20, 26]. Interestingly, some of the six sporadic prion disease subtypes have also been found to co-occur in the same brain with variable frequency, indicating the possibility that prion strain co-occurrence (i.e. generating distinct disease phenotypes upon transmission to susceptible syngenic animals) may also take place in sCJD, as has been suggested for scrapie [4, 5, 10, 28, 31, 35].
Variant CJD (vCJD or MM 2V) represents an additional very distinctive CJD phenotype. It originated from a bovine prion strain [3, 38], and is characterized by a type 2 PrPSc form that differs from the type 2 found in sCJD due to a relatively high representation of the diglycosylated PrPSc glycoform [7, 23].
Given the biological and phenotypic heterogeneity of prion diseases, the correct identification or “typing” of the prion strain/s associated with each case of human TSE has obvious implications for disease diagnosis, epidemiologic surveillance, and future therapeutics. Transmission studies are extremely expensive, time-consuming, and only give information about the inoculum used, which is not necessarily representative of the whole affected brain. Either molecular or histopathological data (or both) may, therefore, provide reliable surrogate markers for strain typing in humans.
Analysis of the physicochemical properties of PrPSc (i.e. PrPSc “typing”) has already proven to be a reliable means of strain typing and performing molecular classification of CJD [29]. Limitations of this approach, however, particularly in the interlaboratory setting, are the association of one PrPSc type with more than one CJD phenotype, which precludes the ultimate case classification if not supported by further analysis (e.g. PrPSc type 1 in MM1 or VV1 types or type 2 in MM 2C or MM 2T types), and the difficulty involved in fully recognizing the sCJD cases with mixed phenotypic features, in particular when one of the co-occurring PrPSc types 1 and 2 expresses itself only focally, as in some MM 1+2C cases, or when the co-occurring sCJD phenotypes are associated with the same PrPSc type (e.g. MV 2K+C or MM 2T+C) [29, 31]. Furthermore, PrPSc typing cannot be performed in cases lacking fresh or frozen brain tissue. As it is based on the recognition of distinctive histopathological features, CJD “histotyping” may not suffer from the limitations listed above. In particular, CJD histotyping has been recently reported to perform better than PrPSc typing in recognizing sCJD subtypes with mixed phenotypical features, especially for MM or MV subjects [12, 31].
The full validation of CJD histotyping requires the standardization and harmonization of practice among laboratories. To achieve this aim, a collaborative study among seven laboratories involved in TSE surveillance in several European countries and the USA was set up to compare the classifications assigned and to assess their reproducibility among different assessors. Laboratories were sent selected sections from cases of sCJD and vCJD with detailed protocols to be followed in determining the histotype according to the classification originally proposed by Parchi et al. [28], which was recently updated with the inclusion of categories for mixed subtypes [31].
Materials and methods
Case and tissue selection
The study was based on tissue samples from subjects affected by various subtypes of human prion disease, including the whole spectrum of currently characterized human prion strains [30]. All cases were selected from a larger pool of pathological material based on the following essential criteria: (1) availability of all necessary brain structures (see below); (2) inclusion of all known sporadic TSE subtypes according to the updated classification of Parchi et al. [31] as well as vCJD; and (3) absence of additional significant pathological comorbidity, such as “intermediate” or “high” Alzheimer’s disease neuropathological changes [19], cerebral haemorrhages or infarcts, or a known history of neurological disorders other than prion disease. PRNP genetics and PrPSc typing had been performed in all selected cases as described previously [10, 31]. PrPSc typing, in particular, was performed in each case in at least six brain samples (frontal, temporal, parietal, and occipital cortices, thalamus, and cerebellum). Local ethical committee approval (Medical Ethics Committee of the Ludwig Maximilians University, Munich) was obtained for the study of autopsy material, with written informed consent for research use provided by the patients during life or by their next of kin after death.
A total of 21 cases were included. Two “atypical” CJD cases (i.e. not fitting any of the subtypes defined by the current CJD classification) were also added to the series. The total number and anatomical representation of the sections included in the study were chosen by the reference group (PP, AG, and HK). The two major aims were on the one hand to keep the number of slides to a minimum and on the other to select all brain regions that highlight the distinctive features of each TSE subtype [11, 30, 31].
All selected cases displayed histopathological lesions and PrP-immunoreactive deposits.
Two sets of 4 μm thick sections were produced. Each set contained haematoxylin and eosin (H&E) stained sections from seven brain areas (frontal and occipital cortices, hippocampus, striatum, thalamus, medulla, and cerebellum) of the 21 cases, as well as sections stained for PrP by immunohistochemistry from the frontal and occipital cortices, hippocampus, and cerebellum of all cases. All sections were stained using the same methodology in one laboratory (LMU, München). Specimens were sent blindly with a combined letter and numerical code together with the operative instructions (see below).
Immunohistochemistry
In brief, after rehydration, the sections were autoclaved in citrate buffer and pretreated with 98 % formic acid for 60 min. Subsequently, the sections were incubated overnight at 4 °C with a monoclonal primary antibody directed against PrP (mab L42, a gift from Martin Groschup [37], diluted 1:50). The reaction product was visualized using the I View DAB detection system (Ventana/Roche, Mannheim, Germany) with the use of DAB as the chromogen.
Construction of assessment instructions and reference assessment of the selected material
Members of the reference group (PP, AG, HK) prepared a draft of the assessment instructions, including a detailed description of the typical histopathological features as well as the major exclusion criteria for each CJD type (Table 1), a diagnostic flow chart to be followed during the assessment (Fig. 1), photographs and definitions of the pathology to be evaluated (Fig. 2; Table 2), and a standardized data sheet (Table 3). The latter aimed to collect information on the dominant vacuole size, on the main pattern of PrP deposition, and on whether or not kuru-type or florid amyloid plaques were seen. In addition, five specific questions concerning the distribution and severity of histopathological lesions in specific neuroanatomical structures were included (Table 3). Finally, an alternative nomenclature, more suitable for a histopathological diagnosis performed in the absence of molecular data, was proposed for each of the sporadic disease variants or subtypes (Table 4).
Table 1.
Diagnostic criteria for pure and mixed human TSE histotypes
| Histotype | Typical features | Major criteria for exclusion |
|---|---|---|
| Pure phenotypes | ||
| MM/MV 1 | Spongiform change characterized by small vacuoles with predominant corticostriatal-thalamic and cerebellar involvementa Relative sparing of hippocampus compared to occipital cortexb Absence of a definite/clear-cut laminar pattern (i.e. predominant involvement of deep layers) of spongiform change and PrP deposition in the cerebral cortexc Synaptic pattern of PrP deposition |
Clusters of large, confluent vacuoles in the grey matter Coarse or perivacuolar PrP deposition in the grey matter PrP plaque-like deposition PrP-positive amyloid plaques |
| VV2 | Spongiform change characterized by small and medium-sized vacuoles Significant spongiform change in subcortical grey matter which is often more severe in striatum than in cerebral neocortex More severe spongiform change in the hippocampal CA1 and subiculum than in the occipital cortex Definite laminar pattern (i.e. predominant involvement of deep layers) of spongiform change and PrP deposition in at least one specimen from the cerebral cortex Plaque-like PrP deposition (best seen in cerebellar granular layer and white matter), usually associated with a perineuronal pattern in deep cortical layers and CA4 Significant cerebellar atrophy (particularly involving the granule cell layer) in comparison to that of occipital cortex |
Clusters of large, confluent vacuoles associated with perivacuolar/coarse PrP deposition in the grey matter Typical fully formed PrP-positive amyloid plaques of the kuru-type or florid plaques in the cerebellum or other areas Absence of plaque-like PrP deposition |
| MV 2K | Widespread cortical and subcortical pathology with plaque-like PrP deposition and amyloid plaques of kuru type mainly localized in the cerebellar granular layer | Clusters of large confluent vacuoles associated to perivacuolar PrP deposition in the grey matter Absence of PrP-positive amyloid plaques of the kuru type in the cerebellum Amyloid plaques of the florid type in the cerebral cortex |
| MM/MV 2C | Corticostriatal distribution of pathology with relative sparing of brainstem and cerebellum Spongiform change mainly comprising grape-like clusters of relatively large confluent vacuoles |
PrP-positive amyloid plaques Synaptic-type PrP staining in the molecular layer of cerebellum |
| MM 2T (sFI) | Moderate to severe selective atrophy of thalamic nuclei (i.e. anterior, dorsomedial, and pulvinar) in the absence of definite spongiform change in the same regions and not associated with severe cortical atrophy Moderate to severe selective atrophy of inferior olivary nuclei Absence of definite spongiform change in the cerebellum Relative sparing of striatum compared to the thalamus |
Definite PrP plaque-like or perivacuolar deposition PrP-positive amyloid plaques |
| VV1 | Corticostriatal distribution of pathology with relative sparing of cerebellum compared to the cerebral cortex Spongiform change comprising medium-sized vacuoles Synaptic PrP deposition (usually faint) Presence of (often) ballooned neurons in the most severely affected areas of the cerebral cortex |
Clusters of large vacuoles in the grey matter Definite coarse or perivacuolar PrP deposition in the grey matter Plaque-like PrP deposition in the grey matter PrP-positive amyloid plaques The cerebellum is more involved (i.e. shows more significant pathological changes) than the frontal cortex |
| vCJD (MM 2V) | Multiple florid plaques in the cerebral cortex (all lobes) and cerebellar cortex Spongiform encephalopathy most marked in the caudate nucleus and putamen Severe neuronal loss and gliosis in the pulvinar of the thalamus PrP-positive florid plaques, small cluster plaques and amorphous pericellular and pericapillary deposits |
Absence of florid plaques in routinely stained sections Lack of PrP-positive florid plaques, small cluster plaques and amorphous pericellular and pericapillary deposits |
| Mixed phenotypes | ||
| MM/MV 1+2C | Fits most criteria for MM/MV 1, but: Also shows clusters of large vacuoles associated with perivacuolar and coarse PrP deposition in the grey matter (most commonly in cerebral cortex or thalamus) or Fits most criteria for MM 2C, but also shows synaptic-type PrP staining in the molecular layer of the cerebellum |
|
| MV 2K+C | Fits most criteria for MV 2K, but also shows clusters of large vacuoles associated with perivacuolar and coarse PrP deposition in the grey matter (most commonly in cerebral cortex or thalamus) | |
| Atypical | Any case which does not fit the criteria for the pure or mixed phenotypes outlined above | |
The vacuolation largely disappears and is replaced by status spongiosus in cases with long duration and severe atrophy and gliosis
In cases with long duration and severe cortical atrophy and astrogliosis, this assessment should not be based on the degree of spongiform change but rather on the extent of neuronal loss and astrogliosis
An apparent predominant PrP deposition in the deep cortical layers may be seen in cases with long duration and severe cortical atrophy and astrogliosis
Fig. 1.

Recommended diagnostic flowchart for diagnosing human sporadic prion disease histotypes and vCJD
Fig. 2.
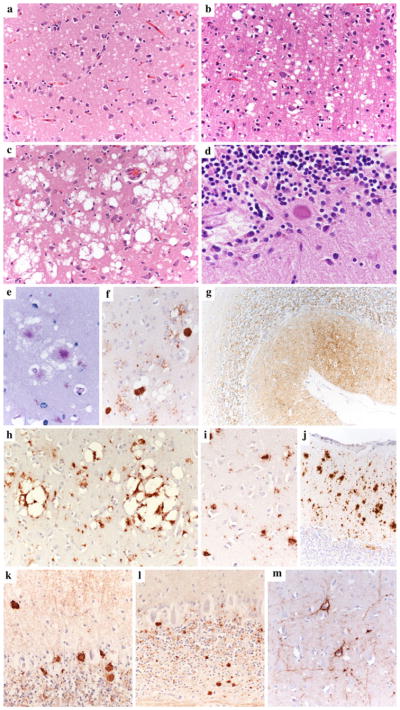
Distinctive histopathological features of sporadic human prion disease subtypes. a Typical spongiform change characterized by small, fine, microvacuoles (H&E stain, sCJD MM1, cerebral cortex); b spongiform change characterized by medium-sized vacuoles (H&E stain, sCJD VV1, cerebral cortex); c spongiform change characterized by relatively large confluent vacuoles (H&E stain, sCJD MM 2C cerebral cortex); d unicentric amyloid plaque of the kuru type (H&E stain, sCJD MV 2K, cerebellum); e, f florid plaques in the cerebral cortex (PAS stain and PrP IHC, vCJD, cerebral cortex); g synaptic pattern of PrP deposition in the cerebellum of sCJD MM1. A delicate diffuse staining is seen in the molecular layer, while the cerebellar glomeruli are stained in the granular cell layers. h Perivacuolar and i coarse PrPSc staining as typically seen in MM 2C and MM/MV 1+2C mixed sCJD cases. The cerebellum in MM 2C is either PrP negative or shows a focal patchy/coarse staining (j). k Plaque-like PrP deposits in the granular and molecular layers of the cerebellum in MV 2K. l Smaller plaque-like PrP deposits in the granular and molecular layers of the cerebellum in sCJD VV2. These plaque-like deposits are not visible in routine H&E or PAS-stained sections. m Perineuronal PrPSc staining in the deep cortical layers of sCJD VV2. All pictures in the panel have the same magnification (×200), except for d (×400) and g (×100)
Table 2.
Definition of “diagnostic” lesions (see also Fig. 1)
| Spongiform change | PrP+ amyloid deposits | Other PrP deposits |
|---|---|---|
|
Small vacuoles (Fig. 1a): Small round to oval vacuoles located in the neuropil (most vacuoles are not larger than the nuclei of activated astrocytes and neuronal cells) |
Of kuru type (Fig. 1d): Unicentric, rounded, amyloid plaques with a saw-like contour |
“Synaptic” pattern (Fig. 1g): Diffuse or focal fine granular staining of the neuropil including focal areas with more dense staining and immunoreactive dot-like structures |
|
Medium-sized vacuoles (Fig. 1b): Small to medium-sized round to oval vacuoles located in the neuropil (many vacuoles are larger than the nuclei of activated astrocytes and neuronal cells) |
Of florid type (Fig. 1e, f): Large fibrillary amyloid plaques surrounded by a halo of spongiform change |
Coarse or perivacuolar pattern (Fig. 1i, h): Diffuse or patchy staining of the neuropil characterized by coarse deposits (Fig. 1i) of irregular size and shape which are typically (but not always) associated with clusters of large confluent vacuoles (Fig. 1h). In the cerebellum (when present), these patchy/coarse deposits are not associated with spongiform change and are mostly found in the molecular layer (Fig. 1j) |
| Clusters of large, confluent vacuoles (Fig. 1c): Morula/grape-like clusters of coalescing large vacuoles |
Plaque-like pattern (Fig. 1k, l): Focal, well-demarcated, rounded/oval extracellular deposits. In the cerebellum they are mainly located in the granular layer. Typically, some deposits are also found in the white matter. Their size varies but at least some of them exceed the size of the nucleus of cerebellar Purkinje cells. They are larger and more numerous in MV 2K (Fig. 1k) than in VV2 (Fig. 1i) |
|
|
Perineuronal pattern (Fig. 1m): Dot-like pattern of staining outlining the contour of the perikarya and/or the neurites of pyramidal neurons |
Table 3.
Assessment sheet
| Section | Anatomical region | Vacuolation size: S,M,C* | Kuru type plaques yes/no | Florid type plaques yes/no | PrP synaptic yes/no | PrP coarse/ peri vacuolar yes/no | PrP plaque-like yes/no | PrP perineuronal yes/no | Comments |
|---|---|---|---|---|---|---|---|---|---|
| 1. Cerebellum (Cbl) | molecular cell layer | ** | |||||||
| granular cell layer | |||||||||
| white matter | |||||||||
| 2. Frontal Cortex (FCtx) | |||||||||
| 3. Occipital Cortex (OCtx) | |||||||||
| 4. Hippocampus (Hipp) | CA4 | ||||||||
| CA1/subiculum | |||||||||
| 5. Striatum (Str) | caudate/putamen |
S=small vacuoles, M=medium size vacuoles, C=Clusters of large confluent vacuoles. It can be either one or more (S or S+M or S+C and so on…)
Grey boxes are not required to be filled in.
| 1) Is there a clear-cut laminar pattern in the cerebral cortex (deep cortical layers more affected by spongiform change/PrP-deposits than the superficial ones)? (YES/NO): | __________ |
| 2) Is there a significant relative sparing of the hippocampus compared to the occipital cortex? (YES/NO): | __________ |
| 3) Is there a significant relative sparing of the cerebellum compared to the frontal cortex? (YES/NO): | __________ |
| 4) Is there a moderate to severe atrophy of the medial thalamic nuclei without spongiform change? (YES/NO): | __________ |
| 5) Is there a moderate to severe atrophy of the inferior olivary nuclei? (YES/NO): | __________ |
| Classification of CJD type (tick one box): |
|
| COMMENTS: _______________ | |
Table 4.
Human prion disease types—terminology
| No. | Molecular types | Histopathological types (Prion disease type with…..) |
|---|---|---|
| 1 | MM/MV 1 | Diffuse synaptic deposits |
| 2 | VV2 | Perineuronal and cerebellar plaque-like deposits |
| 3 | MV 2K | Kuru plaques |
| 4 | MM 2C | Cortical confluent vacuoles |
| 5 | MM 2T (=sFI) | Thalamo-olivary atrophy |
| 6 | VV1 | Corticostriatal synaptic deposits |
| 7 | MM 2V (=vCJD) | Florid plaques |
| 8 | MM/MV 1+2C | Mixed diffuse synaptic deposits and cortical confluent vacuoles |
| 9 | MV 2K+C | Mixed Kuru plaques and cortical confluent vacuoles |
| 10 | Atypical |
Assessors were then invited to a joint meeting to discuss the document drafts and simultaneously assess some exemplary cases using a multi-headed microscope.
The documents were then refined based on the most significant suggestions that emerged during the meeting, and a final version of each document (Tables 1, 2, 3, 4; Figs. 1, 2) was prepared by the reference group to be circulated among the participants.
Instructions to raters
All raters were supplied with the written directives on the criteria to define each CJD type, the definition of the lesions to be assessed, the diagnostic flow-chart to follow, and the standardized assessment sheet. Raters were requested to fill in the data sheet and assign a CJD type diagnosis based on the criteria summarized in Table 1 and the guidance given by the diagnostic flow-chart (Fig. 1).
Joint assessment by the reference group
Following the individual assessment phase, the reference group convened a meeting to jointly analyze the data recorded in the assessment sheets by each evaluator. Inconsistencies in these observations were discussed and pitfalls were sought. In particular, the consistency between the actual observations made by the assessor concerning the type and distribution of histopathological lesions (upper part of the assessment sheet) and the final CJD type-specific diagnosis were carefully evaluated. In other words, the accurate application of diagnostic criteria by each assessor was double-checked. Finally, when there were significant discrepancies among the assessors, the stained sections were re-evaluated under the multi-headed microscope.
Results
Reference group joint assessment
One of the 21 selected cases gave rise to a highly significant inter-rater discrepancy in the assessment. Originally selected as a case of sCJD MV 2K, under the joint reassessment the case was re-classified as MV 2K+C because of the recognition of a tiny area in the cerebral cortex showing large confluent vacuoles and a perivacuolar pattern of PrP deposition.
The joint re-analyses also identified two assessments in which there was an obvious inconsistency between the observed lesions (i.e. those described in the upper part of the assessment sheet) and the final chosen TSE histotype. The first concerned the atypical case showing amyloid plaques and plaque-like PrP deposition in both cortical and cerebellar white matter. One assessor correctly identified this case as an atypical MM1 case with white matter amyloid plaques as described by Kobayashi et al. [13], but then typed it as MM/MV1. The second inconsistency concerned a typical VV2 case. The assessor correctly identified all of the distinctive features of the VV2 sCJD histotype and did not report any atypical features, but then crossed the box “atypical case”. Given the obviousness of these discrepancies, the final diagnosis was changed accordingly for both of these assessments.
Agreement of different raters
Table 5 shows how the 21 cases were “histotyped” by the 13 assessors. Apart from the case which was re-assessed and re-typed by the reference group, the inter-rater histotyping agreement was high overall, ranging from 69 to 100 %. All of the assessors were in full agreement regarding the assigned histotype in nine cases, and showed very strong agreement (i.e. only one or two divergent assessments) in six additional cases. Overall, the agreement was strongest (full agreement) for the two most common sCJD histotypes (i.e. MM/MV1 and VV2) and vCJD, very high for the pure phenotypes MV 2K, MM 2T, and MM 2C as well as the mixed MM/MV 1+2C subtype, whereas the pure phenotype VV1, the mixed MV 2K+C subtype and the “atypical” cases shared the lowest degree of diagnostic concordance. Taken together, 11 out of the 13 raters assessed 86–100 % of the cases correctly.
Table 5.
Results of the histotyping by each assessor (Ass)
| TSE histotyping by reference group | Ass. 1 | Ass. 2 | Ass. 3 | Ass. 4 | Ass. 5 | Ass. 6 | Ass. 7 | Ass. 8 | Ass. 9 | Ass. 10 | Ass. 11 | Ass. 12 | Ass. 13 | Agree (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MM/MV 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 100 |
| MM/MV 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 100 |
| MM 1+2C | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 1 | 1 | 8 | 8 | 8 | 85 |
| MM 1+2C | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 100 |
| VV 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 100 |
| VV 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 100 |
| MV 2K | 3 | 3 | 9e | 3 | 2c | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 85 |
| MV 2K | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 100 |
| MV 2K+C | 9 | 9 | 9 | 9 | 3 | 9 | 9 | 3 | 3 | 3 | 3 | 9 | 3 | 9 | 54 |
| MV 2K+C | 9 | 9 | 9 | 9 | 8b | 9 | 10c | 9 | 9 | 9 | 9 | 9 | 9 | 3 | 77 |
| MM 2C | 4 | 8 | 4 | 4 | 4 | 4 | 4 | 10d | 9f | 4 | 4 | 4 | 4 | 4 | 77 |
| MM 2C | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 9f | 9f | 4 | 4 | 4 | 4 | 4 | 85 |
| MM 2C | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 8h | 8h | 4 | 4 | 4 | 4 | 4 | 85 |
| VV 1 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 1 | 1 | 1a | 1a | 6 | 6 | 6 | 69 |
| VV 1 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 1 | 1 | 6 | 6 | 6 | 6 | 10d | 77 |
| MM 2T | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 100 |
| MM 2T | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 10d | 92 |
| Atypical | 10 | 3g | 10 | 1b | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 85 |
| Atypical | 10 | 6b | 10 | 4 | 10 | 10 | 10 | 6b | 6b | 10 | 10 | 10 | 10 | 10 | 69 |
| vCJD | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 100 |
| vCJD | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 100 |
| Agree (%) | 86 | 95 | 90 | 86 | 100 | 95 | 66 | 66 | 86 | 86 | 100 | 95 | 86 | ||
The assessor stated that VV1 would also be a possible diagnosis
Impossible diagnosis according to the given exclusion criteria
The kuru-type amyloid plaques in cerebellum were missed
Criteria for defining the case as atypical were not provided by the assessor
The assessor considered the presence of a single “doubtful” IHC focus of coarse PrP deposition in the occ. cortex to be relevant
Typed as MV 2K despite no kuru-type amyloid plaques being seen
Typed as MV 2K despite no plaque-like deposits being seen in the cerebellar granular layer
Typed as MM 1+2C despite no synaptic deposits being seen in the molecular layer of the cerebellum
Analyses of divergent assessments
The analyses of the histotype assessments, which were at variance with those of the reference group, showed that the discrepant diagnosis was generated either by the incorrect application of diagnostic criteria or by obvious misjudgement in about 30 % of cases. For example, in three instances, MM 2C cases were typed as MV 2K despite no kuru-type amyloid plaques being seen; in another one, the rater typed an atypical case showing amyloid plaques in the white matter as MV 2K despite the fact that no plaque-like deposits were detected in the cerebellar granular layer, and two assessors typed a MM 2C case as MM 1+2C despite no synaptic deposits being seen in the molecular layer of the cerebellum. Furthermore, in four instances, the two atypical cases were typed either as VV1 or as MM/MV1 despite the presence of plaque-like deposits, which was given as an exclusion criterion for both the VV1 and MM/ MV1 histotypes.
There were only two significant sources of divergent assessment that were unrelated to incorrect application of the diagnostic criteria. The first concerned the recognition of MM 2C features co-occurring with either MM/MV1 or MV 2K, especially when they were focal and limited to one area in one slide, whereas the second pertained to the distinction between the MM/MV1 and the VV1 histotypes. Finally, additional minor sources of incorrect assessment were the lack of recognition of the cerebellar amyloid plaques of the kuru type in two cases by one rater, and the lack of recognition of the rarest phenotypes (the MM 2C, MM 2T, and VV1 variants were typed as atypical cases once each) by three raters.
Analyses of the prion-related pathology assessment
The analyses of the data sheet (Table 3) including the assessment of the prion-related histopathology provided additional relevant information.
Vacuole size
This morphological feature is relevant to distinguish between MM/MV1 and VV1, and it represents, above all, one of the distinctive histological markers for the MM 2C type. As far as the first differential diagnosis was concerned, the presence of medium-sized vacuoles in the cerebral cortex was recognized in 25 out of 26 assessments in the VV1 cases, and in 10 out of 26 assessments in the MM/MV1 cases. More significantly, while 22 out of 26 evaluations detected medium-sized vacuoles in the striatum of VV1, none of the assessors saw the same type of vacuoles in the striatum of MM/MV1 cases. Furthermore, while all participants identified the relative sparing of cerebellum compared to the frontal cortex in the VV1 cases, none of them detected the same characteristic in the MM/MV1 cases. Finally, recognition of large confluent vacuoles was significantly associated with the MM/ MV 2C type (either pure or mixed) with 86.3 % specificity and 96.2 % sensitivity (the specificity increased to 97 % when the two vCJD cases were excluded from the analyses).
Kuru-type plaques in the cerebellum
These were only detected in MV 2K or MV 2K+C (100 % specificity), although one assessor did not identify the plaques in two cases (96.2 % sensitivity).
Florid-type plaques
These were seen only in vCJD by all evaluators (100 % specificity and sensitivity).
Pattern of PrP deposition (IHC)
As expected, none of the recognized patterns of PrP deposition (i.e. synaptic, coarse/perivacuolar, plaque-like, and perineuronal) were specifically associated with a single histotype. Furthermore, the analyses of the inter-rater assessment of PrP deposition revealed significant disagreement among assessors in the definition of cerebellar and, to a lesser extent, cortical deposits in MM 2C cases. Indeed, while 100 % of evaluations recognized coarse and perivacuolar protein deposits in the cerebral cortex, in agreement with previous reference descriptions of this rare subtype [28, 31], some (49 %) also detected plaque-like deposits. Even more strikingly, cerebellar PrP deposits in MM 2C cases were more frequently described as plaque-like than as coarse or perivacuolar (85 vs. 36 %).
Nevertheless, when the analysis was restricted to specific anatomical regions, the coarse/perivacuolar pattern in the cerebral cortex was significantly associated with the MM/MV 2C type (either pure or mixed) with 91 % specificity, and the plaque-like pattern in the granular layer of cerebellum with both VV2 and MV 2K types (93 % specificity among sCJD subtypes).
Similarly, the perineuronal pattern of PrP deposition was significantly associated with both the VV2 and MV 2K subtypes (98 % sensitivity and 92 % specificity among sCJD subtypes). Finally, the synaptic pattern of PrP deposition only appeared useful as a negative marker; indeed, among cases lacking amyloid plaques, the absence of this pattern in the cerebellum strongly indicated either the MM 2C or the MM 2T subtype.
Analyzing the answers given by the assessors to the five questions included in the data sheet (Table 3) gave the following results:
The recognition of a clear-cut laminar pattern in the cerebral cortex (Fig. 3) was significantly correlated with either the VV2 or the MV 2K subtype (96 % sensitivity and 94 % specificity).
The relative sparing of the cerebellum compared to the frontal cortex distinguished the MM 2C, MM 2T, and VV1 groups (Fig. 3) from the other CJD subtypes (93 % sensitivity and 86 % specificity).
The coexistence of moderate to severe thalamic and olivary atrophy (Fig. 3) was a strong diagnostic marker for the MM 2T subtype (100 % sensitivity and 99 % specificity).
Fig. 3.

Distinctive regional distribution of histopathological changes in sCJD histotypes MM 2T, VV1, and VV2. Severe neuronal loss and gliosis in the medial thalamic (a) and inferior olivary nuclei (b) and relative sparing of anterior striatum (c) in sCJD MM 2T. Moderate to severe spongiform change (small to medium-sized vacuoles), gliosis, and neuronal loss in the cerebral cortex (d) and anterior striatum (e), and relative sparing of the cerebellum (f) in sCJD VV1; laminar distribution of spongiform change, with relative sparing of superficial (g) compared to the deep (h) layers in the cerebral neocortex of sCJD VV2. Severe spongiform change in the CA1/subiculum areas of the hippocampus (i) despite the virtually complete sparing of the occipital cortex (j) in a typical VV2 case with a relative short duration (five months). All pictures in the panel have the same magnification (×100) except for a and b (×200)
Discussion
In the present study we tested, for the first time, the inter-rater reliability of histopathological criteria aimed at distinguishing among sporadic human prion disease subtypes as well as vCJD. Most of the neuropathologists responsible for human TSE diagnosis in Europe and the USA were involved in this study. By showing an overall strong agreement among the raters, the results obtained not only fully support the current classification of human prion disease, but also demonstrate that neuropathological examination alone allows reliable classification and strain typing in the absolute majority of cases, with obvious implications for current diagnostic practice and epidemiologic studies. Results were particularly excellent for vCJD, the three most common pure sCJD subtypes (i.e. MM/MV1, VV2, and MV 2K), and MM 2T, a rare but very distinctive disease subtype, also known as sFI. Notably, four out of five of these human sporadic TSE variants have been shown to generate distinct prion strains upon transmission to experimental animals, which points further to the strain phenomenon being the main determinant of phenotypic variability in human prion disease [2, 14–17, 20, 26]. Among the pure and strain-specific sCJD subtypes, results were less satisfactory only for the VV1 group, one of the rarest sCJD variants. Nevertheless, the results of the present study indicate that the diagnostic accuracy of this sCJD type, and its distinction from the MM/MV 1 variant in particular, can be further improved by more strongly emphasizing the combined criteria of (1) the presence of medium-sized vacuoles in the striatum and (2) the relative sparing of cerebellum compared to the frontal cortex. Indeed, by retrospectively applying only these two features as inclusion criteria, we calculated that there was 89 % assessor agreement in the distinction between the two subtypes. Furthermore, it must be also considered here that codon 129 genotyping, which is often performed on blood samples collected in vitam, may also help to corroborate the correct typing of VV1 cases, even in the absence of frozen tissue.
The most significant source of disagreement among raters was, as expected, the recognition of the co-occurrence of phenotypic features of two subtypes when one of the two is only focally expressed. This difficulty was especially evident in the MM/MV 2K+C group, likely because the coarse/perivacuolar pattern (“2C” phenotype) is less distinguishable from the plaque-like pattern (“2K” phenotype) than from the synaptic pattern (MM/MV1 phenotype). The results obtained, showing strong agreement in three out of four cases, are, nevertheless, encouraging and significant, given that histopathological examination is to date the only way to recognize mixed phenotypes associated with the same (or similar) PrPSc type (i.e. 2K+C). Furthermore, it should be borne in mind that in order to reduce the complexity and the cost of the diagnostic protocol in this study, we kept the number of slides to a minimum, while the chance of recognizing focal phenotypic features is known to increase—at least to a certain degree—with the amount of tissue examined. Based on the present results it is therefore recommended that a minimum of four samples from the cerebral cortex (one for each lobe) should be examined histopathologically, as recently proposed by Parchi et al. [31]. Similarly, it is recommended that at least two distinct samples of cerebellum—possibly including the vermis—should be examined, since this will increase the likelihood of the amyloid kuru-type plaques being recognized, even when they are scanty.
The analyses of the assessments of PrP immunostaining indicate that none of the previously defined patterns of PrP deposition are fully specific to one histotype, although each of the three major PrP staining patterns (i.e. synaptic, coarse/perivacuolar, and plaque-like) strongly characterizes some of the histotypes either positively (i.e. it is always present) or negatively (i.e. it is always absent). As a consequence, the present results validate PrP staining as a reliable source of exclusion diagnostic criteria for some disease subtypes. The most significant examples of such exclusion criteria include the presence of either coarse/ perivacuolar or plaque-like deposits in the grey matter of MM/MV1, VV1, or MM 2T, the absence of plaque-like deposits in VV2, the absence of coarse or perivacuolar deposits in MM/MV 2C, the absence of PrP-positive amyloid plaques in the granular layer of cerebellum in MV 2K, and the presence of a synaptic type of staining in the molecular layer of cerebellum in MM 2C.
Similarly, the assessment of the regional distribution of lesions confirmed that some patterns, although not totally specific to one histotype, constitute reliable supporting diagnostic criteria for some disease subtypes. These include, in order of importance, the moderate to severe thalamic and olivary atrophy seen in MM 2T (Fig. 3), the relative sparing of cerebellum compared to the cerebral cortex seen in VV1 (Fig. 3) and MM 2C, and the laminar intracortical distribution of spongiform change seen in VV2 (Fig. 3) or MV 2K.
Finally, it is also important to underline that all 13 assessors from the seven laboratories involved in CJD surveillance worldwide correctly identified the two vCJD cases and distinguished them from all sCJD variants and atypical cases without the need for PrPSc typing. In practical terms, this is also a significant result for human CJD surveillance in Europe and USA.
In conclusion, this study validates “histotyping” as a reliable means of human prion disease classification and provides detailed instructions for histopathological typing that can be used as a reference for future epidemiological studies. When combined with those previously obtained on PrPSc typing [29], the present results indicate that both molecular (i.e. PrPSc typing and codon 129 genotyping) and histopathological analyses are reliable and powerful approaches for human prion disease classification and strain typing. They both guarantee an extremely high interlaboratory accuracy even when applied individually, although the combination of the two approaches, when feasible, remains the gold standard for the diagnosis and classification of human prion disease.
Acknowledgments
This work was supported by the European Commission (contract: FOOD-CT-2004-506579). We wish to thank Prof. Martin Groschup, Friedrich-Löffler-Institut, Greifswald-Insel Riems, for his generous gift of monoclonal antibody L42, as well as Angelika Henn, Ursula Jung, and Sabrina Boninsegna for technical assistance.
Contributor Information
Piero Parchi, Email: piero.parchi@unibo.it, IRCCS Istituto delle Scienze Neurologiche and Dipartimento di Scienze Neurologiche, Universitàdi Bologna, Bologna, Italy.
Laura de Boni, Institut für Neuropathologie, Ludwig-Maximilians-Universität, Munich, Germany.
Daniela Saverioni, IRCCS Istituto delle Scienze Neurologiche and Dipartimento di Scienze Neurologiche, Universitàdi Bologna, Bologna, Italy.
Mark L. Cohen, National Prion Pathology Surveillance Centre, CWRU, Cleveland, OH, USA
Isidro Ferrer, Institut de Neuropatologia, Hospital Universitari de Bellvitage, Universitat de Barcelona, Barcelona, Spain.
Pierluigi Gambetti, National Prion Pathology Surveillance Centre, CWRU, Cleveland, OH, USA.
Ellen Gelpi, Neurological Tissue Bank of the Biobanc-Hospital Clinic-IDIBAPS, Barcelona, Spain.
Giorgio Giaccone, Laboratorio di Neuropatologia, Fondazione IRCCS Istituto Nazionale Neurologico Carlo Besta, Milan, Italy.
Jean-Jacques Hauw, APHP, Laboratoire de Neuropathologie Raymond Escourolle, GH Pitiè-Salpêtrière Université Pierre et Marie Curie (UPMC-Paris 6), Sorbonne-Universités, Paris, France.
Romana Höftberger, Institute of Neurology, AKH 4J, Medical University of Vienna, Vienna, Austria.
James W. Ironside, National CJD Research and Surveillance Unit, Western General Hospital, Edinburgh EH4 2XU, UK
Casper Jansen, Dutch Surveillance Centre for Prion Diseases, University Medical Centre Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands.
Gabor G. Kovacs, Institute of Neurology, AKH 4J, Medical University of Vienna, Vienna, Austria
Annemieke Rozemuller, Dutch Surveillance Centre for Prion Diseases, University Medical Centre Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands.
Danielle Seilhean, APHP, Laboratoire de Neuropathologie Raymond Escourolle, GH Pitiè-Salpêtrière Université Pierre et Marie Curie (UPMC-Paris 6), Sorbonne-Universités, Paris, France.
Fabrizio Tagliavini, Laboratorio di Neuropatologia, Fondazione IRCCS Istituto Nazionale Neurologico Carlo Besta, Milan, Italy.
Armin Giese, Institut für Neuropathologie, Ludwig-Maximilians-Universität, Munich, Germany.
Hans A. Kretzschmar, Email: hans.kretzschmar@med.uni-muenchen.de, Institut für Neuropathologie, Ludwig-Maximilians-Universität, Munich, Germany
References
- 1.Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 2.Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt–Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA. 2010;107:12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 4.Bruce ME, Boyle A, Cousens S, McConnell I, Foster J, Goldmann W, Fraser H. Strain characterization of natural sheep scrapie and comparison with BSE. J Gen Virol. 2002;83:695–704. doi: 10.1099/0022-1317-83-3-695. [DOI] [PubMed] [Google Scholar]
- 5.Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J, Langeveld JP, Parchi P, Safar JG, Zou WQ, Gambetti P. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt–Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132:2643–2658. doi: 10.1093/brain/awp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt–Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011;121:21–37. doi: 10.1007/s00401-010-0760-4. [DOI] [PubMed] [Google Scholar]
- 7.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 8.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 9.Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P. Hereditary prion protein amyloidoses. Clin Lab Med. 2003;23:65–85. doi: 10.1016/s0272-2712(02)00064-1. [DOI] [PubMed] [Google Scholar]
- 10.Head MW, Bunn TJR, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, Williams MC, McCardle L, MacKenzie J, Knight R, Will RG, Ironside JW. Prion protein heterogeneity in sporadic but not variant Creutzfeldt–Jakob Disease: UK cases 1991–2002. Ann Neurol. 2004;55:851–859. doi: 10.1002/ana.20127. [DOI] [PubMed] [Google Scholar]
- 11.Ironside JW, McCardle L, Horsburgh A, Lim Z, Head MW. Pathological diagnosis of variant Creutzfeldt–Jakob disease. APMIS. 2002;110:79–87. doi: 10.1034/j.1600-0463.2002.100110.x. [DOI] [PubMed] [Google Scholar]
- 12.Jansen C, Parchi P, Capellari S, Ibrahim-Verbaas CA, Schuur M, Strammiello R, Corrado P, Bishop MT, van Gool VA, Verbeek MM, Baas F, van Saane V, Spliet WGM, Jansen GH, van Duijn CM, Rozemuller AJM. Human prion diseases in The Netherlands (1998–2009): clinical, genetic and molecular aspects. PLoS ONE. 2012;7:e36333. doi: 10.1371/journal.pone.0036333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi A, Arima K, Ogawa M, Murata M, Fukuda T, Kitamoto T. Plaque-type deposition of prion protein in the damaged white matter of sporadic Creutzfeldt–Jakob disease MM1 patients. Acta Neuropathol. 2008;116:561–566. doi: 10.1007/s00401-008-0425-8. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T. Experimental verification of a traceback phenomenon in prion infection. J Virol. 2010;84:3230–3238. doi: 10.1128/JVI.02387-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, Heinrich C, Tremblay P, De-Armond SJ, Prusiner SB. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci USA. 2003;100:4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, Prusiner SB. Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med. 1999;340:1630–1638. doi: 10.1056/NEJM199905273402104. [DOI] [PubMed] [Google Scholar]
- 17.Moda F, Suardi S, Di Fede G, Indaco A, Limido L, Vimercati C, Ruggerone M, Campagnani I, Langeveld J, Terruzzi A, Brambilla A, Zerbi P, Fociani P, Bishop MT, Will RG, Manson JC, Giaccone G, Tagliavini F. MM2-thalamic Creutzfeldt–Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol. 2012 doi: 10.1111/j.1750-3639.2012.00572. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, Cortelli P, Montagna P, Ghetti B, Goldfarb LG, Gajdusek DC, Lugaresi E, Gambetti P, Autilio-Gambetti L. Fatal familial insomnia and familial Creutzfeldt–Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA. 1994;91:2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nonno R, Di Bari M, Cardone F, Vaccari G, Fazzi P, Dell’Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, Sbriccoli M, Lipp HP, Bruce M, Pocchiari M, Agrimi U. Efficient transmission and characterization of Creutzfeldt–Jakob disease strains in bank voles. PLoS Pathog. 2006;2:e12. doi: 10.1371/journal.ppat.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B, Gambetti P, Kretzschmar HA, Parchi P. Effects of different experimental conditions on the PrPSc core generated by protease digestion. J Biol Chem. 2004;279:16797–16804. doi: 10.1074/jbc.M313220200. [DOI] [PubMed] [Google Scholar]
- 22.Notari S, Strammiello R, Capellari S, Giese A, Cescatti M, Grassi J, Ghetti B, Langeveld JP, Zou WQ, Gambetti P, Kretzschmar HA, Parchi P. Characterization of truncated forms of abnormal prion protein in Creutzfeldt–Jakob disease. J Biol Chem. 2008;283:30557–30565. doi: 10.1074/jbc.M801877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parchi P, Capellari S, Chen SG, Petersen RB, Gambetti P, Kopp N, Brown P, Kitamoto T, Tateishi J, Giese A, Kretzschmar H. Typing prion isoforms. Nature. 1997;386:232–233. doi: 10.1038/386232a0. [DOI] [PubMed] [Google Scholar]
- 24.Parchi P, Capellari S, Chin S, Schwartz HB, Schecter NP, Butts JD, Hudkins P, Burns DK, Powers JM, Gambetti P. A subtype of sporadic prion disease mimicking fatal familial insomnia. Neurology. 1999;52:1757–1763. doi: 10.1212/wnl.52.9.1757. [DOI] [PubMed] [Google Scholar]
- 25.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AAF, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt–Jakob disease. Ann Neurol. 1996;39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 26.Parchi P, Cescatti M, Notari S, Schulz-Schaeffer WJ, Capellari S, Giese A, Zou WQ, Kretzschmar H, Ghetti B, Brown P. Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain. 2010;133:3030–3042. doi: 10.1093/brain/awq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann–Sträussler–Scheinker disease. Proc Natl Acad Sci USA. 1998;95:8322–8327. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt–Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 29.Parchi P, Notari S, Weber P, Schimmel H, Budka H, Ferrer I, Haik S, Hauw JJ, Head MW, Ironside JW, Limido L, Rodriguez A, Ströbel T, Tagliavini F, Kretzschmar HA. Inter-laboratory assessment of PrPSc typing in Creutzfeldt–Jakob disease: a Western blot study within the NeuroPrion Consortium. Brain Pathol. 2009;19:384–391. doi: 10.1111/j.1750-3639.2008.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol. 2011;121:91–112. doi: 10.1007/s00401-010-0779-6. [DOI] [PubMed] [Google Scholar]
- 31.Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A, Zerr I, Roncaroli F, Cras P, Ghetti B, Pocchiari M, Kretzschmar H, Capellari S. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009;118:659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ, Jr, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57:979–988. doi: 10.1097/00005072-199810000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. Sporadic Creutzfeldt–Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology. 1999;53:2173–2176. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- 36.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 37.Vorberg I, Buschmann A, Harmeyer S, Saalmüller A, Pfaff E, Groschup MH. A novel epitope for the specific detection of exogenous prion proteins in transgenic mice and transfected murine cell lines. Virology. 1999;255:26–31. doi: 10.1006/viro.1998.9561. [DOI] [PubMed] [Google Scholar]
- 38.Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 39.Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP, Castellani R, Notari S, Crain B, Schmidt RE, Geschwind M, Dearmond SJ, Cairns NJ, Dickson D, Honig L, Torres JM, Mastrianni J, Capellari S, Giaccone G, Belay ED, Schonberger LB, Cohen M, Perry G, Kong Q, Parchi P, Tagliavini F, Gambetti P. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68:162–172. doi: 10.1002/ana.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]