

Abstract
Exposure to arsenic is associated with an increased risk of lung disease. Novel strategies are needed to reduce the adverse health effects associated with arsenic exposure in the lung. Nrf2, a transcription factor that mediates an adaptive cellular defense response, is effective in detoxifying environmental insults and prevents a broad spectrum of diseases induced by environmental exposure to harmful substances. In this report, we tested whether Nrf2 activation protects mice from arsenic-induced toxicity. We used an in vivo arsenic inhalation model that is highly relevant to low environmental human exposure to arsenic-containing dusts. Two-week exposure to arsenic-containing dust resulted in pathological alterations, oxidative DNA damage, and mild apoptotic cell death in the lung; all of which were blocked by sulforaphane (SF) in an Nrf2-dependent manner. Mechanistically, SF-mediated activation of Nrf2 alleviated inflammatory responses by modulating cytokine production. This study provides strong evidence that dietary intervention targeting Nrf2 activation is a feasible approach to reduce adverse health effects associated with arsenic exposure.
Keywords: Nrf2, Keap1, Arsenic, Antioxidant response
Introduction
Inorganic arsenic is a ubiquitous environmental contaminant found in water and dust. Several epidemiological studies have correlated arsenic exposure to the incidence of human disease (Cohen et al., 2000; Hertz-Picciotto and Smith, 1993; Smith et al., 1992; Steinmaus et al., 2000; Tchounwou et al., 2003), including bladder, skin and lung cancer (Byrd et al., 1996; Chen et al., 1992; Kenyon and Hughes, 2001; Tchounwou et al., 2003). The lung is a major target organ for arsenic-induced toxicities. Human epidemiological studies found that exposure to arsenic contributes to more than 5000 lung cancer cases per year in the United States (Putila and Guo, 2011). In addition, these studies also found a strong association of arsenic exposure with increased incidence of pulmonary malfunctions manifested as chronic cough, bronchitis and shortness of breath (Mazumder et al., 2000; Smith et al., 1998; von Ehrenstein et al., 2005). Consequently, novel strategies are needed to reduce the adverse health effects associated with arsenic exposure in the lung.
Several studies have shown that activation of the transcription factor Nrf2 elicits a cellular defense system that detoxifies drugs and environmental pollutants (Higgins et al., 2009; Kensler et al., 2007; Villeneuve et al., 2010; Zhang, 2006). Nrf2 mediates this response by upregulating antioxidant response element (ARE)-bearing genes (Higgins et al., 2009; Kensler et al., 2007; Villeneuve et al., 2010; Zhang, 2006), including glutamate-cysteine ligase (both subunits GCLC/GCLM), glutathione S-transferase (GST), NAD(P)H quinone oxidoreductase 1 (NQO1), and heme oxygenase 1 (HO-1) (Higgins et al., 2009; Kensler et al., 2007; Villeneuve et al., 2010; Zhang, 2006). Activation of Nrf2 has been reported to confer protection against many human diseases, including cancer, neurodegenerative disease, cardiovascular diseases, and diabetes-related complications (Cho and Kleeberger, 2010; Murthy et al., 2009; Osburn and Kensler, 2008; Vargas and Johnson, 2009; Zheng et al., 2011).
The importance of Nrf2 in defending against arsenic toxicity has been well documented in cell culture systems. Our laboratory has shown that Nrf2-knockout (Nrf2-KO) mouse embryonic fibroblast (MEF) cells are more sensitive to arsenic, compared to Nrf2-wildtype (Nrf2-WT) MEF cells (Wang et al., 2007). Sulforaphane (SF) and tBHQ, two well known Nrf2 activators, also reduce the cytotoxic effects of inorganic arsenic (sodium arsenite) and organic arsenic (monomethylarsonuous acid (MMAIII)) in UROtsa cells (Wang et al., 2007). Other groups have also demonstrated the effects of SF against arsenic toxicity in hepatocytes (Shinkai et al., 2006). Many other Nrf2 activators including lipoic acid, cinnamaldehyde, and oridonin were also able to protect against arsenic-induced toxicity in a variety of cell types (Du et al., 2008; Huerta-Olvera et al., 2010; Wondrak et al., 2010).
Our laboratory recently reported the protective effects of Nrf2 in vivo. Nrf2-KO mice exposed to sodium arsenite through drinking water for six weeks showed more pathological alterations in the bladder, liver and lungs compared to Nrf2-WT mice (Jiang et al., 2009). However, until now, no single study had been done in a rodent model investigating the efficacy of Nrf2 activation in ameliorating inhaled arsenic toxicity in the lung. In this report, we used an in vivo arsenic inhalation model that is highly relevant to low environmental human exposure to arsenic-containing dusts. Two-week exposure of mice to arsenic-containing dust resulted in obvious pathological alterations, oxidative DNA damage, and mild apoptotic cell death in the lung; all of which were blocked by SF in an Nrf2-dependent manner. Mechanistically, SF-mediated activation of Nrf2 alleviated inflammatory responses by reducing proinflammatory cytokine production. This study provides strong evidence that dietary intervention targeting Nrf2 activation is a feasible approach to reduce adverse health effects associated with arsenic exposure.
Materials and methods
Whole animal inhalation exposure
Mice were housed in polycarbonate cages (4/cage), provided AIN-76A diet and water ad libitum and maintained on a 12–12 h light–dark cycle at 22±5 °C and 50±20% relative humidity. At 6–8 weeks of age, Nrf2-WT and Nrf2-KO mice (4 groups (control, SF, AS, SF+As) for both Nrf2-WT and Nrf2-KO mice; 5 mice per group) were used for whole body inhalation exposure. Synthetic dusts that mimic the size of arsenic containing particles in the “real” world were generated by our team. The synthetic dust consists of 10% arsenic trioxide (2–3 μm) mixed with an inert background dust (Arizona Road dust purchased from Powder Technology, Burnsville, MN, median diameter ≈2 μm). A constant PM10 of 100 μg/m3 (an arsenic concentration of 10 μg/m3) for 24 h is a close mimic to the real world exposure. However, in order to reduce stress during exposure, animals were exposed to 4.8 mg/m3 of the synthetic dust for 30 min/day. SF was intraperitoneally (i.p.) injected (10 mg/kg) every other day until the end of experiment (14 days). All 40 mice survived arsenic-dust exposure and/or SF injections.
Bronchoalveolar lavage, cell differential analysis, and cytokine measurement
Following two-week exposure, mice were euthanized by i.p. injections of Avertin (Sigma). The lungs were lavaged three times with 0.5 ml of PBS. The supernatant from the first bronchoalveolar lavage (BAL) fluid was stored at −80 °C until measurement of cytokines. Cytokine concentrations (IL-6 and TNF-α) were measured according to the manufacturers’ instructions (Ready-SET-Go, eBioscience, San Diego, CA 92121). The cells from the three BAL fluid collection from each lung were combined and resuspended in PBS. An aliquot was cytocentrifuged at 1500 rpm for 3 min on glass slides. The slides were then stained with a HEMA3 STAT PACK kit (Fisher Scientific Company, Middletown, VA). For cell differential analyses, at least 200 cells were counted under the microscope. Data were expressed as mean±SD (n=5).
Tissue collection and hematoxylin and eosin (H&E) staining
Following lavage, lungs were isolated. The lung tissue was cut into parts: one part was frozen in liquid nitrogen and stored at −80 °C for total RNA extraction and for immunoblot analysis, and the other part was fixed in 10% buffered formalin and embedded in paraffin. 5 μm sections were cut and stained with H&E.
Apoptotic cell death
An in situ cell death detection kit (Roche, IN, USA) was used for detecting apoptotic cell death in the bladder epithelium according to the manufacturer’s instructions. Briefly, tissue sections were pretreated with proteinase K (15 ug/ml) in 10 mM Tris/HCl (pH 7.8) at 37 °C for 30 min. After washing 3 times with PBS, tissue sections were incubated with TUNEL reaction mixture for 1 h at 37 °C in the dark. Tissue sections were then costained with Hoechst, and analyzed under a fluorescence microscope (Zeiss Observer.Z1 microscope with the Slidebook computer program). (The excitation wavelength: 450–500 nm and the detection wavelength: 515–565 nm). Hoechst stain was visualized under UV.
Immunoblot, immunohistochemistry, and antibodies
Lung tissues were homogenized in 1× sample buffer (50 mM Tris–Cl, 2% SDS, 10% glycerol, and 100 mM DTT), and centrifuged at 10,000 rpm at 4 °C for 15 min to move debris. Lysates were then heated, sonicated, and used for electrophoresis and immunoblot. For immunohistochemical (IHC) analysis, lavaged lungs were paraffin embedded. Tissue sections were cut, baked and deparaffinized. Antigen retrieval was performed by boiling in sodium citrate buffer. After blocking peroxidase and nonspecific binding, tissue sections were incubated with primary and then secondary antibody. The staining was detected using the ABC kit (Vector Lab, Burlingame, CA, USA) and 3,3′-diaminobenzidine (DAKO, Carpinteria, CA, USA). The slides were then counterstained with hematoxylin. Anti-Nrf2, NQO1, γGCS, HO-1, and β-actin were purchased from Santa Cruz (Santa Cruz, CA); and anti-p65 and p-p65 antibodies were from Cell Signaling Technology (Danvers, Massachusetts).
Oxidative DNA damage
A monoclonal antibody against 8-Oxo-7,8-dihydro-2′-deoxyguanosine (8-Oxo-dG) (Trevigen, Gaithersburg, MD) was used for the detection of oxidative DNA damage. Fixed tissue sections were incubated with proteinase K (10 μg/ml) in PBS for 30 min at 37 °C, and then exposed to 2 M HCl for 5 min at room temperature. Following quenching of the endogenous peroxidase activity by 0.3% H2O2, tissue sections were first incubated with anti-8-Oxo-dG at 1:250 dilution at RT for 2 h, followed by sequential incubation with the secondary antibody and the ABC kit. Finally, tissue sections were developed and counterstained with hematoxylin.
qRT-PCR analysis
Total RNA from the lung was extracted using Trizol. Equal amounts of RNA (2 μg) were reverse-transcripted into cDNA using the Transcriptor First Strand cDNA synthesis Kit (Roche). Primers were synthesized by Sigma and the sequences are as follows:
NQO1: GGTAGCGGCTCCATGTACTC (forward); AGACCTGGAAGCC ACAGAAA (reverse)
HO1: GAGCCTGAATCGAGCAGAAC (forward); CTCGGCTTGGATGT GTACCT (reverse)
GCLM: TCCCATGCAGTGGAGAAGAT (forward); AGCTGTGCAACTCC AAGGAC (reverse)
β-actin: AAGGCCAACCGTGAAAAGAT (forward); GTGGTACGACCAGAGGCATAC (reverse).
The qPCR conditions were: one cycle of initial denaturation (95 °C for 3 min), 40 cycles of amplification (95 °C for 10 s, 60 °C for 20 s, and 72 °C for 5 s), 1 cycle of melting curve measurement (95 °C for 5 s, 65 °C for 60 s, and raise temperature to 97 °C in 5–10 acquisition/°C), and a cooling period (40 °C for 30 s). The data presented were relative mRNA levels normalized to β-actin, and the value from the Nrf2-WT control group was set as 1.
Statistics
Two-way Student’s t test was used to determine the significant difference between two samples. Results are expressed as mean±SD. p<0.05 is considered to be significant.
Results
The lungs of mice exposed to arsenic particles and/or SF have elevated levels of Nrf2 and its target genes
Although arsenic has been reported to induce Nrf2 in a variety of cultured cell lines and in mice exposed to arsenic through contaminated drinking water, the effect of inhaled arsenic on the Nrf2 stress response has not been previously investigated. We measured the protein level of Nrf2 in the lungs of Nrf2-WT mice that inhaled arsenic-containing dusts for 14 days. Nrf2 was markedly induced in the lung as measured by immunoblot analysis (Fig. 1A) and immunohistochemistry (Fig. 1C). We used whole cell lysates because previously our laboratory showed that the protein level of Nrf2 in whole cell lysates is proportional to Nrf2 expression in the nucleus (Zhang and Hannink, 2003). Consistent with our previous results, intraperitoneal injection of SF also induced Nrf2 (Figs. 1A and C). Moreover, induction of Nrf2 is most prominent in the group receiving both arsenic and SF (Figs. 1A and C). Similar to the level of Nrf2, expression of its target genes, NQO1, γGCS, and HO-1 was induced in all three treated groups with the highest level seen in the combined treatment group (Fig. 1B). As expected, knockout mice had no detectable Nrf2 levels and basal and inducible levels of NQO1, γGCS, and HO-1 were low, as measured by immunoblot (data not shown) and IHC (Fig. 1C). Furthermore, the mRNA level for NQO1, GCLM, and HO-1 in the treatment groups was lower in Nrf2-KO mice, compared to the corresponding group of Nrf2-WT mice (Fig. 1B). It is worth mentioning that only NQO1, not GCLM and HO-1, had lower basal mRNA levels in Nrf2-KO than Nrf2-WT mice (Fig. 1B).
Fig. 1.

The lungs of mice exposed to arsenic particles and/or SF have elevated levels of Nrf2 and its target genes. (A) The lungs from Nrf2-WT mice were homogenized. Equal amounts of tissue lysate (200 μg total protein per lane) were loaded and subjected to immunoblot analysis. Each lane represents an individual mouse (right panel). The relative band intensity (to β-actin) was plotted (left panel) (B) mRNA levels of NQO1, GCLM, and HO-1 in Nrf2-WT and Nrf2-KO were analyzed by qRT-PCR. mRNA was extracted from three mice per group and used to run qRT-PCR in duplicate. Results are expressed as means±SD (n=3: SD was calculated from the mean of three mice), *p<0.05 versus respective “Con”. (C) IHC staining of lung tissue sections from both Nrf2-WT and Nrf2-KO in untreated (control), SF-injected (SF), arsenic-inhaled (As), and arsenic-inhaled plus SF-injected (As+SF) groups. Scale bar: 100 μm.
Sulforaphane prevents pathological alterations, oxidative damage and cell death in the lungs of mice exposed to arsenic particles
Pulmonary histology indicates that untreated Nrf2-WT and Nrf2-KO mice have a thin layer of alveolar epithelial cells with a minimal amount of infiltrated inflammatory cells (Fig. 2A). Injection of SF did not cause noticeable changes in pulmonary morphology (Fig. 2A). Arsenic exposure resulted in a noticeable thickening of the alveolar septa with increased collagen deposition, proliferation of fibroblasts, and hyperplasia of pneumocytes (Fig. 2A). In addition, some infiltrated lymphocytes, macrophages and plasma cells were present in both arsenic-inhaled Nrf2-WT and Nrf2-KO groups even though the lungs were lavaged (data shown). Significantly, all these arsenic-induced changes were not observed in Nrf2-WT mice treated with SF, indicating that SF is able to prevent the pathological alterations caused by arsenic exposure in the lungs of Nrf2-WT mice (Fig. 2A). In contrast, the arsenic-induced pulmonary changes were not alleviated by SF treatment in Nrf2-KO mice, demonstrating that the protective effect of SF against arsenic-induced pulmonary damage is Nrf2-dependent (Fig. 2A).
Fig. 2.
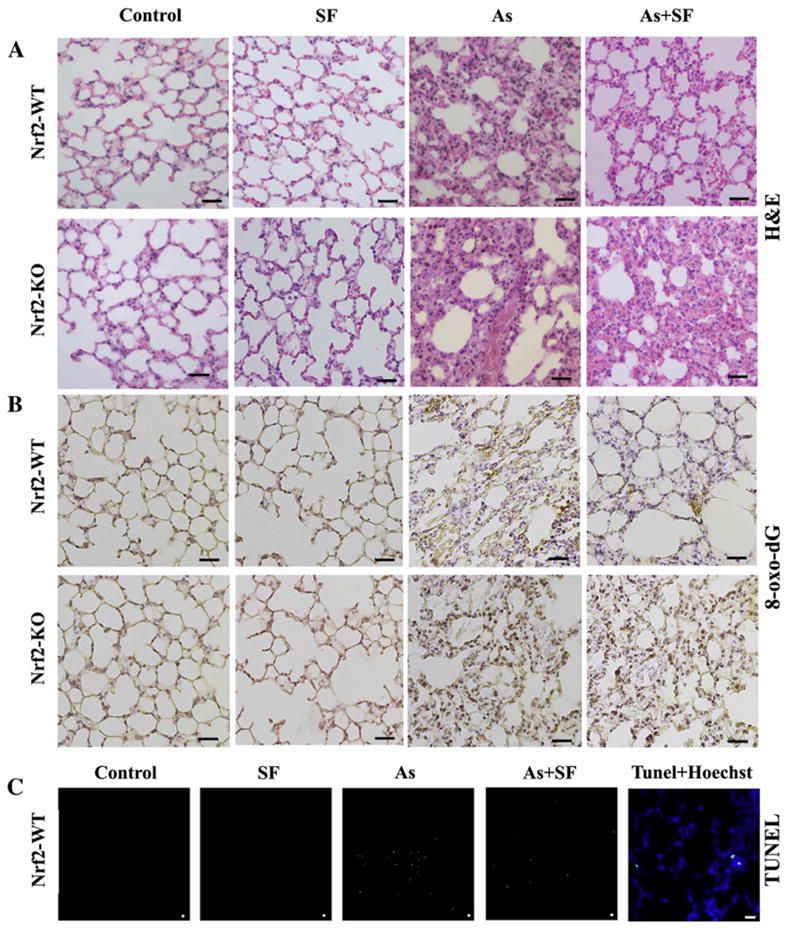
Sulforaphane prevents pathological alterations, oxidative damage and cell death in the lungs of mice exposed to arsenic particles. (A) H&E staining of lavaged lung tissue sections from Nrf2-WT and Nrf2-KO mice. (B) Oxidative damage of lungs was analyzed by IHC staining with an anti-8-oxo-dG antibody. (C) Cell death was measured using TUNEL. The left most panel (Tunel+Hoechst) was a lung tissue section from the arsenic-inhaled group stained with TUNEL plus Hoechst, demonstrating that the TUNEL positive signal is from the nucleus. Each panel is a representative result from three mice per group analyzed. Scale bar: 100 μm.
Next, arsenic-induced DNA damage was measured by IHC staining with an anti-8-Oxo-dG antibody. Arsenic-treated mice in both Nrf2-WT and Nrf2-KO groups had a greater degree of oxidative damage as shown by increased staining. SF itself did not cause DNA damage, but in fact, relieved arsenic-induced damage in Nrf2-WT mice, not in Nrf2-KO mice (Fig 2B). Arsenic also caused apoptotic cell death as measured by TUNEL labeling, which was significantly reduced in the SF-injected Nrf2-WT mice (Fig. 2C). There were no apoptotic cells observed in the control and SF-treated groups (Fig. 2C). Specificity of TUNEL labeling was confirmed by nuclear co-staining of TUNEL with Hoechst (Fig. 2C, TUNEL+Hoechst panel).
Sulforaphane alleviates inflltration of inflammatory cells into the lungs of arsenic-exposed Nrf2-WT mice
The typical response of the lung to inhaled toxic particles is the infiltration of inflammatory cells. Upon arsenic exposure, the total number of inflammatory cells in the BAL fluid was significantly increased in both Nrf2-WT and Nrf2-KO mice (Fig. 3). SF treatment reduced the total cell count of BAL from Nrf2-WT mice, but not from Nrf2-KO mice (Fig. 3). Injection of SF did not elicit any inflammatory response in the lungs of either Nrf2-WT or Nrf2-KO mice (Fig. 3). When the total number of macrophages, neutrophils, and lymphocytes were individually counted and plotted, a similar trend was observed, compared to the total count (Fig. 3). Collectively, these data demonstrate that SF-mediated activation of Nrf2 is able to suppress the pulmonary inflammatory response elicited by arsenic inhalation.
Fig. 3.

Sulforaphane alleviates infiltration of inflammatory cells into the lungs of arsenic-exposed Nrf2-WT mice. Cell differential analysis was performed on the BAL cells from each mouse. Following staining, at least 200 cells were counted under the microscope. The absolute number of total cells, macrophages, neutrophils, or lymphocytes was plotted. Results are expressed as means±SD (n=5), *p<0.05 versus “Con” in Nrf2-WT group, #p<0.05 versus “Con” in Nrf2-KO group, ^p<0.05 “SF+As” versus “As” group.
TNF-α is a multifunctional proinflammatory cytokine that can be induced by arsenic exposure (Das et al., 2005). In this study, inhaled arsenic increased the release of TNF-α into the BAL fluid as detected by ELISA analysis (Fig. 4A). Interestingly, treatment with SF significantly reduced the release of TNF-α into the BAL fluid from Nrf2-WT, but not Nrf2-KO mice (Fig. 4A). TNF-α is not only the target gene of NFκB, but also a strong activator of the NFkB pathway, leading to phosphorylation of p65 on serine 536 (Xing et al., 2011). p65, in turn, regulates an array of cytokines including IL-6 that contributes to inflammation and remodeling of injured lungs (Das et al., 2005). Here, we show that inhalation of arsenic particles activated the NF-κB pathway as demonstrated by the enhancement of phosphorylated p65 (p-p65) in arsenic-inhaled groups while total p65 remained equal in all groups (Fig. 4B). SF repressed arsenic-mediated induction of phosphorylated p65 in Nrf2-WT mice, but had no suppression in Nrf2-KO mice (Fig. 4B). In agreement with the activation of NF-κB in response to arsenic, IL-6 in the BAL fluid was increased in the arsenic-treated group (Fig. 4A). SF treatment was able to prevent the increased production of IL-6 only in Nrf2-WT mice (significant trend), not in Nrf2-KO mice (Fig. 4A). Further analysis using qRT-PCR shows that arsenic exposure resulted in a significant increase in the production of Th2 cytokines, including IL-13 and IL-4, which can be reverted by SF in Nrf2-WT, and not in Nrf2-KO mice (IL-13: statistically significant; IL-4: significant trend) (Fig. 4C). Furthermore, arsenic enhanced production of TGF-β, as well as MCP-1, a chemokine produced by macrophages and epithelial cells that facilitates T lymphocyte infiltration. Arsenic-mediated induction of both TGF-β and MCP-1 was slightly suppressed by SF in Nrf2-WT (significant trend), but not in Nrf2-KO mice. Combined with the data demonstrating an increase in lymphocytes in the BAL fluid from arsenic treated mice (Fig. 3), these results indicate that arsenic triggered predominantly an allergic lung inflammatory response. Remarkably, SF blocked arsenic-induced production of Th2 cytokines (IL-13 and IL-4) and MCP-1 in Nrf2-WT, but not in Nrf2-KO mice, indicating that the anti-inflammatory activity of SF is Nrf2-dependent.
Fig. 4.
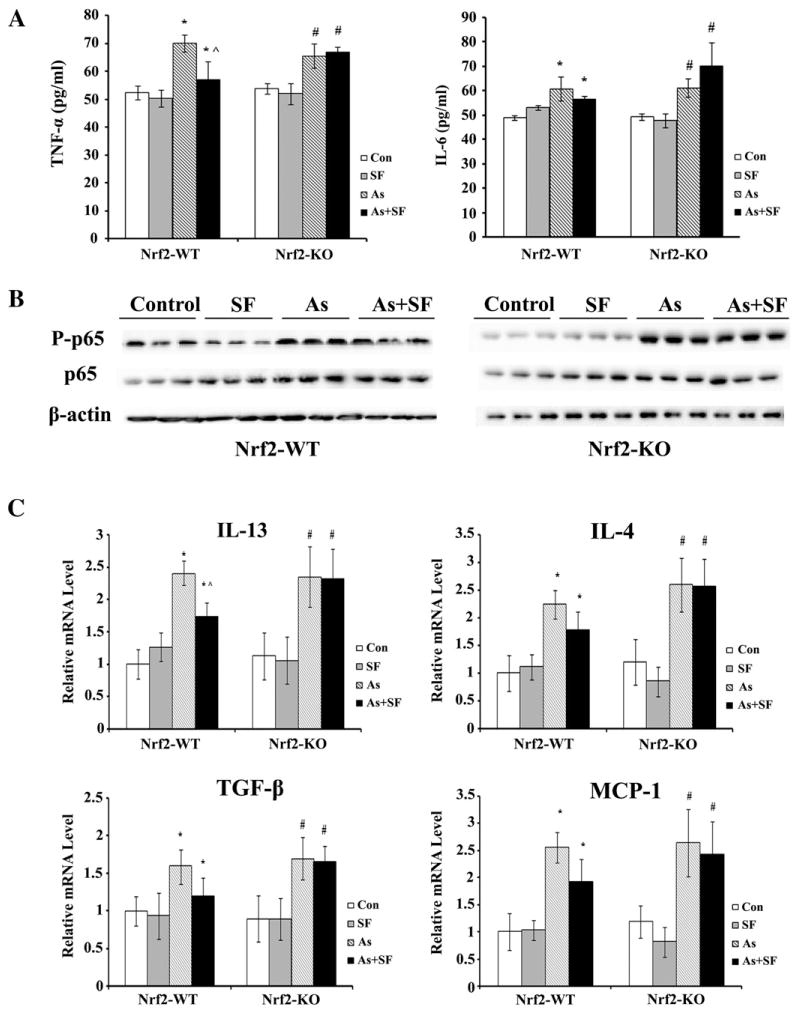
Sulforaphane suppresses arsenic-induced proinflammatory cytokine production in Nrf2-WT mice. (A) The amount of TNF-α or IL-6 in the BAL fluid was measured by ELISA. Results are expressed as means±SD (n=5), *p<0.05 versus “Con” in Nrf2-WT group, #p<0.05 versus “Con” in Nrf2-KO group, ^p<0.05 “SF+As” versus “As” group. (B) The lavaged lung tissues were subjected to immunoblot analysis with the indicated antibodies. Each lane represents the lung tissue lysate from an individual mouse. (C) mRNA expression of IL-13, IL-4, TGF-β and MCP-1 was measured by qRT-PCR. mRNA was extracted from three mice per group and used to run qRT-PCR in duplicate. Results are expressed as means±SD (n=3), *p<0.05 versus “Con” in Nrf2-WT group, #p<0.05 versus “Con” in Nrf2-KO group, ^p<0.05 “SF+As” versus “As” group.
Discussion
Although a large population is still suffering from many diseases associated with arsenic exposure, effective approaches geared toward reducing arsenic toxicity are still lacking. Arsenic exposure is known to induce the inflammatory response in the airway and chronic exposure to arsenic can elicit or exacerbate bronchitis, pneumonia, asthma and COPD (Mazumder et al., 2000; Smith et al., 1998; von Ehrenstein et al., 2005). In this study, we investigate the pulmonary response to arsenic-particle exposure and the effectiveness of targeting Nrf2 activation for intervention. For the first time, we show the efficacy of Nrf2 activation in preventing molecular and pathological alterations in the lung induced by short-term exposure to environmentally relevant doses of arsenic using an in vivo inhalation model. It is astonishing to see such substantial pathological changes in the lungs after only two weeks of exposure to real world doses of arsenic-containing dusts. Remarkably, SF blocked the arsenic-induced pathological changes and tissue damage in Nrf2-WT and not in Nrf2-KO, implicating that this SF-mediated protection is specifically through the activation of Nrf2. This study provides a solid basis that dietary intervention targeting Nrf2 activation is a great approach to prevent or alleviate a variety of human diseases associated with arsenic exposure, which may be inevitable for people living in areas with a high concentration of arsenic in their drinking water or in the air.
Our study also demonstrates that activation of Nrf2 is able to alleviate the allergic lung inflammatory response elicited by arsenic. It has been shown that SF inhibits inflammation through suppression of the inflammatory cytokine production by macrophages (Harvey et al., 2011; Kong et al., 2010; Li et al., 2008; Park et al., 2012; Thejass and Kuttan, 2007; Youn et al., 2010). Consistent with these studies, our results show that SF significantly inhibited the production of inflammatory cytokines including TNF-α, IL-6, and TGF-β (Figs. 4A and C). In our study, SF also inhibited MCP-1, which is predominantly produced by monocytes, macrophages and epithelial cells (Conti and DiGioacchino, 2001; Sebastiani et al., 2002; Starrett and Blake, 2011; van Zoelen et al., 2011), as well as production of Th-2 cytokines, such as IL-13 and IL-4 (Fig. 4C). Therefore, SF appears to inhibit arsenic-induced lung inflammation through multiple mechanisms. (i) SF suppresses the adaptive immune response by down-regulating T cell differentiation toward Th2, (ii) SF inhibits innate immune response by down-regulating inflammatory cytokine production, and (iii) SF appears to interfere with the crosstalk between the innate and adaptive immune response through down-regulation of MCP-1. MCP-1 is produced by innate immune cells, but is crucial in recruiting lymphocytes into the inflammation site to elicit an adaptive immune response. More importantly, this anti-inflammatory activity of SF largely depends on Nrf2 as the therapeutic effects of SF in reducing arsenic-induced lung inflammation was only observed in Nrf2-WT, but not in Nrf2-KO mice.
In this study, we demonstrate our novel finding that activation of Nrf2 confers protection against pulmonary pathological changes and oxidative damage induced by low environmentally relevant doses of inhaled arsenic particles during the two-week exposure period. It is interesting to note that at basal conditions, there was no difference between Nrf2-WT and Nrf2-KO in response to arsenic-induced pathological changes (Fig. 2), oxidative damage (Fig. 2), inflammatory cell in-filtration in the lung (Fig. 3), and cytokine production (Figs. 4A, and C), which is consistent with our previous report when arsenic was ingested by mice. In that study, sodium arsenic was administered through drinking water for six weeks and pathological alterations between Nrf2-WT and Nrf2-KO mice were observed only in the liver and bladder, but not in the lung (Jiang et al., 2009). This may be due to the fact that the basal Nrf2 expression was extremely low as evidenced by the low protein level of Nrf2, NQO1, γ-GCS, and HO-1 (Fig. 1A), as well as the similar basal mRNA level of GCLM and HO-1 (Fig. 1B). Another observation from our study is that the modulation of cytokine production by arsenic was not dramatic, thus the suppression of SF on arsenic-induced cytokine production was moderate, as shown that the effect of SF on the production of certain cytokines was not statistically significant but only showed significant trend. This is likely due to either the low environmental dose of arsenic used or the short duration of exposure. However, pathological analysis and pulmonary inflammatory cell infiltration clearly demonstrate the efficacy of SF in Nrf2-WT mice.
In summary, this study provides proof-of-concept experimental evidence that Nrf2 is a great target for disease prevention. Further work should include identification of more effective and specific Nrf2 activators with low cost, which will allow us to test the effectiveness of oral delivery of Nrf2 activators for intervention of human pathologies associated with exposure to environmental arsenic.
Acknowledgments
This work was supported by the NIH grants 2R01 ES015010 and R01 CA154377 to D.D.Z., P42ES004940 to R.C.L., and P30ES006694, a center grant.
Footnotes
Conflict of interest statement
No conflicts of interest to declare.
References
- Byrd DM, Roegner ML, Griffiths JC, Lamm SH, Grumski KS, Wilson R, Lai S. Carcinogenic risks of inorganic arsenic in perspective. Int Arch Occup Environ Health. 1996;68:484–494. doi: 10.1007/BF00377874. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Chen CW, Wu MM, Kuo TL. Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br J Cancer. 1992;66:888–892. doi: 10.1038/bjc.1992.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol. 2010;244:43–56. doi: 10.1016/j.taap.2009.07.024. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Shirai T, Steineck G. Epidemiology and etiology of premalignant and malignant urothelial changes. Scand J Urol Nephrol Suppl. 2000:105–115. doi: 10.1080/00365590050509869. [DOI] [PubMed] [Google Scholar]
- Conti P, DiGioacchino M. MCP-1 and RANTES are mediators of acute and chronic inflammation. Allergy Asthma Proc. 2001;22:133–137. doi: 10.2500/108854101778148737. [DOI] [PubMed] [Google Scholar]
- Das S, Santra A, Lahiri S, Guha Mazumder DN. Implications of oxidative stress and hepatic cytokine (TNF-alpha and IL-6) response in the pathogenesis of hepatic collagenesis in chronic arsenic toxicity. Toxicol Appl Pharmacol. 2005;204:18–26. doi: 10.1016/j.taap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Du Y, Villeneuve NF, Wang XJ, Sun Z, Chen W, Li J, Lou H, Wong PK, Zhang DD. Oridonin confers protection against arsenic-induced toxicity through activation of the Nrf2-mediated defensive response. Environ Heal Perspect. 2008;116:1154–1161. doi: 10.1289/ehp.11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, Feller-Kopman D, Wise R, Biswal S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3:78ra32. doi: 10.1126/scitranslmed.3002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Picciotto I, Smith AH. Observations on the dose–response curve for arsenic exposure and lung cancer. Scand J Work Environ Health. 1993;19:217–226. doi: 10.5271/sjweh.1480. [DOI] [PubMed] [Google Scholar]
- Higgins LG, Kelleher MO, Eggleston IM, Itoh K, Yamamoto M, Hayes JD. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol Appl Pharmacol. 2009;237:267–280. doi: 10.1016/j.taap.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Huerta-Olvera SG, Macias-Barragan J, Ramos-Marquez ME, Armendariz-Borunda J, Diaz-Barriga F, Siller-Lopez F. Alpha-lipoic acid regulates heme oxygenase gene expression and nuclear Nrf2 activation as a mechanism of protection against arsenic exposure in HepG2 cells. Environ Toxicol Pharmacol. 2010;29:144–149. doi: 10.1016/j.etap.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Jiang T, Huang Z, Chan JY, Zhang DD. Nrf2 protects against As(III)-induced damage in mouse liver and bladder. Toxicol Appl Pharmacol. 2009;240:8–14. doi: 10.1016/j.taap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kenyon EM, Hughes MF. A concise review of the toxicity and carcinogenicity of dimethylarsinic acid. Toxicology. 2001;160:227–236. doi: 10.1016/s0300-483x(00)00458-3. [DOI] [PubMed] [Google Scholar]
- Kong JS, Yoo SA, Kim HS, Kim HA, Yea K, Ryu SH, Chung YJ, Cho CS, Kim WU. Inhibition of synovial hyperplasia, rheumatoid T cell activation, and experimental arthritis in mice by sulforaphane, a naturally occurring isothiocyanate. Arthritis Rheum. 2010;62:159–170. doi: 10.1002/art.25017. [DOI] [PubMed] [Google Scholar]
- Li W, Khor TO, Xu C, Shen G, Jeong WS, Yu S, Kong AN. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem Pharmacol. 2008;76:1485–1489. doi: 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder DN, Haque R, Ghosh N, De BK, Santra A, Chakraborti D, Smith AH. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int J Epidemiol. 2000;29:1047–1052. doi: 10.1093/ije/29.6.1047. [DOI] [PubMed] [Google Scholar]
- Murthy NS, Mukherjee S, Ray G, Ray A. Dietary factors and cancer chemoprevention: an overview of obesity-related malignancies. J Postgrad Med. 2009;55:45–54. doi: 10.4103/0022-3859.43549. [DOI] [PubMed] [Google Scholar]
- Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659:31–39. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kim JW, Lee CM, Kim YD, Chung SW, Jung ID, Noh KT, Park JW, Heo DR, Shin YK, Seo JK, Park YM. Sulforaphane inhibits the Th2 immune response in ovalbumin-induced asthma. BMB Rep. 2012;45:311–316. doi: 10.5483/bmbrep.2012.45.5.311. [DOI] [PubMed] [Google Scholar]
- Putila JJ, Guo NL. Association of arsenic exposure with lung cancer incidence rates in the United States. PLoS One. 2011;6:e25886. doi: 10.1371/journal.pone.0025886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani S, Albanesi C, De PO, Puddu P, Cavani A, Girolomoni G. The role of chemokines in allergic contact dermatitis. Arch Dermatol Res. 2002;293:552–559. doi: 10.1007/s00403-001-0276-9. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Sumi D, Fukami I, Ishii T, Kumagai Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006;580:1771–1774. doi: 10.1016/j.febslet.2006.02.031. [DOI] [PubMed] [Google Scholar]
- Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, Wood R, Kosnett MJ, Smith MT. Cancer risks from arsenic in drinking water. Environ Heal Perspect. 1992;97:259–267. doi: 10.1289/ehp.9297259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Goycolea M, Haque R, Biggs ML. Marked increase in bladder and lung cancer mortality in a region of Northern Chile due to arsenic in drinking water. Am J Epidemiol. 1998;147:660–669. doi: 10.1093/oxfordjournals.aje.a009507. [DOI] [PubMed] [Google Scholar]
- Starrett W, Blake DJ. Sulforaphane inhibits de novo synthesis of IL-8 and MCP-1 in human epithelial cells generated by cigarette smoke extract. J Immunotoxicol. 2011;8:150–158. doi: 10.3109/1547691X.2011.558529. [DOI] [PubMed] [Google Scholar]
- Steinmaus C, Moore L, Hopenhayn-Rich C, Biggs ML, Smith AH. Arsenic in drinking water and bladder cancer. Cancer Invest. 2000;18:174–182. doi: 10.3109/07357900009038249. [DOI] [PubMed] [Google Scholar]
- Tchounwou PB, Patlolla AK, Centeno JA. Carcinogenic and systemic health effects associated with arsenic exposure—a critical review. Toxicol Pathol. 2003;31:575–588. doi: 10.1080/01926230390242007. [DOI] [PubMed] [Google Scholar]
- Thejass P, Kuttan G. Immunomodulatory activity of Sulforaphane, a naturally occurring isothiocyanate from broccoli (Brassica oleracea) Phytomedicine. 2007;14:538–545. doi: 10.1016/j.phymed.2006.09.013. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Verstege MI, Draing C, de Beer R, van’t Veer C, Florquin S, Bresser P, van der Zee JS, te Velde AA, von Aulock S, van der Poll T. Endogenous MCP-1 promotes lung inflammation induced by LPS and LTA. Mol Immunol. 2011;48:1468–1476. doi: 10.1016/j.molimm.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17. doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve NF, Lau A, Zhang DD. Regulation of the nrf2-keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010;13:1699–1712. doi: 10.1089/ars.2010.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ehrenstein OS, Mazumder DN, Yuan Y, Samanta S, Balmes J, Sil A, Ghosh N, Hira-Smith M, Haque R, Purushothamam R, Lahiri S, Das S, Smith AH. Decrements in lung function related to arsenic in drinking water in West Bengal, India. Am J Epidemiol. 2005;162:533–541. doi: 10.1093/aje/kwi236. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi JA, Zhang DD. Nrf2 protects human bladder urothelial cells from arsenite and monomethylarsonous acid toxicity. Toxicol Appl Pharmacol. 2007;225:206–213. doi: 10.1016/j.taap.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondrak GT, Villeneuve NF, Lamore SD, Bause AS, Jiang T, Zhang DD. The cinnamon-derived dietary factor cinnamic aldehyde activates the Nrf2-dependent antioxidant response in human epithelial colon cells. Molecules. 2010;15:3338–3355. doi: 10.3390/molecules15053338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Gong K, Feng W, Nozell SE, Chen YF, Chatham JC, Oparil S. O-GlcNAc modification of NFkappaB p65 inhibits TNF-alpha-induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS One. 2011;6:e24021. doi: 10.1371/journal.pone.0024021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, Seo JA, Lim KM, Kwak MK, Hwang DH, Lee JY. Sulforaphane suppresses oligomerization of TLR4 in a thiol-dependent manner. J Immunol. 2010;184:411–419. doi: 10.4049/jimmunol.0803988. [DOI] [PubMed] [Google Scholar]
- Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Whitman SA, Wu W, Wondrak GT, Wong PK, Fang D, Zhang DD. Therapeutic potential of nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60:3055–3066. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]