

Abstract
DNA double strand breaks (DSBs), induced by ionizing radiation (IR) and endogenous stress including replication failure, are the most cytotoxic form of DNA damage. In human cells, most IR-induced DSBs are repaired by the non-homologous end joining (NHEJ) pathway. One of the most critical steps in NHEJ is ligation of DNA ends by DNA ligase IV (LIG4), which interacts with, and is stabilized by, the scaffolding protein X-ray cross-complementing gene 4 (XRCC4). XRCC4 also interacts with XRCC4-like factor (XLF, also called Cernunnos); yet, XLF has been one of the least mechanistically understood proteins and precisely how XLF functions in NHEJ has been enigmatic. Here, we examine current combined structural and mutational findings that uncover integrated functions of XRCC4 and XLF and reveal their interactions to form long, helical protein filaments suitable to protect and align DSB ends. XLF-XRCC4 provides a global structural scaffold for ligating DSBs without requiring long complementary DNA ends, thus ensuring accurate and efficient ligation and repair. The assembly of these XRCC4-XLF filaments, providing both DNA end protection and alignment, may commit cells to NHEJ with general biological implications for NHEJ and DSB repair processes and their links to cancer predispositions and interventions.
Keywords: Non-homologous end joining, DNA double strand break, DNA repair, XRCC4, XLF
Introduction
The genome is under constant attack from DNA damage generated internally and through exposure to DNA damaging agents in our environment. Critical maintenance of genomic integrity results from the balance between incurred DNA damage and its repair. Inability to faithfully repair DNA damage in a timely and accurate manner can lead to mutations, chromosomal translocations, genomic instability and cell death (Ciccia and Elledge, 2010). The observations of constant damage and robust repair pathways imply i) that the impact of environmental and endogenous DNA damage depends largely on the nature of repair pathways and their ability to respond to a given type of damage, and ii) that agents that impact repair may have more profound impacts on cells and organisms than those that simply damage DNA, as seen for cadmium inhibition of mismatch repair (McMurray and Tainer, 2003).
DNA double strand breaks (DSBs) are widely considered the most cytotoxic form of DNA damage. In mammalian cells, the major pathway for the detection and repair of DSBs is non-homologous end joining (NHEJ) (reviewed in (Lieber, 2010; Mahaney et al., 2009)), which, for convenience, can be divided into three stages. First, DSBs are detected by the Ku70/80 heterodimer with subsequent tethering by the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs) (DeFazio et al., 2002; Gottlieb and Jackson, 1993). Subsequent autophosphorylation of DNA-PKcs results in conformational changes (Hammel et al., 2010b) that may promote DNA-PKcs dissociation and access of downstream repair factors (reviewed in (Dobbs et al., 2010; Meek et al., 2008; Neal and Meek, 2011)). In the second step, non-ligatable end groups are removed and DNA ends are processed by enzymes that include the Artemis endonuclease (Goodarzi et al., 2006; Rooney et al., 2003), polynucleotide kinase/phosphatase (PNKP) (Chappell et al., 2002; Koch et al., 2004), and/or the gap filling DNA polymerases μ and λ (Mahajan et al., 2002; Nick McElhinny and Ramsden, 2004). Third, the processed DNA ends are ligated by DNA ligase IV (LIG4) in complex with X-Ray Cross Complementing gene 4 (XRCC4) (Critchlow et al., 1997; Grawunder et al., 1998a; Grawunder et al., 1998b) and XLF (XRCC4-like factor, also called Cernunnos) (Ahnesorg et al., 2006; Buck et al., 2006) (Fig. 1). The NHEJ pathway is also critical for V(D)J recombination, the site specific rearrangement of immunoglobulin and T-cell receptor genes that is required for the development of mature T and B cells in the vertebrate immune system (Helmink and Sleckman, 2012; Malu et al., 2012). Consequently, disruption of NHEJ proteins (Ku, DNA-PKcs, XRCC4, XLF, LIG4 and Artemis) manifests as defects in DSB repair, radiation sensitivity and severe combined immunodeficiency (SCID) (Buck et al., 2006; Dai et al., 2003; Gu et al., 1997; Li et al., 1995; Moshous et al., 2001; Riballo et al., 1999; Taccioli et al., 1998).
Figure 1. A model for non-homologous end joining.

NHEJ is proposed to occur in three stages, (i) end detection and tethering, (ii) end processing to remove non-ligatable end groups and (iii) ligation of the DNA ends. In the first step, the DSBs are detected by the Ku70/80 heterodimer. Ku is required for recruitment of DNA-PKcs, which displaces Ku form the DNA end (Yoo and Dynan, 1999) and promotes DNA end-tethering and formation of a synaptic complex (DeFazio et al., 2002). Autophosphorylation of DNA-PKcs induces conformational changes (Hammel et al., 2010b) that may induce its dissociation from DNA ends (Dobbs et al., 2010) and regulate subsequent processing of DNA ends (reviewed in (Meek et al., 2008)) but when this occurs relative to end processing and recruitment of the XRCC4-Lig4 complex is not clear. Processing of the DNA ends is thought to involve Artemis (which interacts with DNA-PKcs (Ma et al., 2002)), PNKP (which interacts with CK2 phosphorylated XRCC4 (Koch et al., 2004)) and DNA polymerases of the pol X family (Nick McElhinny and Ramsden, 2004), however, precisely when each protein is recruited to and subsequently released from the NHEJ repair complex is not known (indicated by dashed lines and question marks). In the final step, the processed DNA ends are ligated by the LIG4-XRCC4 complex. XRCC4 and XLF may promote DNA end ligation by regulating the catalytic activity of LIG4 and by the formation of end bridging filaments, as described in the text. In the figure, Ku80 is shown in orange and Ku70 in red (orientation relative to DSB ends from (Walker et al., 2001)), DNA-PKcs is in blue, Artemis (Art) in grey, dimeric XRCC4 in green, dimeric XLF in purple, LIG4 in black and PNKP in pink.
Critical to elucidation of the mechanism of NHEJ is a thorough understanding of the dynamic interactions of the NHEJ proteins with each other and with their substrate, damaged DNA. Small angle X-ray scattering (SAXS) is emerging as a powerful approach to investigate dynamic interactions between proteins and protein-nucleic acids, as well as conformational changes within proteins and protein complexes in solution (Perry et al., 2010; Rambo and Tainer, 2010). SAXS is also valuable for determining accurate structures and assemblies in solution (Putnam et al., 2007) and experimentally distinguishing flexibility from switching between discrete conformations (Rambo and Tainer, 2011). Moreover, SAXS is amenable to high throughput under near physiological conditions (Hura et al., 2009). SAXS is especially powerful in combination with high-resolution structural techniques such as crystallography (Putnam et al., 2007). We recently used SAXS, along with atomic modeling to map interactions between Ku, DNA-PKcs and DNA and showed that autophosphorylation of DNA-PKcs promotes conformational changes that facilitate end processing in the initial stages of NHEJ (Dobbs et al., 2010; Hammel et al., 2010b). In this review, we describe our current understanding of the roles of XRCC4 and XLF in NHEJ, highlighting recent structural studies from our group (Hammel et al., 2011; Hammel et al., 2010a) as well as other laboratories (Andres and Junop, 2011; Andres et al., 2012; Ropars et al., 2011; Wu et al., 2011) that reveal how XRCC4 and XLF combine to form functionally-important long super-helical filaments suitable to position and align DNA ends for ligation in the final stage of NHEJ. The emerging structural biochemistry of XLF-XRCC4 reveals the power of SAXS with crystallography to define the biologically important and functional assembly of macromolecules in solution.
XRCC4 and XLF play important structural roles in NHEJ and enhance DNA ligation
XRCC4 was named for its ability to complement a Chinese Hamster Ovary (CHO) cell line (XR-1) that is deficient in DNA DSB repair (Giaccia et al., 1990). Absence of XRCC4 in cell lines leads to radiation sensitivity and defects in DSB repair and V(D)J recombination, while disruption of XRCC4 or its binding partner LIG4 in mice leads to neuronal apoptosis and embryonic lethality, suggesting critical roles in development (Barnes et al., 1998; Gao et al., 1998; Li et al., 1995). In 2003, Jeggo and colleagues identified a patient with immunodeficiency and cellular defects in DSB repair and V(D)J recombination, indicative of a defect in NHEJ (Dai et al., 2003). However, cells from this patient (termed 2BN) appeared competent for the known NHEJ factors, suggesting the existence of an additional NHEJ component. In 2006, XLF was identified as an XRCC4-interacting protein and the missing factor in 2BN cells, as well as cells from other patients displaying growth retardation, microcephaly, radiation sensitivity and immune deficiency (Ahnesorg et al., 2006; Buck et al., 2006).
Neither XRCC4 nor XLF have known enzymatic activities and are thus assumed to play structural roles in NHEJ. Although they share no significant amino acid similarity, XRCC4 and XLF share several structural similarities. Both are composed of a globular head domain, an elongated α-helical stalk and an unstructured C-terminal region (CTR) that is absent in the crystal structures (Fig. 2A/B). XRCC4 and XLF form stable homodimers mediated by interactions between the head domains and/or the tops of the stalk regions (Ahnesorg et al., 2006; Andres et al., 2007; Junop et al., 2000; Li et al., 2008) (Fig. 2C/D). The coiled-coil stalk region of XRCC4 (Fig. 2C) interacts directly with residues within and between the C-terminal tandem breast cancer susceptibility gene C-terminal (BRCT) domains of LIG4 (Critchlow et al., 1997; Grawunder et al., 1998c; Sibanda et al., 2001; Wu et al., 2009) forming a highly stable complex (Critchlow et al., 1997; Modesti et al., 2003). In contrast, XLF interacts weakly, if at all with LIG4 (Deshpande and Wilson, 2007; Lu et al., 2007), and the coiled-coil domain folds back upon itself (Andres et al., 2007; Li et al., 2008) placing the CTR close to the head domains (Hammel et al., 2010a) (Fig. 2D). Similarly, in the XRCC4 homodimer, the CTR is predicted to lie close to the head domains through flexibility of the CTR (Hammel et al., 2010a) (Fig. 2C).
Figure 2. Structures of XRCC4 and XLF.

A: Cartoon showing the major regions of XRCC4. Boundaries between the head, stalk and C-terminal (CTR) domains are from (Junop et al., 2000; Sibanda et al., 2001). Phosphorylation sites (shown in red) are taken from Phosphosites.org (search term http://www.phosphosite.org/proteinAction.do?id=18166&showAllSites=true).
B: Cartoon showing major regions of XLF. Boundaries between head (orange) and stalk (grey) domains are from (Andres et al., 2007; Li et al., 2008). Phosphorylation sites (shown in red) are from (Yu et al., 2008).
C: Crystal structure of XRCC4 with positions of head (orange) and stalk (green) domains from (Junop et al., 2000; Sibanda et al., 2001) and the predicted position of the unstructured CTR (light-magenta) from SAXS (Hammel et al., 2011; Hammel et al., 2010b). Phosphorylation sites are in red, as in panel A.
D: Crystal structure of XLF with predicted position of XLF-CTR (light-magenta) from SAXS (Hammel et al., 2011; Hammel et al., 2010b). Phosphorylation sites are in red, as above. The extreme CTR of XLF, shown to interact with Ku (Yano et al., 2011) and the region shown to support filament formation (Hammel et al., 2011) are indicated.
Interaction with XRCC4 stabilizes LIG4 protein in cells (Bryans et al., 1999), and all of the cellular LIG4 likely exists in complex with XRCC4 (Critchlow et al., 1997; Robins and Lindahl, 1996). In contrast, the molar ratio of XRCC4 dimers to LIG4 monomers in the nuclei of human cells was estimated as ~3:1 (Mani et al., 2010), suggesting that XRCC4 homodimers may exist separately from LIG4. Although XRCC4 and XLF lack enzymatic activity, both have been reported to stimulate DNA end rejoining by LIG4. One way that this may occur is through direct effects on LIG4 catalytic activity. LIG4 exists in an adenylated form in cells (Robins and Lindahl, 1996) and during the enzymatic reaction, enzyme-bound AMP is transferred to the 5′ phosphate of DNA (Ellenberger and Tomkinson, 2008). LIG4 requires re-adenylation to participate in the next catalytic cycle and this re-adenylation step is rate limiting in vitro (Wang et al., 2007). Both XRCC4 and XLF have been reported to stimulate LIG4 activity by promoting its re-adenylation (Modesti et al., 1999; Riballo et al., 2009) but how this is achieved is unknown. Another way that XRCC4 and XLF may enhance end joining is by promoting DNA end bridging or alignment via structural roles. Indeed, XLF exerts a greater effect on end joining of non-cohesive ends compared to cohesive ends, consistent with a role in end alignment (Gu et al., 2007; Tsai et al., 2007).
Although XRCC4 exists primarily as a homodimer, tetramers and higher order structures have also been observed (Junop et al., 2000; Modesti et al., 1999). Moreover, the CTRs of both XRCC4 and XLF interact with double-stranded (ds) DNA in a DNA length- and protein concentration-dependent manner in vitro, suggesting cooperativity of DNA binding (Lu et al., 2007; Modesti et al., 1999; Yu et al., 2008). These properties led Lieber and colleagues to propose that XRCC4 and XLF align parallel to dsDNA (Lu et al., 2007), but precisely how this was accomplished was unclear. A recent flurry of structural studies now reveal that XRCC4 and XLF combine to form long super-helical filaments, suitable to position and align DNA ends for ligation (Andres et al., 2012; Hammel et al., 2011; Ropars et al., 2011; Wu et al., 2011), thus providing a probable structural basis for the scaffolding roles of XRCC4 and XLF and their ability to stimulate end joining in NHEJ.
XRCC4 and XLF interact to form helical filaments
Using size exclusion chromatography (SEC) combined with SAXS and atomic modeling, we showed that XRCC4 homodimers interact in a head-to-head fashion (Hammel et al., 2010a), and not via their stalks as previously suggested (Junop et al., 2000; Modesti et al., 2003). Moreover, alternating homodimers of XRCC4 and XLF formed long protein filaments of variable lengths in solution (Hammel et al., 2010a). Notably, modeling predicted a tilt between the stalks of the XRCC4-XLF dimers, suggesting that the filaments adopt a helical, curvilinear structure. The non-parallel alignment of the stalks was also predicted to accommodate binding of the BRCT domains of LIG4 without steric hindrance (Hammel et al., 2010a).
The X-ray crystal structures of the XRCC4-XLF complex, composed of the head and stalk domains of both proteins, confirmed the SAXS-based predicted head-to-head interaction of XRCC4 and XLF homodimers (Andres et al., 2012; Hammel et al., 2011; Ropars et al., 2011; Wu et al., 2011) (Fig. 3). Small conformational changes in the head domains allow L115 of XLF to form a “leucine-lock” that fits into a hydrophobic pocket, generated by M59, M61, F106 and L108 as well as K65 and K99 of XRCC4. In addition, R64 and L65 of XLF and R107, E55 and D58 of XRCC4 form a secondary and complementary interaction interface (Fig. 3). The interaction interface between the XRCC4-XLF head domains is between 600 – 1300Å2 (Andres et al., 2012; Hammel et al., 2011; Ropars et al., 2011; Wu et al., 2011), which is smaller than that observed for XLF or XRCC4 homodimers or for the XRCC4-LIG4 BRCT interaction interface (Andres et al., 2007; Junop et al., 2000; Li et al., 2008; Sibanda et al., 2001; Wu et al., 2009). Consistent with this, the XRCC4-XLF interaction is weaker than that between the respective homodimers or between XRCC4 and LIG4 (Li et al., 2008; Malivert et al., 2010; Modesti et al., 2003; Riballo et al., 2009).
Figure 3. The XRCC4-XLF interface.
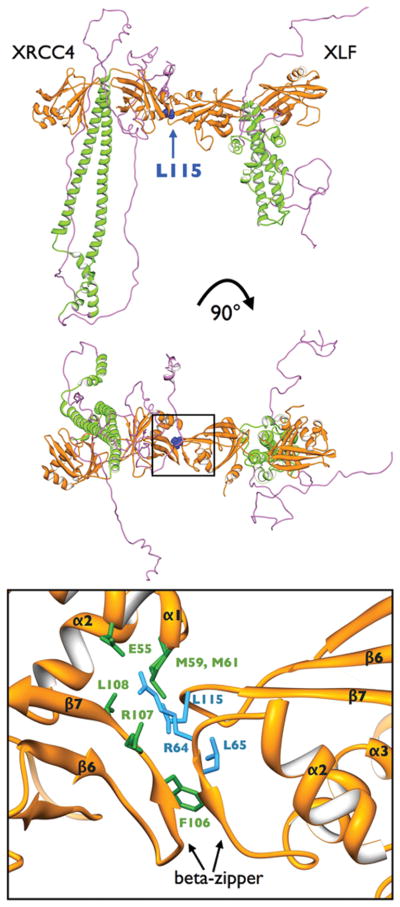
Top: Structures of XRCC4 homodimers (left) and XLF homodimers (right) coloured as in Figure 2, showing the location of XLF-L115 in the XRCC4-XLF interface.
Middle: Structure above, rotated by 90°.
Lower: Close-up of XRCC4-XLF interface showing locations of E55, M59, M6, F106, R107 and L108 for XRCC4 (green) and R64, R65 and L65 (blue). The formation of a beta-zipper structure upon complexation is highlighted (see (Hammel et al., 2011) for details). The head domains of XRCC4 and XLF are shown in gold as above.
Mutational studies highlight the importance of the head-to-head interface between XRCC4 and XLF homodimers. Mutation of XLF L115 abolishes binding to XRCC4 (Andres et al., 2007; Andres et al., 2012; Hammel et al., 2011), and mutation of the hydrophobic pocket in XRCC4 disrupts the interaction with XLF (Ropars et al., 2011). Similarly, mutagenesis experiments showed that XLF-R64 and L65 are required for interaction with XRCC4 and implicated several positively charged residues in XRCC4 in the interaction with XLF (Andres et al., 2007; Malivert et al., 2010; Ropars et al., 2011). The XLF-XRCC4 interaction is functionally important for NHEJ, as XLF-R64E, L65D and L115D mutants that do not interact with the XRCC4-LIG4 complex were unable to restore V(D)J recombination in XLF-deficient fibroblasts (as assessed by joining of an inversional, chromosomal substrate that would require joining of both coding and signal ends) (Malivert et al., 2010). Moreover, XRCC4 mutants that do not interact with XLF failed to restore joining of extrachromosomal V(D)J recombination substrates in XRCC4-deficient CHO cells (XR-1) (Roy et al., 2012). In this case, the defect was specific for DNA coding ends, indicating a possible requirement for the XLF-XRCC4 interaction in end alignment (Roy et al., 2012) (discussed further below). Furthermore, the XLF mutants that were unable to bind XRCC4 did not rescue bleomycin sensitivity of XLF-deficient cells, and γ-H2AX foci persisted in these cells following IR (Malivert et al., 2010). Similarly, XRCC4 mutants that were defective in XLF binding only partially rescued the radiosensitivity of XR-1 cells and their over-expression acted as a partial dominant negative (Roy et al., 2012). In addition, residues R57 and C123 which maintain the integrity of XLF β7-sheet required for XRCC4 interaction (Andres et al., 2012; Hammel et al., 2011; Ropars et al., 2011; Wu et al., 2011) are mutated in XLF patients, and these mutations (R57G and C123R) are predicted to destabilize head domain interactions (Malivert et al., 2010). Collectively these studies reveal that the head-to-head mediated interaction of XRCC4 and XLF is critical for NHEJ in vivo.
Notably, the biological unit in the XLF-XRCC4 crystal structures contained long filaments of alternating dimers of XRCC4 and XLF, ranging from 720 to over 800 Å in length (Andres et al., 2012; Hammel et al., 2011; Ropars et al., 2011; Wu et al., 2011), confirming observations made from earlier SAXS data (Hammel et al., 2010a). These left-handed, super-helical filaments (Fig. 4A) were predicted to adopt flexible, tube-like structures with an internal diameter ranging from 70 to 120 Å depending on the bend of the super-helical bundle (Andres et al., 2012; Hammel et al., 2011; Ropars et al., 2011; Wu et al., 2011). In these tube-like assemblies, the head domains of XRCC4 and XLF face inwards and are tilted ~30° to form the rim of a tube or channel, and the coiled-coils/stalks radiate outwards (Fig. 4AB). Filament formation in solution was dynamic and protein concentration-dependent (Hammel et al., 2011; Wu et al., 2011), and required XLF L115 as well as L64 and R65 (Hammel et al., 2011), again supporting the importance of head domain interactions. XRCC4-XLF filaments with a diameter of ~100Å, composed of thinner 50 Å filaments wrapped together, were also observed by transmission electron microscopy (Ropars et al., 2011). Although multimeric complexes were observed with XRCC4 or XLF alone, the combination of XRCC4 and XLF greatly enhanced filament formation (Ropars et al., 2011) and filament length (Hammel et al., 2011), and the CTR of XLF promoted the formation of filaments (Hammel et al., 2011), suggesting that XLF is critical for filament formation.
Figure 4. Models for XRCC4-XLF filaments in NHEJ.

A: Model of filaments composed of XRCC4 (blue) and XLF (magenta). Two parallel filaments are shown as observed in the crystal and in solution (Hammel et al., 2011). On the right, the parallel filaments are rotated by 90° to show the central channel or pore (see text for details).
B: Model of XRCC4-XLF super-helical bundles, coloured as in panel A, with dsDNA (green) wrapping around the filament as proposed by (Hammel et al., 2011) (top) or within the filament as proposed by (Andres et al., 2012) (bottom).
C: Possible positions of the Ku heterodimer (grey) and DNA-PKcs (peach) relative to DNA-protein filaments (adapted from (Hammel et al., 2011)).
D: Possible models for the position of XRCC4-XLF filaments relative to the DSB: In all panels, XRCC4 is shown in blue, XLF in magenta, Ku in grey and dsDNA in green as in panels A–C. The positions of the DSB ends are marked by triangles. Possible access points for DNA-PKcs, processing enzymes and/or LIG4 are indicated by the arrows. In all models, it is possible that filament dynamics and interactions with other proteins are regulated by protein phosphorylation, possibly of the CTRs of XRCC4 and XLF. We speculate that filaments promote synapsis of DNA ends and protect ends from nuclease degradation, which may affect pathway choice as well as DNA fidelity, see text for details. (i) XRCC4/XLF filaments form proximal to the DSB, supporting and aligning dsDNA either side of the DSB. How DNA-PKcs, processing enzymes (for example PNKP) and LIG4 gain access to the DNA ends is presently unknown. One possibility is that XRCC4-XLF filaments form only after DNA-PKcs autophosphorylation and release (see Fig. 1). Also, since cells contain excess of XRCC4 over LIG4, it is possible that XRCC4 in filaments is devoid of LIG4 and that ligation is carried out by a soluble pool of XRCC4-LIG4, perhaps in complex with PNKP. It has been suggested that the presence of LIG4 near the DNA ends may disrupt the XRCC4/XLF filaments, thereby providing a possible mechanism to both terminate filament formation and provide LIG4 with access to the DNA ends. Protein phosphorylation may also play a role in remodeling the filament ends (see text for details). (ii) The Ku-DNA-PKcs complex forms at the DSB ends and the XRCC4-XLF filament forms distal to the break. This model is attractive in that the DNA ends would be accessible for binding by DNA-PKcs. Subsequent DNA-PKcs autophosphorylation and dissociation/remodeling might then allow processing enzymes such as PNKP and Artemis to gain access to the DSB ends (see Fig. 1). This model would also be consistent with two pools of XRCC4, one in filamentous form and one that interacts with LIG4. (iii) Filaments perform a bridging role, spanning two Ku-DNA complexes. Remodeling of filament ends, possibly by protein phosphorylation, could allow access of processing enzymes and LIG4 to DNA termini. (iv) Given the dimensions of the internal channel, another possibility is that the filaments could form around two dsDNA molecules, allowing processing and ligation to occur at the exposed DNA termini.
Individual filaments were found to associate in parallel via the head domains of XRCC4, and alignment of multiple filaments was stabilized by interactions with XRCC4 stalk domains to form multi-filament bundles (Hammel et al., 2011; Wu et al., 2011) (Fig. 4B). The crystal lattice formed by XRCC4 and XLF suggested that multiple parallel filaments interact to form an extensive lattice that resembled the cogs of a gear (Wu et al., 2011). The authors noted that this gear-cog lattice strongly resembled the “cartwheel” structures formed by centriole assembly protein SAS-6 (van Breugel et al., 2011), supporting the idea that the XRCC4-XLF complex may serve a similar scaffolding function in DNA repair. Assembly of these XRCC4-XLF super helical bundles was dynamic and enhanced by the presence of dsDNA (Hammel et al., 2011). Based on our collective results from crystallography, hydrogen-deuterium exchange mass spectrometry (HDX), SAXS and computer modeling, we proposed that XRCC4 and XLF interact to form long, super-helical filaments that interact in parallel to form a grooved U-shaped channel, ~65 Å wide (Fig. 4B). Based on hybrid structural determination, we propose that the dsDNA winds around the outside of the super-helical bundle and suggest that the super-helical filament bundles form an alignment channel for DNA to facilitate DSB ligation (Fig. 4B, upper panel). The filament structure itself was predicted to be flexible and the internal pore is predicted to be large enough to enclose dsDNA, Ku, DNA-PKcs or an entire nucleosome, leading others to speculate that the protein filaments might wrap around dsDNA (Andres et al., 2012; Wu et al., 2011) (Fig. 4B, lower panel). Whether filaments form around dsDNA or whether DNA wraps around the filaments, or some combination of both, remains to be directly determined but will be important to understanding filament function. If filaments do indeed form around nucleosomes, how filament assembly is coordinated with nucleosome remodeling and phosphorylation of H2AX must also be considered.
Integration of XRCC4-XLF filaments with other NHEJ proteins
Another critical, yet unanswered question is the location of LIG4 within the filament structures. LIG4 is composed of amino-terminal DNA binding and catalytic domains connected to the C-terminal tandem BRCT domains by a flexible linker. The BRCT domains interact directly with residues 155–195 of the XRCC4 coiled-coil stalk (Sibanda et al., 2001; Wu et al., 2009), and the flexible linker region is predicted to provide the catalytic domain with considerable range of motion (Perry et al., 2010). Indeed, the cryo-EM structure of XRCC4-LIG4 positions the catalytic domain of LIG4 near the XRCC4 head domain (Recuero-Checa et al., 2009). Together, these studies suggest that the LIG4 catalytic domain resides near the DNA binding site(s) and alignment channel of the filaments, thus providing LIG4 with access to its substrate. Given that human cells contain more XRCC4 than LIG4 (Mani et al., 2010), it seems unlikely that each subunit of XRCC4 in the filament contains a bound LIG4 molecule. Rather, we propose that LIG4 may only be present at the ends of the filaments, where it can carry out its enzymatic function. Interestingly, the LIG4 tandem BRCT domain disrupted the large protein-DNA networks observed for XLF-XRCC4-DNA and disrupted DNA end bridging by XLF-XRCC4 in vitro (Andres et al., 2012). The BRCT domains also had a similar effect on XRCC4 filaments in vitro (Hammel et al., 2010a). Thus, we hypothesize that XRCC4 homodimers directly involved in end joining might be complexed to LIG4, while XRCC4 molecules involved in alignment of DNA ends by XLF-XRCC4 filaments may be free of LIG4. This would provide a mechanism for forming the XRCC4-LIG4 complex only at the ends of the filaments and supports a model in which LIG4 is not present within the filaments. In contrast, Roy et al. proposed that filament bundles span the DSB, and that phosphorylation-dependent remodeling facilitates positioning of XRCC4/LIG4 units within filaments, over the DSB (Roy et al., 2012) (Fig. 4C/D).
XRCC4 also interacts with PNKP and the related proteins aprataxin (Clements et al., 2004) and APLF (aprataxin and PNKP-like factor) (Kanno et al., 2007). PNKP is a 3′-DNA phosphatase, 5′-DNA kinase that directly removes non-ligatable groups from DNA termini and replaces them with ligatable 5′-phosphates and 3′-hydroxyl groups (Weinfeld et al., 2011). Aprataxin, which is mutated in ataxia oculomotor apraxia 1, removes AMP formed from abortive ligation reactions from DNA termini (Clements et al., 2004; Rass et al., 2008; Reynolds et al., 2009), while APLF is reported to have exonuclease activity and enhances NHEJ (Kanno et al., 2007; Li et al., 2011; Rulten et al., 2011). Each of these proteins contains a fork-head associated (FHA) domain that interacts with phosphorylated T233 of XRCC4 (Clements et al., 2004; Kanno et al., 2007; Koch et al., 2004). XRCC4-T233, located in the CTR, is phosphorylated by CK2 in vitro (Koch et al, 2004), and is constitutively phosphorylated in vivo (Mani et al., 2010). The effects of CK2 phosphorylation of XRCC4 on filament structure and how the interaction of phosphorylated XRCC4 with the FHA domains of PNKP, aprataxin and APLF affects filament assembly and dynamics are important questions that need to be addressed. Moreover, PNKP, APLF and aprataxin carry out their enzymatic functions at DNA ends, therefore, like LIG4, mechanisms must exist to control the access of these enzymes to DSB ends rather than placing them throughout the filaments.
The Ku heterodimer plays a critical role in recruiting multiple NHEJ factors to DSBs (Gu and Lieber, 2008). Both XRCC4 and XLF interact with Ku (Costantini et al., 2007; Mari et al., 2006; Nick McElhinny et al., 2000; Yano et al., 2008), and Ku is required to recruit XRCC4 and XLF to sites of UV laser-induced DNA damage in vivo (Yano et al., 2008). Similarly, Ku enhances recruitment of XRCC4-LIG4 and XLF to DNA in vitro (Nick McElhinny et al., 2000; Yano et al., 2008). Surprisingly, XRCC4 was not required to recruit XLF to DNA damage sites, although it helped to stabilize/retain XLF at breaks (Yano et al., 2008). Understanding how filaments coordinate with Ku will therefore be important to understanding their function. As a first step towards this goal, we determined the overall arrangement of the Ku-DNA-XLF-XRCC4 assembly in solution using SAXS (Hammel et al., 2011). Our results suggest that the Ku-DNA complex is positioned on one side of the XLF-XRCC4 assembly, providing a possible mechanistic basis for the Ku and DNA-dependent recruitment of the XLF-XRCC4 complex to DSBs in vivo (Yano et al., 2008). However, whether Ku binds to DNA ends and then translocates inwards allowing the filaments to form on the free end of the DNA (i.e. proximal to the break), or remains bound to the DNA termini, allowing filaments to form internal to DNA-bound Ku (i.e. distal to the break) (Fig. 4C/D), remains to be determined. These and other potential models for filament positioning relative to dsDNA end orientation and other NHEJ proteins are shown in Fig. 4D.
Since Ku also recruits DNA-PKcs to DNA ends (Gottlieb and Jackson, 1993; Yoo et al., 1999), the location of Ku relative to the filaments is critical for understanding how DNA-PKcs accesses DNA ends and functions at DSBs. Upon recruitment, DNA-PKcs undergoes autophosphorylation-dependent conformational changes that regulate its interaction with DNA ends (Dobbs et al., 2010; Hammel et al., 2010b; Neal and Meek, 2011). The timing of DNA-PKcs recruitment and release, how it is coordinated with filament formation, and how it might regulate the function of the filaments are all unknown and merit investigation (Fig. 4D).
Potential functions of XRCC4-XLF filaments in DNA end bridging, protection and alignment in vivo
While there is no direct evidence for the presence of XRCC4-XLF filaments in vivo, there are strong suggestions in the literature that XLF and XRCC4 have functions in addition to their direct effects on LIG4 activity that are compatible with a structural role for XRCC4-XLF filaments in DNA end bridging, end alignment, end protection and synapsis. While the majority of the cellular LIG4 exists in complex with XRCC4 (Critchlow et al., 1997; Robins and Lindahl, 1996), the excess ratio of XRCC4 to LIG4 in cells (Mani et al., 2010) suggests that cells contain free XRCC4 in addition to XRCC4 that is conjugated with LIG4. Furthermore, XRCC4 mutants that are impaired in their ability to interact with LIG4 were still able to enhance V(D)J recombination in vivo (although minimally) and partially rescued the IR sensitivity of XR-1 cells (Modesti et al., 2003), indicating functions for XRCC4 in addition to its role in regulating LIG4. However, all XRCC4 mutants that were completely deficient in interaction with LIG4 could not support V(D)J joining in living cells. Interestingly, mutants that interacted normally with LIG4 but were impaired in tetramerization also displayed NHEJ deficits in vivo (Modesti et al., 2003), supporting the idea that XRCC4 oligomers are important for repair in vivo.
In vitro studies also support a role for XLF and XRCC4 in the alignment of DNA ends. XRCC4-LIG4 has been shown to bridge DNA ends in vitro (Chen et al., 2000), and XLF enhances intermolecular ligation as well as ligation of non-cohesive ends, suggesting that it too may function to bridge DNA ends (Gu et al., 2007; Lu et al., 2007; Tsai et al., 2007). Moreover, XLF stimulates end joining of partially complementary ends in a non-linear fashion, suggesting a cooperative mechanism (Akopiants et al., 2009) that is also consistent with filament formation. Most recently, elegant biochemical studies by Junop and colleagues showed that together XRCC4 and XLF tether or bridge two dsDNA molecules. Moreover, XRCC4 or XLF mutants that do not interact are unable to bridge DNA, and truncation of the CTRs of either XRCC4 or XLF, which abolished DNA binding, also abolished DNA bridging (Andres et al., 2012). In systems such as FEN1 complexes, the mechanism for specific binding to DNA at an end is clear from structures of DNA complexes with implications for a superfamily of nucleases (Grasby et al., 2012; Tsutakawa et al., 2011). We may therefore expect illumination on XLF-XRCC4-LIG4 specificity from structural data on DNA complexes. Given that DNA ligases may need to undergo dynamic switching for ligation as seen for human DNA ligase III (Cotner-Gohara et al., 2010), the filament evidently provides a scaffold that flexibly positions ligase to allow functionally-important conformational changes while also maintaining control of the DNA ends to aid efficient ligation.
The importance of the XRCC4-XLF head-to-head interface in vivo is also shown by the observation that XRCC4 mutants that are impaired in their ability to interact with XLF only partially complement the radiation sensitivity of XRCC4-deficient XR-1 cells (Roy et al., 2012). But, perhaps the most persuasive argument for the importance of filaments comes from work on V(D)J recombination. In this process, the RAG endonuclease complex induces two DSBs, each with two distinct termini: covalently sealed coding ends, and blunt, ligatable signal ends. Coding ends are joined rapidly into coding joints; however the blunt ended signal ends are retained in a synaptic complex by the RAG proteins prior to joining by the NHEJ machinery. Roy et al. showed that XRCC4 mutations that disrupt the interaction with XLF caused reduced frequency of coding end joining but did not affect signal end joining (Roy et al., 2012). To explain this phenotype, the authors proposed that coding end rejoining requires additional protein factors to help maintain DNA end-bridging (Roy et al., 2012). This DNA end-bridging would be dispensable for signal end joining because the RAG proteins maintain the signal ends, synapsed in the post-cleavage complex. Moreover, it was noted that several recent studies have shown that disruption of other DNA repair genes, specifically ATM, MRN and 53BP1, that are not considered “core” NHEJ proteins, also result in disruption of coding end joining (Bredemeyer et al., 2006; Difilippantonio et al., 2008; Helmink et al., 2009). Thus, these factors may all play a role in promoting synapsis and/or bridging of DNA coding ends to facilitate their ligation (Roy et al., 2012). Further supporting this hypothesis, Alt and colleagues have shown that ATM and 53BP1 are functionally redundant with XLF (Liu et al., 2012; Oksenych et al., 2012; Zha et al., 2011). In addition, Roy et al. have proposed an early role for filaments in end joining, whereby XLF-XRCC4 filaments form across Ku bound DNA ends, serving as a molecular splint to prevent dislocation of DNA ends prior to assembly of a functional NHEJ complex (Roy et al., 2012). Their data suggest that phosphorylation regulates transition from this bridging complex to a functional repair complex (see below).
Together, these data make a strong case that XRCC4-XLF filaments function to promote end bridging and synapsis of DNA ends prior to ligation. However, filaments are unlikely to be essential for NHEJ as signal ends are joined normally in mutants with disruption of the XRCC4-XLF interaction (Roy et al., 2012). Furthermore, while loss of XRCC4 in mice is embryonic lethal (Gao et al., 1998), XLF deficiency results in a milder cellular phenotype, that includes genome instability, radiosensitivity and defects in V(D)J recombination (Zha et al., 2007). This suggests that unlike XRCC4-LIG4, XLF is not absolutely required for NHEJ and thus, filaments likely facilitate end joining rather than carry out an essential function in NHEJ. However, as discussed above, redundancy with ATM, MRN and 53BP1 may also account for the milder phenotype of XLF-deficient cells.
Another possible role for XRCC4-XLF filaments is in DNA end protection. Indeed, both DNA-PKcs and XLF appear to protect DNA ends from resection (Dai et al., 2003; Liu et al., 2012; Meek et al., 2008; Oksenych et al., 2012; Zha et al., 2011). Such protection is also seen for MRE11 by BRCA2 at stalled replication forks (Schlacher et al., 2011) and by ATM at damaged sites (Rahal et al., 2010), suggesting that protection of DNA ends is a critical aspect of DNA protection and repair complexes. Thus, for NHEJ it is possible that XRCC4-XLF filaments do not form until DNA-PKcs has been released and, at this point, the filaments function to restrict DNA end resection. This would be consistent with reports that XLF promotes retention of 3′ overhangs in vitro (Tsai et al., 2007) and that end resection during V(D)J recombination may be restricted by XLF (Buck et al., 2006; Dai et al., 2003).
As a general theme, stable binding at damaged DNA sites can not only protect damage but also control pathway choice as seen for Atl1 taking base damage into the nucleotide excision repair (NER) pathway (Tubbs et al., 2009) by recruiting TFIIH and initiating removal of a 27 nucleotide patch (Fuss and Tainer, 2011). In fact, single residue defects in NER protein XPD that impact specifically binding or processing of the damaged site can result in cell death or mutation (Fan et al., 2008). It will be interesting to test if XLF-XRCC4 filament binding and protection of DNA ends provides a commitment to NHEJ. In NER with CKD7, the MRE11 complex with ATM, and NHEJ with DNA-PK, a common theme involves the coordinated binding of a signaling and effector kinase with damage recognition and processing proteins (Fuss and Tainer, 2011).
Effect of phosphorylation on filament assembly and function
The CTRs of both XRCC4 and XLF are both flexible unstructured regions that were not present in the available crystal structures. Both CTRs are predicted to lie close to the head domains, and thus, close to the walls of the inner pore of the filament bundles (Fig. 2–4). Moreover, both CTRs are phosphorylated in vitro and in vivo, suggesting that protein phosphorylation may play an important role in the regulation of filament function. The XRCC4 CTR is phosphorylated at multiple sites by DNA-PK in vitro (Lee et al., 2004; Yu et al., 2003) and, in vivo, serine 318 and other sites in the CTR are phosphorylated following DNA damage (Yu, Ye and Lees-Miller, unpublished) (Fig. 2). In vitro phosphorylation of the XRCC4 CTR by DNA-PK blocks DNA binding by XRCC4; however, the CTR of XRCC4 is dispensable for V(D)J recombination and radioresistance (Leber et al., 1998; Modesti et al., 1999). DNA-PK also phosphorylates the CTR of XLF in vitro, and IR results in ATM and DNA-PK-dependent phosphorylation of XLF on serines 245 and 251 (Yu et al., 2008). Although phosphorylation at these sites does not affect DNA binding by XLF in vitro (Yu et al., 2008), the CTR of XLF was shown to promote filament formation (Hammel et al., 2011). Interestingly, Roy et al. demonstrated that phosphorylation of XRCC4 and/or XLF by DNA-PK results in loss of DNA end-bridging by XRCC4-XLF in vitro, implying that phosphorylation of the CTR disrupts filaments (Roy et al., 2012). Moreover, CHO cells co-expressing non-phosphorylatable forms of XLF and XRCC4 were more resistant to the DSB inducing agent zeocin than wild-type cells, indicating that phosphorylation may de-stabilize filament bundles, and, significantly, that phosphorylation of the CTRs of XRCC4 and XLF act redundantly in vivo (Roy et al., 2012). Based on these observations, the authors proposed a model whereby Ku initially targets nucleation of XRCC4-XLF filament bundles that span the DSB. If Ku targets formation of filaments starting at each end of a DSB, then perhaps the two filaments are tethered by filament bundle formation. Targeting of DNA-PKcs by Ku at the break would result in local phosphorylation at the break site, reducing filament complexity, and allowing end processing factors access to the ends, and perhaps positioning XRCC4-LIG4 units over the break (Fig. 4D). As discussed above, how XRCC4-XLF filaments form in the context of the complete NHEJ reaction and the role of post-translational modifications such as phosphorylation is the focus of ongoing work in many laboratories. For the MRE11 complex with NBS1, a flexible C-terminal NBS1 connection provides a functional link (Williams et al., 2009) that is also regulated by phosphorylation (Falck et al., 2012). By analogy with NBS1 functions in the MRE11 complex, XLF-XRCC4 may not only be an adaptor to recruit LIG4 but may also provide a scaffold for flexibly tethered ligase and impact on the functional architecture and coordination of NHEJ.
The finding that phosphorylation of the CTRs of XRCC4 and XLF is functionally redundant may help to explain some of the discrepancies in the literature on the importance of the XRCC4-CTR and the XLF-CTR in NHEJ. Whereas the XRCC4-CTR and the XLF-CTR are both required for DNA binding in vitro (Andres et al., 2007; Yu et al., 2008) and the XLF-CTR is required for recruitment of XLF to sites of UV-laser induced damage in vivo (Yano et al., 2011), both CTRs are dispensable for cell survival following IR as well as V(D)J recombination in vivo (Leber et al., 1998; Malivert et al., 2009; Modesti et al., 1999). However, it may be that the CTR of either XRCC4 or XLF can compensate for loss of the other. Future experiments taking this into consideration will be required to better determine the in vivo function of XLF and XRCC4 CTRs.
Evolutionary conservation and other considerations
We speculate that the function of XRCC4-XLF filaments, like that of DNA-PKcs, may be specific to mammalian cells, as the interactions between the yeast homologues of XLF, XRCC4 and Ku appear to be inverted. Whereas XLF and XRCC4 interact through their head domains and the XLF-Ku interaction seems to depend on the C-terminal region of XLF in mammals (Yano et al., 2011), in S. cerevisiae, the N-terminal region of Lif1 (XRCC4 homolog) interacts with the C-terminal region of Nej1 (XLF homolog) (Deshpande and Wilson, 2007), and the Nej1-Ku interaction is dependent on the N-terminal region of Nej1 (Chen and Tomkinson, 2011). Thus, if filaments exist in lower organisms such as yeast, their structures may differ from those in mammals. It is interesting to postulate that if these filaments, like DNA-PKcs, are specific to higher organisms then perhaps XLF-XRCC4 filaments and DNA-PKcs function coordinately.
Although putative homologues of Ku as well as ATP-dependent DNA ligases have been reported in bacteria, bacterial NHEJ also apparently proceeds in the absence of XRCC4, XLF and DNA-PKcs (Pitcher et al., 2007; Shuman and Glickman, 2007). Yet, as shown by results in mycobacteria that have a robust NHEJ repair pathway requiring Ku but with ligase fused to polymerase, these pathogens have a shocking 50% error rate for NHEJ (Akey et al., 2006; Aniukwu et al., 2008; Gong et al., 2005) that may reflect the importance of the extra eukaryotic components for fidelity. As a general theme, filaments provide geometric control of DNA pieces, as seen for bacterial pili that promote bacterial uptake of DNA for transformation (Hartung et al., 2011).
General implications and perspectives
For base-excision repair (BER), crystal structures and designed mutations show how the APE1 nuclease acts to coordinate the orderly transfer of unstable DNA damage intermediates between the excision and synthesis steps of DNA repair (Mol et al., 2000). For the overall BER pathway, combined structural and function results reveal conformational controls, coordinated handoffs, and biological activities including links to cancer (Hitomi et al., 2007). For NHEJ, structures have proven challenging, but recent breakthroughs have opened the door to an increasing depth of understanding. These advances come in part from the increasing use of combined methods needed to understand dynamic and flexible complexes, such as SAXS and crystallography (Putnam et al., 2007), combined with genetics and biochemistry, providing an understanding of complex dynamic structures with biological and potential therapeutic impacts (Rambo and Tainer, 2010; Perry et al., 2010). Such combined structural and mutational results have now elucidated the molecular architecture of XRCC4 revealing that its NHEJ functions are aided by XLF interactions. The discovery that XLF and XRCC4 interact dynamically to form extended super-helical filaments supports and extends earlier speculation on their functions as scaffolding proteins. It furthermore raises intriguing questions regarding the roles of these filaments in NHEJ as well as the spatial and temporal relationship of these filaments to dsDNA and other components of the NHEJ pathway. Moreover, XLF-XRCC4 appear to provide at least two modes of regulation: differential localization, and direct physical interactions controlling the geometry of the DNA ends.
Although the concept of protein-DNA filaments is new to NHEJ, dynamic assembly of proteins and protein-DNA complexes to form macromolecular filaments is well established in other aspects of DSB repair. Specifically, in the alternative DSB repair pathway, homologous recombination repair (HR), Rad51 forms an extended nucleoprotein filament via its polymerization motif (that also interacts with BRCA2 (Shin et al., 2003)) on long single stranded DNA 3′ extensions, that is essential for invasion of the undamaged sister chromatid and initiation of accurate DSB repair (Holthausen et al., 2010; San Filippo et al., 2008). At replication forks BRCA2 and RAD51 filaments protect DNA ends from MRE11-mediated degradation (Schlacher et al., 2011) as MRE11 can bind and process replication forks as well as DSBs (Williams et al., 2008). Indeed, the whole MRE11/RAD50/NBS1 complex, which plays a key role in many DNA end associated processes, provides an early binding flexible scaffold at replication forks and DSB ends for HR and end joining (Williams et al., 2010). Moreover, RAD50 includes extended filament-like coiled-coils that interact via a Zn hook (Hopfner et al., 2002). Similarly, HR in bacteria and archaea involves formation of nucleoprotein filaments, mediated by RecA and RadA, respectively (Chang et al., 2009; De Vlaminck et al., 2012).
We propose that in NHEJ, XRCC4-XLF filaments serve to both protect DNA ends and to maintain DNA end alignment, preventing inappropriate end resection and ligation, and thus genomic instability, a hallmark of cancer (Hanahan and Weinberg, 2011). These filaments may therefore partly explain how DNA-PK and NHEJ distinguishes collapsed replication forks (one-ended breaks) from IR or V(D)J recombination induced DSBs (two-ended breaks). Targeting of XRCC4-XLF filament stability therefore has potential to lead to new approaches for the modulation of DSB repair and development of new cancer therapeutics.
Acknowledgments
We thank members of the Lees-Miller laboratory for helpful comments and discussion and apologize to authors whose work we were unable to cite due to space limitations. Work in the author’s laboratories is supported by National Institutes of Health program project grant P01 CA92584 (Structural Cell Biology of DNA Repair Machines to JAT) and the Canadian Institutes of Health Research (SPLM). BLM and SPLM are supported by the Alberta Heritage Foundation for Medical Research/Alberta Innovates-Health Solutions.
Abbreviations
- ATM
ataxia-telangiectasia mutated
- BRCA
breast and ovarian cancer susceptibility gene
- BER
base excision repair
- BRCT
BRCA C-terminal
- CHO
Chinese Hamster Ovary
- CTR
C-terminal region
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- ds
double stranded
- DSB
DNA double strand break
- HDX
hydrogen deuterium exchange mass spectrometry
- HR
homologous recombination
- IR
ionizing radiation
- LIG4
DNA ligase IV
- NHEJ
non-homologous end joining
- SAXS
small angle X-ray scattering
- ACID
severe combined immunodeficiency
- SEC
size exclusion chromatography
- XLF
XRCC4 like factor
- XRCC4
X-ray cross-complementing gene 4
References
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Akey D, Martins A, Aniukwu J, Glickman MS, Shuman S, Berger JM. Crystal structure and nonhomologous end-joining function of the ligase component of Mycobacterium DNA ligase D. J Biol Chem. 2006;281:13412–13423. doi: 10.1074/jbc.M513550200. [DOI] [PubMed] [Google Scholar]
- Akopiants K, Zhou RZ, Mohapatra S, Valerie K, Lees-Miller SP, Lee KJ, Chen DJ, Revy P, de Villartay JP, Povirk LF. Requirement for XLF/Cernunnos in alignment-based gap filling by DNA polymerases lambda and mu for nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res. 2009;37:4055–4062. doi: 10.1093/nar/gkp283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres SN, Junop MS. Crystallization and preliminary X-ray diffraction analysis of the human XRCC4-XLF complex. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:1399–1402. doi: 10.1107/S1744309111033549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. Crystal Structure of Human XLF: A Twist in Nonhomologous DNA End-Joining. Mol Cell. 2007;28:1093–1101. doi: 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniukwu J, Glickman MS, Shuman S. The pathways and outcomes of mycobacterial NHEJ depend on the structure of the broken DNA ends. Genes Dev. 2008;22:512–527. doi: 10.1101/gad.1631908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewll I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Current Biology. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- Bryans M, Valenzano MC, Stamato TD. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat Res. 1999;433:53–58. doi: 10.1016/s0921-8777(98)00063-9. [DOI] [PubMed] [Google Scholar]
- Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Plebani A, Stephan JL, Hufnagel M, le Deist F, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Chang YW, Ko TP, Lee CD, Chang YC, Lin KA, Chang CS, Wang AH, Wang TF. Three new structures of left-handed RADA helical filaments: structural flexibility of N-terminal domain is critical for recombinase activity. PLoS One. 2009;4:e4890. doi: 10.1371/journal.pone.0004890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. Embo J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Sung P, Tomkinson AE. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem. 2000;275:26196–26205. doi: 10.1074/jbc.M000491200. [DOI] [PubMed] [Google Scholar]
- Chen X, Tomkinson AE. Yeast nej1 is a key participant in the initial end binding and final ligation steps of nonhomologous end joining. J Biol Chem. 2011;286:4931–4940. doi: 10.1074/jbc.M110.195024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, Bieganowski P, Brenner C, Moreira MC, Taylor AM, Caldecott KW. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst) 2004;3:1493–1502. doi: 10.1016/j.dnarep.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst) 2007;6:712–722. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Cotner-Gohara E, Kim IK, Hammel M, Tainer JA, Tomkinson AE, Ellenberger T. Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry. 2010;49:6165–6176. doi: 10.1021/bi100503w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol. 1997;7:588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- Dai Y, Kysela B, Hanakahi LA, Manolis K, Riballo E, Stumm M, Harville TO, West SC, Oettinger MA, Jeggo PA. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc Natl Acad Sci U S A. 2003;100:2462–2467. doi: 10.1073/pnas.0437964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlaminck I, van Loenhout MT, Zweifel L, den Blanken J, Hooning K, Hage S, Kerssemakers J, Dekker C. Mechanism of Homology Recognition in DNA Recombination from Dual-Molecule Experiments. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.03.029. [DOI] [PubMed] [Google Scholar]
- DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. Embo J. 2002;21:3192–3200. doi: 10.1093/emboj/cdf299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Wilson TE. Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA Repair (Amst) 2007;6:1507–1516. doi: 10.1016/j.dnarep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, Kruhlak MJ, Callen E, Livak F, Nussenzweig MC, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst) 2010;9:1307–1314. doi: 10.1016/j.dnarep.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Forment JV, Coates J, Mistrik M, Lukas J, Bartek J, Jackson SP. CDK targeting of NBS1 promotes DNA–end resection, replication restart and homologous recombination. EMBO Rep. 2012 doi: 10.1038/embor.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Denko N, MacLaren R, Mirman D, Waldren C, Hart I, Stamato TD. Human chromosome 5 complements the DNA double-strand break-repair deficiency and gamma-ray sensitivity of the XR-1 hamster variant. Am J Hum Genet. 1990;47:459–469. [PMC free article] [PubMed] [Google Scholar]
- Gong C, Bongiorno P, Martins A, Stephanou NC, Zhu H, Shuman S, Glickman MS. Mechanism of nonhomologous end-joining in mycobacteria: a low-fidelity repair system driven by Ku, ligase D and ligase C. Nat Struct Mol Biol. 2005;12:304–312. doi: 10.1038/nsmb915. [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Harer C, Marchetti C, Morrice N, Jeggo PA, et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. Embo J. 2006;25:3880–3889. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- Grasby JA, Finger LD, Tsutakawa SE, Atack JM, Tainer JA. Unpairing and gating: sequence-independent substrate recognition by FEN superfamily nucleases. Trends Biochem Sci. 2012;37:74–84. doi: 10.1016/j.tibs.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998a;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Kulesza P, Lieber MR. Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J Biol Chem. 1998b;273:24708–24714. doi: 10.1074/jbc.273.38.24708. [DOI] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Leiber MR. DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr Biol. 1998c;8:873–876. doi: 10.1016/s0960-9822(07)00349-1. [DOI] [PubMed] [Google Scholar]
- Gu J, Lieber MR. Mechanistic flexibility as a conserved theme across 3 billion years of nonhomologous DNA end-joining. Genes Dev. 2008;22:411–415. doi: 10.1101/gad.1646608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lu H, Tsai AG, Schwarz K, Lieber MR. Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Res. 2007;35:5755–5762. doi: 10.1093/nar/gkm579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, Davidson L, Cheng HL, Sekiguchi JM, Frank K, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–665. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney B, Weinfeld M, et al. XRCC4 interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double-strand break repair. J Biol Chem. 2011;286:32638–32650. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF Regulates Filament Architecture of the XRCC4.Ligase IV Complex. Structure. 2010a;18:1431–1442. doi: 10.1016/j.str.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010b;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hartung S, Arvai AS, Wood T, Kolappan S, Shin DS, Craig L, Tainer JA. Ultrahigh resolution and full-length pilin structures with insights for filament assembly, pathogenic functions, and vaccine potential. J Biol Chem. 2011;286:44254–44265. doi: 10.1074/jbc.M111.297242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Bredemeyer AL, Lee BS, Huang CY, Sharma GG, Walker LM, Bednarski JJ, Lee WL, Pandita TK, Bassing CH, et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J Exp Med. 2009;206:669–679. doi: 10.1084/jem.20081326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Holthausen JT, Wyman C, Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA Repair (Amst) 2010;9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junop MS, Modesti M, Guarne A, Ghirlando R, Gellert M, Yang W. Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. Embo J. 2000;19:5962–5970. doi: 10.1093/emboj/19.22.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno S, Kuzuoka H, Sasao S, Hong Z, Lan L, Nakajima S, Yasui A. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. Embo J. 2007;26:2094–2103. doi: 10.1038/sj.emboj.7601663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CA, Agyei R, Galicia S, Metalnikov P, O’Donnell P, Starostine A, Weinfeld M, Durocher D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. Embo J. 2004;23:3874–3885. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber R, Wise TW, Mizuta R, Meek K. The XRCC4 gene product is a target for and interacts with the DNA-dependent protein kinase. J Biol Chem. 1998;273:1794–1801. doi: 10.1074/jbc.273.3.1794. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Jovanovic M, Udayakumar D, Bladen CL, Dynan WS. Identification of DNA-PKcs phosphorylation sites in XRCC4 and effects of mutations at these sites on DNA end joining in a cell-free system. DNA Repair (Amst) 2004;3:267–276. doi: 10.1016/j.dnarep.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Li S, Kanno S, Watanabe R, Ogiwara H, Kohno T, Watanabe G, Yasui A, Lieber MR. Polynucleotide kinase and aprataxin-like forkhead-associated protein (PALF) acts as both a single-stranded DNA endonuclease and a single-stranded DNA 3′ exonuclease and can participate in DNA end joining in a biochemical system. J Biol Chem. 2011;286:36368–36377. doi: 10.1074/jbc.M111.287797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chirgadze DY, Bolanos-Garcia VM, Sibanda BL, Davies OR, Ahnesorg P, Jackson SP, Blundell TL. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. Embo J. 2008;27:290–300. doi: 10.1038/sj.emboj.7601942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang W, Dubois RL, Yamamoto K, Wolner Z, Zha S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc Natl Acad Sci U S A. 2012;109:3903–3908. doi: 10.1073/pnas.1120160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Pannicke U, Schwarz K, Lieber MR. Length-dependent binding of human XLF to DNA and stimulation of XRCC4.DNA ligase IV activity. J Biol Chem. 2007;282:11155–11162. doi: 10.1074/jbc.M609904200. [DOI] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Mahajan KN, Nick McElhinny SA, Mitchell BS, Ramsden DA. Association of DNA polymerase mu (pol mu) with Ku and ligase IV: role for pol mu in end-joining double-strand break repair. Mol Cell Biol. 2002;22:5194–5202. doi: 10.1128/MCB.22.14.5194-5202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malivert L, Callebaut I, Rivera-Munoz P, Fischer A, Mornon JP, Revy P, de Villartay JP. The C-terminal domain of Cernunnos/XLF is dispensable for DNA repair in vivo. Mol Cell Biol. 2009;29:1116–1122. doi: 10.1128/MCB.01521-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malivert L, Ropars V, Nunez M, Drevet P, Miron S, Faure G, Guerois R, Mornon JP, Revy P, Charbonnier JB, et al. Delineation of the Xrcc4-interacting region in the globular head domain of cernunnos/XLF. J Biol Chem. 2010;285:26475–26483. doi: 10.1074/jbc.M110.138156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malu S, Malshetty V, Francis D, Cortes P. Role of non-homologous end joining in V(D)J recombination. Immunol Res. 2012 doi: 10.1007/s12026-012-8329-z. [DOI] [PubMed] [Google Scholar]
- Mani RS, Yu Y, Fang S, Lu M, Fanta M, Zolner AE, Tahbaz N, Ramsden DA, Litchfield DW, Lees-Miller SP, et al. Dual modes of interaction between XRCC4 and polynucleotide kinase/phosphatase: implications for nonhomologous end joining. J Biol Chem. 2010;285:37619–37629. doi: 10.1074/jbc.M109.058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari PO, Florea BI, Persengiev SP, Verkaik NS, Bruggenwirth HT, Modesti M, Giglia-Mari G, Bezstarosti K, Demmers JA, Luider TM, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci U S A. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT, Tainer JA. Cancer, cadmium and genome integrity. Nat Genet. 2003;34:239–241. doi: 10.1038/ng0703-239. [DOI] [PubMed] [Google Scholar]
- Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- Modesti M, Hesse JE, Gellert M. DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. Embo J. 1999;18:2008–2018. doi: 10.1093/emboj/18.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesti M, Junop MS, Ghirlando R, van de Rakt M, Gellert M, Yang W, Kanaar R. Tetramerization and DNA ligase IV interaction of the DNA double-strand break repair protein XRCC4 are mutually exclusive. J Mol Biol. 2003;334:215–228. doi: 10.1016/j.jmb.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- Neal JA, Meek K. Choosing the right path: does DNA-PK help make the decision? Mutat Res. 2011;711:73–86. doi: 10.1016/j.mrfmmm.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Ramsden DA. Sibling rivalry: competition between Pol X family members in V(D)J recombination and general double strand break repair. Immunol Rev. 2004;200:156–164. doi: 10.1111/j.0105-2896.2004.00160.x. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol Cell Biol. 2000;20:2996–3003. doi: 10.1128/mcb.20.9.2996-3003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, Patel H, Su A, Guo C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A. 2012;109:2455–2460. doi: 10.1073/pnas.1121458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JJ, Cotner-Gohara E, Ellenberger T, Tainer JA. Structural dynamics in DNA damage signaling and repair. Curr Opin Struct Biol. 2010 doi: 10.1016/j.sbi.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher RS, Brissett NC, Doherty AJ. Nonhomologous end-joining in bacteria: a microbial perspective. Annu Rev Microbiol. 2007;61:259–282. doi: 10.1146/annurev.micro.61.080706.093354. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. Cell Cycle. 2010;9:2866–2877. doi: 10.4161/cc.9.14.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–137. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC. Molecular mechanism of DNA deadenylation by the neurological disease protein aprataxin. J Biol Chem. 2008;283:33994–34001. doi: 10.1074/jbc.M807124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recuero-Checa MA, Dore AS, Arias-Palomo E, Rivera-Calzada A, Scheres SH, Maman JD, Pearl LH, Llorca O. Electron microscopy of Xrcc4 and the DNA ligase IV-Xrcc4 DNA repair complex. DNA Repair (Amst) 2009;8:1380–1389. doi: 10.1016/j.dnarep.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Reynolds JJ, El-Khamisy SF, Katyal S, Clements P, McKinnon PJ, Caldecott KW. Defective DNA ligation during short-patch single-strand break repair in ataxia oculomotor apraxia 1. Mol Cell Biol. 2009;29:1354–1362. doi: 10.1128/MCB.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, Kysela B, Beamish H, Plowman N, Arlett CF, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- Riballo E, Woodbine L, Stiff T, Walker SA, Goodarzi AA, Jeggo PA. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res. 2009;37:482–492. doi: 10.1093/nar/gkn957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins P, Lindahl T. DNA ligase IV from HeLa cell nuclei. J Biol Chem. 1996;271:24257–24261. doi: 10.1074/jbc.271.39.24257. [DOI] [PubMed] [Google Scholar]
- Rooney S, Alt FW, Lombard D, Whitlow S, Eckersdorff M, Fleming J, Fugmann S, Ferguson DO, Schatz DG, Sekiguchi J. Defective DNA repair and increased genomic instability in Artemis-deficient murine cells. J Exp Med. 2003;197:553–565. doi: 10.1084/jem.20021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, Faure G, Marquez JA, Pietrement O, Guerois R, Callebaut I, et al. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc Natl Acad Sci U S A. 2011;108:12663–12668. doi: 10.1073/pnas.1100758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Andres SN, Vergnes A, Neal JA, Xu Y, Yu Y, Lees-Miller SP, Junop M, Modesti M, Meek K. XRCC4’s interaction with XLF is required for coding (but not signal) end joining. Nucleic Acids Res. 2012;40:1684–1694. doi: 10.1093/nar/gkr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, Poirier G, Reina-San-Martin B, Caldecott KW. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41:33–45. doi: 10.1016/j.molcel.2010.12.006. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, et al. Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J. 2003;22:4566–4576. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S, Glickman MS. Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol. 2007;5:852–861. doi: 10.1038/nrmicro1768. [DOI] [PubMed] [Google Scholar]
- Sibanda BL, Critchlow SE, Begun J, Pei XY, Jackson SP, Blundell TL, Pellegrini L. Crystal structure of an Xrcc4-DNA ligase IV complex. Nat Struct Biol. 2001;8:1015–1019. doi: 10.1038/nsb725. [DOI] [PubMed] [Google Scholar]
- Taccioli GE, Amatucci AG, Beamish HJ, Gell D, Xiang XH, Torres Arzayus MI, Priestley A, Jackson SP, Marshak Rothstein A, Jeggo PA, et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity. 1998;9:355–366. doi: 10.1016/s1074-7613(00)80618-4. [DOI] [PubMed] [Google Scholar]
- Tsai CJ, Kim SA, Chu G. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc Natl Acad Sci U S A. 2007;104:7851–7856. doi: 10.1073/pnas.0702620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011;145:198–211. doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breugel M, Hirono M, Andreeva A, Yanagisawa HA, Yamaguchi S, Nakazawa Y, Morgner N, Petrovich M, Ebong IO, Robinson CV, et al. Structures of SAS-6 suggest its organization in centrioles. Science. 2011;331:1196–1199. doi: 10.1126/science.1199325. [DOI] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lamarche BJ, Tsai MD. Human DNA ligase IV and the ligase IV/XRCC4 complex: analysis of nick ligation fidelity. Biochemistry. 2007;46:4962–4976. doi: 10.1021/bi0621516. [DOI] [PubMed] [Google Scholar]
- Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci. 2011;36:262–271. doi: 10.1016/j.tibs.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PY, Frit P, Meesala S, Dauvillier S, Modesti M, Andres SN, Huang Y, Sekiguchi J, Calsou P, Salles B, et al. Structural and functional interaction between the human DNA repair proteins DNA ligase IV and XRCC4. Mol Cell Biol. 2009;29:3163–3172. doi: 10.1128/MCB.01895-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Ochi T, Matak-Vinkovic D, Robinson CV, Chirgadze DY, Blundell TL. Non-homologous end-joining partners in a helical dance: structural studies of XLF-XRCC4 interactions. Biochem Soc Trans. 2011;39:1387–1392. doi: 10.1042/BST0391387. suppl 1382 p following 1392. [DOI] [PubMed] [Google Scholar]
- Yano K, Morotomi-Yano K, Wang SY, Uematsu N, Lee KJ, Asaithamby A, Weterings E, Chen DJ. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–96. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano KI, Morotomi-Yano K, Lee KJ, Chen DJ. Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS Lett. 2011;585:841–846. doi: 10.1016/j.febslet.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Dynan WS. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999;27:4679–4686. doi: 10.1093/nar/27.24.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Kimzey A, Dynan WS. Photocross-linking of an oriented DNA repair complex. Ku bound at a single DNA end. J Biol Chem. 1999;274:20034–20039. doi: 10.1074/jbc.274.28.20034. [DOI] [PubMed] [Google Scholar]
- Yu Y, Mahaney BL, Yano K, Ye R, Fang S, Douglas P, Chen DJ, Lees-Miller SP. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 2008;7:1680–1692. doi: 10.1016/j.dnarep.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang W, Ding Q, Ye R, Chen D, Merkle D, Schriemer D, Meek K, Lees-Miller SP. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair (Amst) 2003;2:1239–1252. doi: 10.1016/s1568-7864(03)00143-5. [DOI] [PubMed] [Google Scholar]
- Zha S, Alt FW, Cheng HL, Brush JW, Li G. Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci U S A. 2007;104:4518–4523. doi: 10.1073/pnas.0611734104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Wesemann DR, Yuen G, Patel H, Goff PH, et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011;469:250–254. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]