

Abstract
Detailed knowledge regarding the influence of hepatic transport proteins on drug disposition has advanced at a rapid pace over the past decade. Efflux transport proteins located in the basolateral and apical (canalicular) membranes of hepatocytes play an important role in the hepatic elimination of many endogenous and exogenous compounds, including drugs and metabolites. This review focuses on the role of these efflux transporters in hepatic drug excretion. The impact of these proteins as underlying factors for disease is highlighted, and the importance of hepatic efflux proteins in the efficacy and toxicity of drugs is discussed. In addition, a brief overview of methodology to evaluate the function of hepatic efflux transport proteins is provided. Current challenges in predicting the impact of altered efflux protein function on systemic, intestinal and hepatocyte exposure to drugs and metabolites are highlighted.
Keywords: ABC transporter, efflux transporter, liver, hepatocyte, Dubin-Johnson-Syndrome, PFIC
Introduction
The liver plays an important role in the synthesis and secretion of bile acids, in the metabolism and transport of cholesterol, and in the metabolism and efflux of endogenous and exogenous substances by cytochrome P450 enzymes and phase II conjugation systems. Until the latter part of the twentieth century, it was generally assumed that passive diffusion, governed by the physicochemical properties of molecules, determines the uptake and efflux of drugs/metabolites across the basolateral and apical membranes of hepatocytes. However, it is now well established that transport proteins play a key role in the uptake clearance of many drugs from hepatic sinusoidal blood, and in the excretion of parent compounds and/or metabolites from the hepatocyte into bile, or the systemic circulation. The aim of this review is to summarize the current knowledge of hepatic basolateral and canalicular efflux proteins. For transporters involved in hepatic uptake processes, we refer the reader to the recently published review by Hagenbuch et al. (1) Comprehensive reviews are referenced for details on specific efflux transport proteins.
1. Major Hepatic Efflux Transporters
The hepatocyte is highly polarized with respect to transport systems expressed on the basolateral/sinusoidal or apical/canalicular membrane domain. Both membrane leaflets are separated by tight junctions that seal off the lumen of the bile canaliculi from the systemic circulation. The large pores (fenestrae) of the sinusoidal endothelial cells allow contact of the basolateral membrane of hepatocytes and sinusoidal plasma. Basolateral uptake is the first step in hepatic clearance of compounds from the systemic circulation; this explains the high abundance of uptake transporters in the basolateral including the sodium-dependent taurocholate cotransporting polypeptide (NTCP), the organic anion transporting polypeptides (OATPs), organic anion transporters (OATs), and organic cation transporters (OCTs). In contrast, efflux transporters located in both the canalicular and basolateral membrane leaflets mediate excretion into bile or into the systemic circulation, respectively. (Figure 1)
Figure 1. Localization of drug transport proteins in the liver.

This figure was modified from Ho and Kim. (130)
Apical/Canalicular Transporters
Substances are excreted into bile primarily by ATP-dependent proteins of the ABC transporter superfamily [e.g., multidrug-resistance proteins (MDR), and the multidrug-resistance-associated proteins (MRPs)]. More recently, the multidrug and toxin extrusion exchanger 1 (MATE 1) has been identified as an apical efflux transporter for cations in hepatocytes.
BSEP
Bile salt export pump (BSEP) (ABCB11), a member of the ABC transporter superfamily, is located exclusively in the canalicular membrane of hepatocytes. BSEP has a narrow substrate spectrum and transports primarily monoanionic, conjugated bile acids. It exhibits high affinity for bile acids (taurochenodeoxycholate > taurocholate > taurodeoxycholate > tauroursodeoxycholate > glycocholate). Interestingly, only a few drugs have been described as BSEP substrates; pravastatin has been shown to be transported by both human and rat BSEP/Bsep, and vinblastine has been identified as a substrate of mouse Bsep. (2) In humans, BSEP appears to be the sole transporter for monoanionic bile acids across the apical membrane; a decrease in BSEP function may lead to reduced bile acid secretion, enhanced intracellular accumulation of bile acids, cholestasis, and, in the worst case, progressive liver injury. Indeed, BSEP deficiency results in several genetic forms of cholestasis, such as progressive familial intrahepatic cholestasis type 2 (PFIC2) and benign recurrent intrahepatic cholestasis type 2 (BRIC2). In addition, this protein has been implicated in drug-induced cholestasis mediated by interactions between drugs and endogenous compounds, and intrahepatic cholestasis of pregnancy. (3)
P-glycoprotein (P-gp)
P-glycoprotein (MDR1, ABCB1) was one of the first transporters recognized as having an important role in drug distribution and elimination. P-gp was first identified by Juliano and Ling in 1976 in Chinese hamster ovary cells that were selected for colchicine resistance, and that displayed resistance to a wide range of amphiphilic drugs. P-gp functions as an efflux pump for a wide range of substances including amphiphilic bulky cationic drugs, as well as for steroid hormones, hydrophobic peptides, and glycolipids. (see (4) for review) P-gp is expressed in many biological interfaces, including the intestine, liver, the blood-brain and placental barriers, and the kidney, suggesting that this protein plays an important role in the distribution of xenobiotics and endogenous substances. In the liver, P-gp is expressed in the canalicular membrane of hepatocytes. However, hepatic P-gp expression levels are approximately sevenfold lower than in the intestine. (5) Furthermore, high interindividual differences in expression levels (50-fold) have been reported. (6) The relative contribution of hepatic P-gp to the overall disposition of substrate drugs in humans has been questioned.
MRP2
MRP2 (ABCC2) is an organic anion transporter expressed in the apical membrane of polarized cells such as hepatocytes, renal cells, enterocytes and placental cells. It plays a key role in the biliary elimination of glucuronide and sulfate conjugates, and divalent bile acids. (reviewed in (7)) MRP2 is involved in the biliary excretion of glutathione, which is a major osmolyte and a factor that determines bile-salt-independent bile flow. (8) MRP2 maintains a steep concentration gradient of GSH between blood and bile (GSH in bile = 10–15 mmol/l; GSH in blood = 100 μmol/l), thereby drawing water into the bile. This was supported by the observation that animals lacking functional MRP2 (e.g., TR- or Esai hyperbilirubinemic rats (EHBR) rats) exhibited a bile flow rate approximately twofold lower than that in control animals. (9) As discussed below, MRP2 deficiency is associated with Dubin-Johnson-syndrome (DJS) and results in hyperbilirubinemia, due to impaired extrusion of bilirubin glucuronide into bile via MRP2. As a compensatory mechanism, upregulation of MRP3 increases transport of bilirubin glucuronide across the sinusoidal membrane. (10)
BCRP
The breast cancer resistance protein BCRP (ABCG2) was first described in multidrug-resistant cell lines, in which it was shown to confer resistance to cytotoxic compounds such as mitoxantrone, topotecan, irinotecan, and doxorubicin. Given its expression in blood-tissue barriers such as the blood-brain barrier, the intestine, and the placental barrier, BCRP is believed to have a major role in protecting physiologic barriers. Because of its broad substrate specificity, including specificity to anticancer agents and environmental carcinogens, BCRP has been associated with multidrug resistance and tumor development/progression. In the liver, BCRP is expressed in the canalicular membrane of hepatocytes. Numerous studies have demonstrated the important role of BCRP in the biliary elimination of substrates. In Bcrp knockout mice, the biliary excretion rates of methotrexate, the HMG-CoA-reductase inhibitors pitavastatin and rosuvastatin, and fluoroquinolones were significantly lower than in wild-type mice, as published in a recent review. (11)
MATE
The discovery of MATE proteins explained how organic cations are excreted across the apical membrane in the liver and kidney. MATE proteins were identified originally as bacterial transporters. However, in 2005, two human MATE transporter proteins, MATE1 and MATE2, were identified on the basis of gene sequence similarity. (12) In contrast to other canalicular drug efflux transporters, MATE proteins belong to the solute carrier family 47 (SLC47) and function as secondary transport systems that utilize the electrochemical gradient of cations across the membrane for substrate transport, which may occur in both directions. Human MATE proteins mediate the excretion of compounds into bile and urine on the basis of H+/organic cation antiport. MATE1 demonstrates predominant expression in the canalicular membrane of hepatocytes, but it can also be found in the kidney, skeletal muscle, adrenal gland, and testis. MATE2, on the other hand, is almost exclusively expressed in the luminal membrane of proximal renal tubular epithelial cells. Substrates of MATE proteins include the organic cations creatinine, guanidine, and thiamine, as well as numerous drugs such as metformin, cimetidine, oxaliplatin, acyclovir, and fexofenadine. In the liver, organic cation transporter 1 (OCT1) may function in concert with MATE1 to mediate the hepatic uptake and biliary excretion, respectively, of cationic drugs and their metabolites. (13)
Basolateral/sinusoidal transporters
Basolateral efflux transporters mediate the removal of endogenous and xenobiotic compounds from the hepatocyte into sinusoidal blood. Transporters suggested to be involved in basolateral efflux processes include MRP3, MRP4, MRP5, and OSTα/β.
MRP3
MRP3 (ABCC3), an organic anion transporter expressed in the liver, kidney, intestine, adrenals, and pancreas, is localized to the basolateral membrane in polarized epithelial cells. (14) In the human liver, prominent MRP3 staining of cholangiocytes and weak staining of hepatocytes around the portal tract have been shown. However, hepatic MRP3 protein expression is highly variable (up to 85-fold) and highly inducible. (15) For example, in humans, MRP3 expression is upregulated in the absence of functional MRP2, such as in patients with DJS (16), and under cholestatic conditions when the biliary excretion of organic anions via the canalicular membrane of the hepatocyte is impaired. (17) Originally, it was hypothesized that human MRP3 plays a role in hepatocellular bile acid homeostasis based on a report that rat Mrp3 transported conjugated bile acids with rather high affinity. (18) However, it was later discovered that human MRP3 transports bile acids only with low affinity. (19) The clinical relevance of human MRP3 as an important compensatory bile acid efflux pump remains to be established. In Mrp3 knockout mice, there was no evidence of the expected differences in serum bile acid concentrations and hepatic injury after bile duct ligation. (20) However, because of a high affinity for glucuronide conjugates (e.g., morphine-3-glucuronide, bilirubin-glucuronide, etoposide-glucuronide, and acetaminophen-glucuronide), it has been suggested that MRP3 has a defense related function and contributes to the excretion of toxic anions. In this context, MRP3 may act as a switch to change the excretion route from bile to urine under pathological conditions.
MRP4
MRP4 (ABCC4) is expressed widely in normal tissues and has been implicated in the transport of antiviral agents (e.g., azidothymidin, adefovir, and ganciclovir), anticancer agents (e.g. methotrexate, 6-mercaptopurin, and camptothecins) and cardiovascular agents (loop diuretics, thiazides, and angiotensin II receptor antagonists), as well as of endogenous substances such as steroid hormones, prostaglandins, bile acids, and the cyclic nucleotides cAMP and cGMP. Sulfated conjugates of bile acids and steroids have high affinity for MRP4. (21) Although no specific disease has been linked directly to altered MRP4 activity, the sinusoidal expression of this protein in hepatocytes, its ability to transport bile acids, and its increased expression in human and rat livers under cholestatic conditions (22, 23) support the hypothesis that MRP4 is an important component of the protective system of hepatocytes. In support of this hypothesis – and in contrast to the findings in Mrp3 knockout mice – cholestasis induced by bile duct ligation in Mrp4 knockout mice resulted in increased liver toxicity as compared with bile-duct ligated wild-type mice. (24)
MRP5
MRP5 (ABCC5), another member of the ABCC subfamily, is expressed primarily in the colon, liver, kidney and brain. In mice and humans, Mrp5/MRP5 is expressed at relatively low levels in healthy liver; however, MRP5 mRNA and protein are upregulated in patients with primary biliary cirrhosis. (25). Currently, it is not known whether MRP5 participates in hepatic drug transport.
OSTα/β
OSTα/β was identified in 2001 as a novel organic solute and steroid transporter in the little skate Leucoraja erinacea (26); in 2003, mouse and human orthologs were identified and cloned. (27) In vitro transfection experiments have shown that OSTα and OSTβ function as heteromeric proteins, and that c-expression of the two is required for delivery to the plasma membrane and functional activity. In human tissues, mRNA of OSTα and OSTβ is expressed widely; the highest expression levels are in tissues involved in steroid and bile acid homeostasis such as the small intestine, liver, colon, kidney, testes, ovaries, and adrenal gland. OSTα/β substrates include steroid hormones and endogenous compounds such as estrone sulfate and dehydroepiandrosterone sulfate, bile acids, and PGE2, as well as the cardiac glycoside digoxin. (28) Ballatori et al. showed that OSTα/β-mediated transport is bidirectional, ATP-independent, and unaffected by changes in transmembrane electrolyte concentrations and changes in pH gradients, suggesting that the transport takes place via facilitated diffusion. (29) Depending on the extent of the electrochemical gradient, either efflux or uptake of substrates can take place. To date, no human disease has been associated with impaired OSTα/β function. However, the expression of this protein in organs involved in bile acid homeostasis, including the intestine and liver, suggests that this transporter might be involved in diseases associated with bile acid malabsorption or cholestasis. The participation of OSTα/β in bile acid transport in the intestine has been demonstrated in knockout animal models, but its role in bile acid disposition in the liver is less clear.
2. Efflux Transport Proteins as Underlying Factors for Disease
The importance of hepatic efflux proteins in liver function is best exemplified by two diseases in humans, both of with associated with reduced/missing function of drug efflux carriers: PFIC and DJS. The elucidation of the biochemical mechanisms underlying these diseases, highlighted in the following section, played a key role in the early discovery and characterization of hepatic transport proteins and improved our understanding of the function of these proteins in the hepatic disposition of endogenous and exogenous substances.
Progressive Familial Intrahepatic Cholestasis (PFIC)
Bile formation is a major function of the liver. After synthesis from cholesterol, bile acids are secreted via the biliary tree into the intestine, where they are involved in ingestion of fatty acids and fat-soluble vitamins. Along the small intestine, transport proteins facilitate bile acid reabsorption across the intestinal epithelial cells into the portal circulation and back to the liver. From the portal blood, bile acids are taken up into hepatocytes and the process of enterohepatic circulation is repeated. It is well established that hepatic bile acid secretion and enterohepatic circulation require the coordinated action of distinct uptake and efflux transporters on the apical and basolateral membranes of enterocytes and hepatocytes.
Initially, the electrochemical gradient across the canalicular membrane of the hepatocyte was thought to be the driving force for bile acid secretion into bile; however, this gradient was found to be insufficient to act as a driving force for the high concentrations of bile acids achieved in bile. The membrane potential across the canalicular lumen of approximately −35 mV would account for a gradient of only 1:3, whereas in vivo cell-to-bile acid concentration gradients of 1:10 to 1:100 are reached. In the early 1990s, studies in isolated membrane vesicles from rat and human liver demonstrated that canalicular bile acid transport is an ATP-dependent process. (30, 31) In 1995, a gene known as sister of P-gp (Spgp) was cloned by Childs et al. from a pig cDNA library using low stringency screening with a probe sequence from the multidrug-resistance gene MDR1. (32) MDR1 and Spgp shared 61% amino acid identity. Tissue expression of the Spgp mRNA was detected almost exclusively in canalicular microvilli and in subcanalicular smooth membrane vesicles of rat liver, suggesting that it may be a candidate for ATP-dependent bile acid transport. Indeed, Gerloff et al. demonstrated bile acid transport in Xenopus leavis oocytes injected with rat Spgp, and in membrane vesicles from Sf9 cells transfected with rat Spgp cDNA. (33) With these systems, it was shown that Spgp functions as an ATP-dependent bile acid transporter, and the protein was renamed the “bile salt export pump” (BSEP/Bsep). Human BSEP, the functional expression of which observed and characterized in 2002, also acts as a bile acid transporter.
In the late 1990s, a link was identified between PFIC type 2 and BSEP mutations, and this disease was mapped to chromosome 2q24 where BSEP is encoded. (34) PFIC is a very heterogeneous group of autosomal recessive liver disorders leading to intrahepatic cholestasis. On the basis of clinical, biochemical, and histological features, PFIC can be divided into three types that are associated with mutations in ATP8A1 (PFIC type 1), ABCB11 (PFIC type 2), and ABCB4 (PFIC type 3). (35) The findings in patients with mutations in the ABCB11/BSEP gene are characterized by normal γ-glutamyltransferase activity, decreased biliary bile acid concentrations, and the absence of bile duct proliferation. The importance of BSEP function is highlighted by the fact that patients with PFIC type 2 secrete <1% of bile acids into bile as compared with normal individuals. (36) These findings also suggest that there is no backup system for BSEP in the canalicular membrane. Without treatment, PFIC results in cirrhosis, which rapidly progresses to hepatic failure and often requires liver transplantation before the patient reaches adolescence. (37) Several premature termination, missense, and frameshift mutations have been identified in patients with PFIC type 2; these mutations impair trafficking to the canalicular membrane and/or transport activity. (38) (Figure 2) A recent study in BSEP patients demonstrated that the D482G mutation is associated with slower disease progression, consistent with data that the D482G BSEP protein retains some functional activity. (39) Furthermore, these patients develop cirrhosis later in life and thus require liver transplantation later than other PFIC type 2 patients. (40).
Figure 2. Schematic representation of progressive familial intrahepatic cholestasis (PFIC) type 2.<.

br>Left panel: Under normal conditions BSEP transports bile acids into the bile. Right panel: Mutation of the ABCB11/BSEP gene results in proteasomal degradation and/or expression of a protein with low/no function, leading to reduced transport of bile acids into bile and consequently accumulation of deleterious bile acids in the hepatocyte. The broken lines represent minimal or no transport. ER, endoplasmatic reticulum.
Dubin-Johnson Syndrome
First described in 1954, DJS is a rare autosomal recessive disorder, characterized by impaired secretion of organic anions such as bilirubin glucuronide, a black liver due to bilirubin accumulation, and conjugated hyperbilirubinemia in plasma. (41) It had long been assumed that this syndrome was the result of a canalicular secretion defect; however, the underlying mechanism for this disorder was not identified until 1997, when a mutation in the ABCC2 gene was determined to be responsible for impaired expression and function of the multidrug resistance associated protein 2 (MRP2, Figure 3). (42) One year earlier, Kartenbeck et al. had used an antibody directed against MRP1 (a protein related to MRP2) to demonstrate the absence of canalicular immunostaining in the liver section of a DJS patient. This antibody demonstrated lateral and canalicular staining in liver samples from healthy individuals, but only lateral staining in those from patients with DJS. (43) Subsequently, it was determined that this antibody crossreacts with MRP2, detecting the apically localized MRP2 and as possibly the lateral/basolateral MRP1as well.
Figure 3. Schematic representation of Dubin-Johnson-Syndrome.
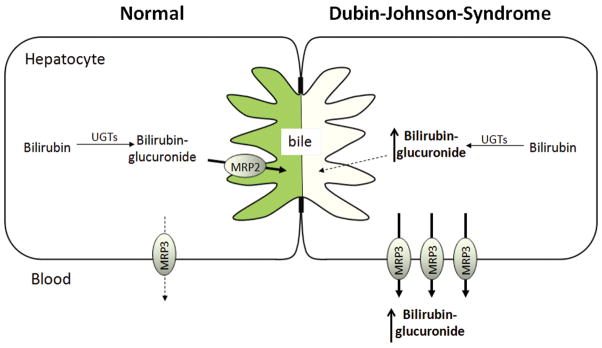
Left panel: Under normal conditions, MRP2 excretes bilirubin-glucuronide into bile. Right panel: Loss-of-function of MRP2 results in accumulation of bilirubin-glucuronide in the hepatocyte. Increased MRP3 on the basolateral membrane of the hepatocytes transports bilirubin-glucuronide into the sinusoidal blood, resulting in conjugated hyperbilirubinemia in the systemic circulation. The broken lines represent minimal or no transport.
The important discovery that loss-of-function of MRP2 is the mechanism underlying DJS was facilitated by the description and characterization of rat strains with inherited, conjugated hyperbilirubinemia, which resemble the findings in human DJS. The mutant TR-/GY (Groningen Yellow) Wistar rat strain was first identified by Jansen et al. (9) A similar mutant strain, EHBR, was identified in Sprague-Dawley rats in 1986 in Japan by Mikami et al. (44) Both mutant rat strains are deficient in the canalicular transport of a variety of anionic conjugates such as bilirubin glucuronide, and glutathione and sulfate conjugates of endogenous and exogenous compounds. Interestingly, recent studies have revealed that TR- rats also exhibit low expression levels of Bcrp protein. (45) Even before members of the ATP superfamily were cloned and functionally characterized, these mutant rat strains provided information on the functional capabilities of a postulated transport protein referred to as canalicular multispecific organic anion transporter (cMOAT), non-bile acid organic anion transporter, glutathione S-conjugate export pump, or leukotriene export pump. The biliary excretion of the radiolabeled glutathione S-conjugate of leukotriene C4 (LTC4), a compound later identified as a high-affinity MRP2 substrate, was significantly lower in TR- rats. (46) Transport measurements carried out on inside-out canalicular membrane vesicles prepared from these mutant rat strains contributed further to our knowledge of substrate specificity. For example, ATP-dependent LTC4 transport was significantly impaired in canalicular membrane vesicles prepared from transport-deficient TR- rats. (47)
The identification of the ABCC2 gene was facilitated by the prior identification of MRP1, which conferred multidrug-resistance in a lung cancer cell line (48); PCR, based on conserved domains of ABCC1, demonstrated a lack of amplification of a 347-bp fragment in TR- but not in normal Wistar rat hepatocytes. Furthermore, the sequence of the amplified fragment was MRP1-like but differed significantly from the cDNA encoding rat Mrp1, indicating the absence of an Abcc transporter isoform in this rat strain. (49) Subsequently, in 1996, cloning and sequencing of rat Mrp2 revealed a nucleotide deletion in the Abcc2 gene of TR- rats. (50) Human cDNA was cloned in the same year from a cisplatin-resistant cell line. (51) Since then, ABCC2/Abcc2 proteins have been expressed and characterized in detail. (7) The contribution of Mrp2 in the pathogenesis of the DJS-like phenotype in rats was confirmed by infecting EHBR rats with an adenovirus carrying ABCC2, which compensated for the genetic defect and corrected the canalicular transport deficiency. (52)
3. Hepatic Efflux Transport Proteins as Determinants of Pharmacokinetics, Efficacy and Side Effects of Drugs
Drug-induced enteropathy and diarrhea
Biliary excretion is one route of elimination for many endogenous compounds such as bilirubin and hormones, as well as for some xenobiotics and their metabolites. Interestingly, in certain cases, the biliary excretion of drugs and/or metabolites may be responsible for injury to the small intestine (enteropathy) and diarrhea. Nonsteroidal anti-inflammatory agents (NSAIDs), irinotecan, and mycophenolic acid are examples of drugs associated with a high risk of drug-induced enteropathy and diarrhea. The role of hepatic efflux transporters in the adverse events associated with these drugs is discussed in the following section.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are associated with gastric injury and are being recognized increasingly as a cause of enteropathy. Recent data using video capsule imaging studies demonstrated an incidence of intestinal damage of ~60% in subjects taking NSAIDs for a 2-week period. (53, 54) The gastric side effects of NSAIDs are well understood, but the pathophysiological mechanisms of enteropathy, characterized by inflammation and lesions in the small intestine, blood loss, increased permeability, and bile acid malabsorption, are far less clear.
Most NSAIDs, including ibuprofen, diclofenac, ketoprofen, and indomethacin, undergo UGT-mediated biotransformation to form a reactive acyl glucuronide, which can then form adducts with proteins. This acyl glucuronide formation has been suggested to be the major toxicokinetic determinant. Drug conjugates are excreted into bile and move into the distal regions of the intestine, where they are deconjugated by bacterial glucuronidases, leading to high local concentrations of the parent compound. (55) Several studies have suggested that intestinal colonization with different types of bacteria plays a significant role in the development of NSAID-associated enteropathy, and that the deconjugation capacity of bacteria might be an important factor in causing intestinal damage. Medications that influence the number and/or types of intestinal bacteria, such as antibiotics or proton pump inhibitors, might exacerbate NSAID-induced intestinal damage. (56, 57)
In the 1970s, biliary excretion and enterohepatic circulation were identified as important factors in the development of NSAID enteropathy; bile-duct ligation in rats prevented intestinal damage. (58, 59) This was substantiated by the observation that NSAIDs with limited enterohepatic circulation did not cause intestinal damage in animal models. (60) Further experiments in MRP2-deficient EHBR and TR- rats demonstrated the key role of Mrp2 in the development of this side effect; rats deficient in Mrp2 exhibited lower levels of adduct formation and ulceration. (61) Mrp2-deficient rats excreted 50% less indomethacin glucuronide and 98% less diclofenac glucuronide into bile as compared with wild-type rats. (61) Transfer of diclofenac glucuronide-containing bile from diclofenac-treated wild-type rats into the intestine of TR- rats significantly increased intestinal damage, demonstrating that biliary excretion is involved in the pathogenesis of small-intestine injury. (62)
Irinotecan, (CPT-11, Figure 4) a topoisomerase 1 inhibitor, is used widely as an anticancer agent, particularly in the treatment of colorectal cancer, lung cancer and malignant melanoma. Clinical use of irinotecan is associated with myelotoxicity and dose-limiting severe diarrhea in 5–40% of patients. (63) After administration by intravenous injection, irinotecan is hydrolyzed by hepatic carboxylesterases to the active SN-38 form, followed by inactivation by CYP-mediated oxidation or glucuronidation. Extensive amounts of irinotecan, SN-38 and SN-38 glucuronide (SN-38G) are excreted into the bile and intestine via several ATP-binding cassette transporters, including P-gp, MRP2, and BCRP (Figure 4). (64, 65) In the human intestine, active SN-38 can be formed from irinotecan via intestinal carboxylesterases or by deconjugation of SN-38G by bacterial β-glucuronidases, and then reabsorbed resulting in a secondary peak in the systemic concentration-time profile. (66) It has been hypothesized that repeated exposure of SN-38 to enterocytes by enterohepatic circulation is the cause of diarrhea.
Figure 4. Drug efflux transporters and toxicity in liver and intestine.

Left panel liver and intestine: In the plasma or hepatocyte, the prodrug irinotecan (CPT-11) is converted to SN-38 by carboxylesterases (CE). SN-38 is further glucuronidated and excreted into bile. In the intestine, SN-38G is converted back to SN-38 by enteric bacterial β-glucuronidases and is available for reabsorption. It has been hypothesized that repeated exposure of enterocytes to SN-38 is hypothesized to be the cause of severe diarrhea. Right panel (liver and intestine): Bile acids are taken up from the systemic circulation by sodium-dependent taurocholate cotransporting peptide (NTCP) or OATPs, or synthesized from cholesterol. The efflux of bile acids across the canalicular membrane into bile is mediated primarily by the bile salt export pump BSEP (which transports predominantly monovalent bile acids) and MRP2 (which transports sulfate and glucuronide conjugates of bile acids). Under cholestatic conditions, MRP3, MRP4, and OSTα/β act to efflux bile acids across the basolateral membrane into the blood. In the intestine, the apical bile salt transporter ASBT and OATPs mediate the reuptake of bile acids. From the enterocyte, bile acids are excreted by MRP3, MRP4, and OSTα/β into the mesenteric circulation, and they ultimately flow into the portal vein. Inhibition of the major bile acid transporter BSEP results in accumulation of cytotoxic bile acids, potentially causing liver injury.
Numerous investigators have attempted to identify the environmental and genetic factors that affect the pharmacokinetic profile of SN-38 in an effort to predict side effects and individualize dosage regimens for patients. A major breakthrough in therapy was the evidence linking the low glucuronidation capacity of SN-38 to an increased risk of irinotecan-induced neutropenia. SN-38 is glucuronidated predominantly by UGT1A1. UGT1A1*28, which is characterized by seven TA repeats in the TATA element of UGT1A1 promoter (the wild-type genotype has six repeats), demonstrated lower expression levels and function and has been associated with irinotecan hemotoxicity. On the basis of these findings, the U.S. Food and Drug Administration (FDA) approved a label change for Camptosar® (irinotecan hydrochloride) to include recommended dosage adjustments for patients with the UGT1A1*28 allele. (67)
Although there is strong evidence linking glucuronidation capacity with neutropenia, the association with diarrhea is less clear; approximately one half of irinotecan-treated patients with severe diarrhea have the UGT1A1*28 genotype. (68) This suggests that there are other mechanisms, apart from UGT activity, that affect the pharmacokinetic and side effect profiles of irinotecan.
The hepatic uptake transporter OATP1B1 has been shown to transport SN38, the active metabolite of irinotecan; several clinical studies have highlighted the importance of this protein in the disposition of SN38. (69, 70) Furthermore, the drug efflux transporters P-gp, MRP2, and BCRP have been implicated in the disposition and toxicity of irinotecan, SN-38, and SN38G because of their involvement in biliary excretion. (64, 65) Two studies in patients with lung cancer demonstrated an association between the P-gp 3435TT genotype (which results in a lower plasma AUC for SN38G) and a higher incidence of diarrhea. (71, 72)
It has been suggested that functional variants in MRP2 (such as ABCC2 3972C>T) have profound effects on the pharmacokinetics, efficacy, and toxicity of irinotecan. (72, 73) De Jong et al. demonstrated that patients carrying the haplotype ABCC2*2 (low activity) exhibited a lower irinotecan clearance and less severe diarrhea if they did not carry the UGT1A1*28 allele, suggesting that MRP2 and glucuronidation play an important role in intestinal side effects. (73) One hypothesis that could explain this observation is that the reduction in MRP2-mediated biliary excretion due to the ABCC2*2 haplotype leads to decreased intestinal formation of SN-38 locally. However, the presence of an UGT1A1*28 allele, and consequently the reduced detoxification and increased exposure to SN-38 in the enterocytes, could counteract this protection. (73)
Mycophenolate mofetil (MMF), the prodrug of mycophenolic acid (MPA), is a commonly used immunosuppressant for the prevention of transplant rejection. Like irinotecan, MMF therapy is associated with severe side effects including gastrointestinal complications and bone marrow suppression leading to dose reduction or drug discontinuation.
After oral administration, MMF is hydrolyzed rapidly to the active MPA by esterases located in the gut wall, the liver, and the systemic circulation. In the liver, MPA is metabolized predominantly to the inactive MPA-phenyl-glucuronide (MPAG) and, to a lesser extent, to the active MPA-acyl-glucuronide (AcMPAG). (74, 75) Although the kidney is the major organ involved in MPA elimination, MPA-glucuronides undergo biliary excretion, intestinal deconjugation, and reabsorption as MPA resulting in a secondary peak in the MPA concentration-time profile, which contributes between 10 and 60% to the AUC of MPA. (76, 77) Although there does not appear to be an association between plasma AUC and diarrhea, (78) increased intestinal exposure to MPA due to biliary excretion of MPA glucuronides seems to be associated with the onset of diarrhea. Experiments in Mrp2-deficient EHBR and TR- rats, and co-treatment with the MRP2 inhibitor cyclosporin A, demonstrated that MRP2 is the major transporter involved in the biliary excretion of MPAG and AcMPAG. (79, 80) These observations prompted a series of studies investigating the role of MRP2 polymorphisms in the development of diarrhea. The studies focused on the ABCC2 C-24T promoter variant, which has been associated with altered expression and activity of MRP2. (81) To date, two studies – one in pediatric heart transplant patients and adult renal transplant patients reported a significantly higher incidence of diarrhea in patients expressing the ABCC2 C-24T promoter variant. However, three studies in patients after renal transplant failed to show this relationship. (82–86) These controversial data suggest that other factors are involved in the development of intestinal injury during MPA therapy, including UGT-mediated glucuronidation and/or local bioactivation by the intestinal microflora.
Drug-induced Liver Injury
Drug-induced liver injury (DILI) is a serious adverse event that often results in the withdrawal of drugs from the market and is one of the leading causes for the failure of drug candidates in development. Although several mechanisms may be involved in the pathogenesis of DILI including reactive metabolite formation, mitochondrial toxicity, and immunologic reactions, hepatobiliary transporters are postulated to be important contributors to the hepatotoxicity associated with some medications. In principle, either of two mechanisms may account for the involvement of transporters in the pathogenesis of DILI: (i) direct transport of potentially hepatotoxic compounds or metabolites, or (ii) interaction of drugs or metabolites with the excretion of potentially hepatotoxic bile acids.
The hepatobiliary disposition of drugs and bile acids is governed by hepatic uptake, metabolism, and excretion. It has become increasingly clear that modifications in these processes, either by drug-drug interactions, disease states, or genetic variation, can greatly influence the disposition of potentially toxic endogenous and exogenous compounds. Currently, little information is available regarding the involvement of basolateral uptake transporters such as OATPs, OCTs, and OATs in the development of DILI. However, several drugs associated with DILI are substrates for these uptake transporters; therefore, enhanced expression or function of these uptake transporters might increase hepatocyte concentrations of hepatotoxic compounds.
In contrast to the relative paucity of information relating to uptake transporters in the development of DILI, more information is available on the impact of efflux transporters, especially BSEP. As described above, BSEP primarily governs the biliary excretion of bile acids under normal physiologil conditions. Inherited dysfunction of BSEP leads to cholestatic syndromes (PFIC, BRIC) and cholestatic liver injury. Several drugs associated with DILI are potent inhibitors of BSEP, including cyclosporin A, bosentan, troglitazone, sulindac, rifamycin, and glibenclamide. (87, 88) The metabolites of a drug may also contribute to its hepatotoxic potential. For example, troglitazone sulfate exhibits 10-fold more potent inhibition of Bsep-mediated taurocholate transport than troglitazone in canalicular plasma membrane vesicles isolated from rat liver. (88) Extensive hepatocellular accumulation of troglitazone sulfate due to impaired canalicular and/or basolateral efflux, may increase the susceptibility of some patients to liver injury mediated by troglitazone. (89) The results of a recent study that screened 200 compounds for BSEP inhibition suggested a strong association between the pharmacologic interference of this transporter and liver toxicity (Figure 4). (90) However, the low incidence of DILI suggests that other factors – genetic or environmental – are involved in the pathogenesis of DILI, predisposing some patients to develop this deleterious side effect. MRP3 and MRP4 are two basolateral proteins that are involved in bile acid efflux from hepatocytes, especially when biliary excretion is impaired. Studies in humans and bile duct-ligated rats have demonstrated that these transporters are upregulated under cholestatic conditions. (14, 23) The development of cholestatic liver injury after treatment with a BSEP inhibitor might be dependent on the expression and function of these basolateral efflux transporters that normally counteract increases in intracellular concentrations of potentially hepatotoxic bile acids.
In addition to inhibition of bile acid transport, the hepatic disposition of toxic drugs and/or metabolites may be an underlying cause of liver injury. For example, patients with an MRP2 polymorphism associated with reduced function (C-24T) have a five- to sixfold higher risk of developing hepatotoxicity after taking diclofenac, probably due to increased intracellular accumulation of reactive metabolites and higher levels of toxic protein adducts. (91) Trabectedin, a promising anticancer drug, exhibits dose-limiting hepatotoxicity. Mrp2 and other hepatic efflux proteins protect against trabectedin-mediated hepatotoxicity as shown in studies in sandwich-cultured rat hepatocytes. (92) The importance of Cyp3a-generated metabolites in trabectedin hepatotoxicity, as well as the protection afforded by drug efflux proteins, was confirmed by studies in transporter knockout mice; only mild hepatotoxicity was observed in Cyp3a/Abcb1a/1b/Abcc2−/− mice in contrast to the severe toxicity seen in Abcb1a/1b/Abcc2−/− mice. (93)
Statins
HMG-CoA reductase inhibitors, commonly referred to as “statins”, are prescribed for the treatment of hypercholesterolemia, a major risk factor for cardiovascular disease; statins are among the most widely used drugs worldwide. The major target of statins is inhibition of the hepatic synthesis of mevalonate, the rate-limiting step in cholesterol biosynthesis. Although statins are generally well tolerated, plasma-concentration dependent myopathy is a side effect characterized by muscle pain, fatigue, and cramping, which ranges from mild myalgia to life-threatening rhabdomyolysis. High interindividual variability in plasma concentrations is a characteristic of statins; plasma concentrations and toxicity may be influenced by hepatic drug transporters. Expression of OATP1B1, an influx transporter localized on the basolateral membrane of human hepatocytes, influences the pharmacokinetic profile as well as the myotoxicity of statins. Individuals expressing the c.521CC genotype exhibited higher plasma AUCs after administration of simvastatin, atorvastatin, and pitavastatin compared to those expressing the c.521TT genotype, probably due to decreased hepatic uptake of statins. Furthermore, the c.521T>C SNP has been associated with simvastatin-induced myopathy and a slight reduction in cholesterol-lowering efficacy (comprehensively reviewed in (94)).
Statins are also substrates of drug efflux transporters, including BCRP. Clinical implications have been reported in patients with the c.421C>A SNP, which reduces the transport function of BCRP. (95, 96) Subjects expressing at least one variant BCRP 421A allele were more likely to attain LDL cholesterol target concentrations after treatment with rosuvastatin. (97) A plausible mechanism is decreased hepatic efflux, leading to higher intracellular concentrations and increased efficacy in patients carrying this variant genotype (Figure 5). However, BCRP also is expressed in the apical membrane of enterocytes where it limits the absorption of rosuvastatin from the small intestine. (98) Carriers of the 421 allele also had higher AUCs and higher peak concentrations of rosuvastatin and atorvastatin in plasma. (99, 100) These studies suggest that the greater efficacy of rosuvastatin in patients carrying the variant BCRP allele might be attributable to a combination of increased plasma rosuvastatin concentrations and increased hepatocyte exposure.
Figure 5. Drug efflux transporters and efficacy.

Top right panel: Statins are taken up into hepatocytes by OATP-mediated transport or passive diffusion. They inhibit the main enzyme of cholesterol synthesis, HMG-CoA-reductase. Excretion of statins into bile is mediated primarily by BCRP. Lower panel: Effect of OATP and BCRP inhibition on the concentration-time profiles of statins in liver and blood (modified from (108)). Top left panel: Morphine is metabolized to morphine-3- and morphine-6-glucuronide followed by canalicular or basolateral efflux. In the systemic circulation, morphine-6-glucuronide contributes to the antinociceptive effect of morphine; morphine-glucuronides (morphine-G) are excreted primarily via the kidney.
Morphine, MRP3, and antinociception
Morphine is a potent opiate analgesic that is used clinically to treat severe acute and chronic pain. A major obstacle in therapy is the development of drug tolerance, which necessitates dose increases and further elevates the risk of side effects. Furthermore, plasma concentrations of morphine and its metabolites vary considerably between patients, and it is necessary to titrate the dose according to the pain. In humans, morphine primarily undergoes hepatic metabolism to morphine-3-glucuronide (M3G; 60%) and morphine-6-glucuronide (M6G; 6–10%), which is mediated by UDP-glucuronosyl transferase 2B7 (UGT2B7); (101) a large amount of the dose is excreted in urine, predominantly in the form of the glucuronide metabolites. (102) In humans, M6G is the major active metabolite of morphine, and is more potent than the parent compound, while M3G has no analgesic properties and even antagonizes the effect of morphine. (103) Compared to the parent compound, the glucuronide metabolites are more polar, which limits diffusion through membranes. Morphine-glucuronide conjugates must be transported into the systemic circulation by efflux proteins on the basolateral membrane of hepatocytes; MRP3 has been suggested as a possible transport protein (Figure 5). (16, 104) Indeed, higher amounts of M3G were recovered in liver and bile of Mrp3 knockout mice; plasma M3G concentrations were reduced 50-fold, suggesting that Mrp3 is involved in the excretion of morphine-glucuronides from hepatocytes into the systemic circulation (M6G is not formed in mice). (105) Human MRP3 also transports M6G, which may influence morphine disposition and analgesic potency. Recently, several MRP3 polymorphisms have been identified; however, these variant alleles had very low frequencies and displayed inter-ethnic variability. (106) As mentioned previously, MRP3 expression in human liver is low, but quite variable (up to 80-fold). (107) These differences in MRP3 expression and/or function may influence interindividual differences in morphine pharmacokinetics and pharmacodynamics.
4. Methods to Study the Function of Drug Efflux Transporters
As highlighted in the previous sections, efflux transporters play a major role in the hepatic physiology/pathophysiology as well as the pharmacological behavior of drugs. Hepatic efflux transporters can significantly influence the efficacy, side effects and toxicity of some drugs. However, we do not yet have a comprehensive understanding of the role of hepatic efflux transporters in clinical pharmacology, in part, because of current limitations in the model systems and techniques available to study hepatic efflux transporters in humans.
In vivo pharmacokinetic/pharmacodynamic studies in humans are the gold standard for identifying clinically significant contributions of transport proteins to drug disposition. Unfortunately, the complexity of the hepatobiliary system, with broad substrate overlap of transporters and limited access in vivo, makes it difficult to identify the functions of specific efflux proteins in the human liver. Studies in patients with polymorphisms in transporter genes might help to elucidate the contributions of single efflux proteins. However, pharmacokinetic analyses based on plasma drug concentrations in clinical studies provide information only on the overall systemic clearance; differentiation between altered hepatic uptake and canalicular efflux is not possible without additional data (e.g., hepatocyte concentrations and/or biliary excretion data). Whereas variations in uptake activity of transporters might have a profound influence on systemic concentrations, variations in canalicular efflux might significantly affect liver concentrations without having an effect on systemic exposure. (108) This is especially relevant for drugs for which the target sites for effect or toxicity are within the hepatocyte. Therefore, quantitative estimations of liver concentrations in vivo are necessary in order to investigate variations in efflux caused by drug-drug interactions or transporter polymorphisms. Imaging approaches such as positron emission tomography, magnetic-resonance imaging, and gamma scintigraphy have provided important insights into the function of hepatic efflux proteins. (109, 110) Furthermore, methods have been developed to quantify the biliary excretion of drugs in humans by sampling duodenal fluid using, for example, nasobiliary or oroenteric tubes. (111, 112) However, these sampling approaches are not used routinely because they are somewhat invasive and require specialized, trained personnel and equipment.
Historically, single-pass in situ or isolated perfused liver studies have been carried out to investigate the physiology and pathophysiology of the liver. In contrast to in vitro models such as isolated hepatocytes and liver slices, the isolated perfused liver preserves hepatic architecture, cell polarity, and bile flow. Furthermore, this model enables sampling of bile as well as inflow and outflow perfusate simultaneously; also liver tissue is available at the end of the study. This provides a rich data set amenable to pharmacokinetic modeling, making the isolated perfused liver system extremely useful for mechanistic studies of hepatobiliary transport. Unfortunately, this approach has limited applicability in humans. More recently, hepatocytes cultured between two layers of gelled collagen (“sandwich-configuration”) have been used to study hepatobiliary transport. Suspended hepatocytes, which are used primarily for initial uptake studies, are not a suitable model for studying the role of hepatic efflux proteins in drug/metabolite disposition because hepatocytes rapidly lose polarity after enzymatic/mechanical disruption (113). By contrast, sandwich-cultured hepatocytes regain mature hepatocyte morphology, reestablish cell polarity, form canalicular networks, and maintain transporter and metabolic enzyme expression, and they are a suitable in vitro model for studying hepatocyte efflux of drugs/metabolites. (114) Sandwich-cultured hepatocytes from preclinical species and humans have been used to assess biliary clearance as a measure to improve predictions of hepatic clearance. (114) However, species-related differences in transport proteins may confound the translation of in vivo data generated in preclinical species to humans.
Historically, vesicle transport assays using either canalicular or basolateral liver plasma membranes isolated from hepatic tissue obtained from preclinical species or humans were used to study carrier-mediated transport of endogenous compounds. This approach was used to identify and characterize bile acid and bilirubin glucuronide transport across the canalicular membrane, which led to the discovery of BSEP and MRP2. Naturally, these membranes contain multiple transport proteins, and ATP-dependent basolateral efflux data generated with basolateral liver plasma membranes may be confounded by uptake transporters. Therefore, these systems are not useful for identifying specific substrates of individual ATP-dependent binding cassette (ABC) efflux proteins. With the help of rapidly evolving molecular biology techniques and identification of individual transport proteins, this tissue-based vesicle assay system has been largely replaced by membrane vesicles generated from vector-transfected or virus-infected bacterial, insect, and more recently mammalian cells (e.g., HEK293, MDCKII, and LLC-PK1) expressing a single ABC-transporter. Given their high-throughput capability, these membrane vesicles have now become part of a standard approach to identify substrates and inhibitors of hepatic efflux transport proteins. As our understanding of the roles of uptake and efflux transporters in facilitating the vectorial transport of xenobiotics across the hepatocyte has evolved, the use of polarized mammalian cells (e.g., MDCKII and LLC-PK1) has become more widely used to identify substrates and inhibitors of hepatic transport proteins. The combined expression of uptake and efflux carriers enabled the analysis of vectorial transport, which is a key step in hepatobiliary elimination. (115, 116) Double-transfected polarized cell lines were developed in the early 2000s and are now valuable tools in studying transcellular transport. Triple- and quadruple transfected cell lines OATP1B1/MRP2/MRP3 or MRP4, as well as OATP1B1/OATP1B3/OATP2B1/MRP2 have been developed to predict hepatobiliary processes more accurately. (117, 118) Recently, a triple-transfected cell line expressing uptake and efflux transporters and a drug-metabolizing enzyme has been described to study transporter-metabolism interplay. (119) To date, the level of recombinant expression cannot be controlled, and this may limit the use of these cell systems to quantitatively predict hepatobiliary transport. Furthermore, the expression of endogenous transporters in these cells might mask the activity of the recombinant transporters and confound data interpretation. (120) The ability of in vitro systems to accurately predict which transport proteins are responsible for hepatic efflux in vivo depends not only on the substrate and its relative affinity for individual transport proteins but also on the relative expression levels of the proteins as compared with the in vivo situation. Quantitative absolute proteomics of drug transport proteins will help to overcome this problem in the future and might enable the establishment of scaling factors for in vitro-in vivo correlation. For OATP transporters, Shitara et al. established a method to estimate the transporter contribution to overall substrate uptake by comparing the ratio of uptake clearances of the test and the reference compounds in overexpressing cell systems and in hepatocytes, and calculating a proportionality (relative activity, R-) factor. (121) The results are based on the assumption that the reference compound is specific for the respective transport protein; however, given to the overlapping substrate spectrum of transport proteins, is hardly ever the case. (122) Given the fact that uptake and metabolism are key steps in hepatobiliary elimination, it is debatable whether a similar approach could be established for efflux transporters.
In addition to in vivo and in vitro systems, computational modeling and simulation has become increasingly important in drug development. Quantitative Structure-Activity Relationship (QSAR) studies have been used to identify substrate and inhibitor characteristics for certain drug transporters. These studies can be used to screen compound databases and predict whether new molecular entities are substrates or inhibitors of certain transporters. In addition, physiologically-based pharmacokinetic modeling approaches facilitate the translation of data generated from in vitro systems to the in vivo situation, and enable predictions of the impact of altered clearance pathways on systemic drug disposition and the pharmacokinetics of drugs/metabolites in the target tissue. (123)
5. Conclusions, Future Directions and Challenges
As highlighted in the previous sections, hepatic efflux transport proteins may markedly affect the pharmacokinetics and/or pharmacodynamics of some drugs and hence their therapeutic efficacy and/or toxicity. Our understanding of the role of hepatic efflux transport proteins in human disease and pharmacotherapy has been evolving for more than half a century. Numerous systems, including in vitro, in vivo, and in silico models, have been used successfully to study the role of hepatic efflux transporters (Figure 6); the majority of the in vitro systems were developed in the past decade. Each system has strengths and limitations; however, to date, no single system can perfectly predict the contribution of hepatic efflux transporters to overall drug disposition in humans in vivo. In the following section, some of the current challenges and future directions in studying hepatobiliary efflux transport are examined.
Figure 6.

Timeline of important discoveries and model systems in the field of drug efflux transport proteins.
Cellular models
Currently, the availability of high quality human hepatocytes is a major limitation in studying hepatic transport processes. Although techniques for cryopreservation have improved substantially over the past decade, there are considerable qualitative differences between batches. Improved hepatocyte-derived cells might hold great promise for overcoming these problems. For example, the human hepatoma HepaRG cell line closely resembles human hepatocytes with respect to morphology as well as enzyme and transporter expression. (124) If they are not terminally differentiated, HepaRG cells can be propagated indefinitely, thereby providing a consistent supply of cells. Some challenges remain regarding the use of HepaRG cells to evaluate the hepatic efflux of drugs and generated metabolites because this model represents a mixed population (hepatocytes and biliary epithelial cells). Inducible pluripotent stem cells (iPS) and human embryonic stem cells are promising sources for large quantities of high quality, high purity hepatocytes. Current strategies to differentiate iPS cells into hepatocytes using growth factors achieve differentiation rates of up to 60–80%. (125) The use of stem cell-derived hepatocytes as a substitute for primary human hepatocytes for studying and predicting hepatobiliary efflux of drugs/metabolites will depend on their comparability to mature hepatocytes with respect to morphology as well as transporter and enzyme expression. Further, improvements in differentiation strategies are necessary to achieve 100% differentiation into hepatocytes and to avoid costly isolation and purification processes.
Specific probes and inhibitors
Although in vitro models such as membrane vesicles and transfected cells facilitated the identification of key transport proteins involved in hepatobiliary drug efflux, predicting the contribution of any single efflux transporter to overall disposition remains a challenge. Moreover, the broad substrate overlap between hepatic efflux transporters complicates efforts to determine the role of any single transport protein. The search continues for specific metabolically stable probes and/or inhibitors that do not affect metabolism; such compounds would be useful tools, both in vivo and in vitro, to study the involvement of single proteins. However, to date such probes have remained elusive.
In vitro – in vivo correlations
Despite the outstanding advances made in recent years in the field of drug transport and disposition, there is a disproportionate amount of in vitro data relative to in vivo data, and preclinical findings relative to clinical findings. Further research is needed to characterize, validate, and refine existing model systems and techniques, and to develop new approaches to assess hepatic efflux transport in humans. In order to accurately predict the pharmacokinetics and pharmacodynamics of therapeutic agents in patients, a more holistic understanding of the interplay among hepatic uptake, metabolism, intracellular disposition, efflux, and the functional redundancy between transport proteins is needed. Also, further information is required about the effects of disease states, genetic polymorphisms, and environmental factors (e.g., alcohol consumption, diet, and smoking) on hepatic efflux transport proteins. Modeling and simulation will be critical in obtaining improved in vitro-in vivo correlations.
In vivo-imaging techniques
Noninvasive imaging techniques such as multiphoton microscopy and radionuclide/fluorescent imaging may become powerful tools in elucidating drug disposition and predicting drug-drug interactions in vivo. Positron emission tomography and magnetic-resonance imaging (MRI) have been employed to determine drug disposition and transport in vivo in animals as well as in humans. However, some limitations of these techniques exist including low resolution and slow image acquisition (PET), the fact that there are only a limited number of suitable contrast agents (MRI), and the need for specialized facilities/equipment. At present, imaging agents must be metabolically stable because it is not possible to differentiate between parent compound and generated metabolites. Recently, multiphoton imaging has enabled minimally invasive deep-tissue in vivo imaging. (126) This technique has been used in mice to study the hepatic disposition of 6-carboxyfluorescein diacetate using an intravital hepatic imaging chamber. (127) Given that the tissue penetration depth is currently only about 500 μm, the applicability of intravital imaging in humans is limited to specific tissues such as skin. The application of novel technologies to quantify drugs and metabolites in vivo a non-invasive manner could lead to major breakthroughs in our ability to investigate the role of efflux proteins in the hepatobiliary disposition of drugs in patients.
Mass spectrometry imaging
Recently, mass spectrometry has emerged as a tool for the direct analysis of small molecule pharmaceuticals and endogenous compounds in biological tissue. These techniques allow for comprehensive assessment of the spatial distribution of parent drug and metabolite(s) in tissues without the need to label compounds (autoradiography) or homogenize tissue, which would result in a loss of information about distribution of the drug in tissue. A recent review by Castellino et al. discusses these techniques in depth. (128)
Conclusion
Over the past decade, rapid technological advances have accelerated the rate of knowledge acquisition about drug transport proteins. Discussions led by the International Transporter Consortium have helped to focus research efforts on important questions in this field, particularly those relating to drug development issues. (129) Currently, we have only a limited ability to make accurate predictions of the influence of hepatic efflux transporters on the pharmacokinetics/pharmacodynamics of drugs, and only a rudimentary knowledge regarding the possible effects of changes in hepatic drug efflux (due to a disease or drug interactions) on pharmacotherapy. However, great advances in this field are on the horizon, as technical innovations in cell biology, engineering, chemistry, and mathematical modeling provide more sophisticated and robust approaches addressing these important questions. Such information is prerequisite to the ultimate goal of delivering personalized medicine.
Acknowledgments
This work was supported, in part, by a grant from the National Institutes of Health, National Institute of General Medical Sciences [R01 GM41935 to KLRB] and by Deutsche Forschungsgemeinschaft Grant Ko4185/1-1 [KK]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- BRIC
benign recurrent intrahepatic cholestasis
- BSEP
bile salt export pump
- DJS
Dubin-Johnson-Syndrome
- EHBR
Esai hyperbilirubinemic rat
- MRI
magnetic-resonance imaging
- OATP
organic anion transporting polypeptide
- OAT
organic anion transporter
- OCT
organic cation transporter
- MRP
multidrug-resistance associated protein
- PET
positron emission tomography
- PFIC
progressive familial intrahepatic cholestasis
- P-gp
P-glycoprotein
- Sf9
Spodoptera frugiperda
References
- 1.Hagenbuch B. Drug uptake systems in liver and kidney: a historic perspective. Clin Pharmacol Ther. 2010;87:39–47. doi: 10.1038/clpt.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stieger B. The role of the sodium-taurocholate cotransporting polypeptide (NTCP) and of the bile salt export pump (BSEP) in physiology and pathophysiology of bile formation. Handb Exp Pharmacol. 2011:205–259. doi: 10.1007/978-3-642-14541-4_5. [DOI] [PubMed] [Google Scholar]
- 3.Nicolaou M, Andress EJ, Zolnerciks JK, Dixon PH, Williamson C, Linton KJ. Canalicular ABC transporters and liver disease. J Pathol. 2012;226:300–315. doi: 10.1002/path.3019. [DOI] [PubMed] [Google Scholar]
- 4.Cascorbi I. P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol. 2011:261–283. doi: 10.1007/978-3-642-14541-4_6. [DOI] [PubMed] [Google Scholar]
- 5.von Richter O, Burk O, Fromm MF, Thon KP, Eichelbaum M, Kivisto KT. Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther. 2004;75:172–183. doi: 10.1016/j.clpt.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Schuetz EG, Furuya KN, Schuetz JD. Interindividual variation in expression of P-glycoprotein in normal human liver and secondary hepatic neoplasms. J Pharmacol Exp Ther. 1995;275:1011–1018. [PubMed] [Google Scholar]
- 7.Jemnitz K, Heredi-Szabo K, Janossy J, Ioja E, Vereczkey L, Krajcsi P. ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab Rev. 2010;42:402–436. doi: 10.3109/03602530903491741. [DOI] [PubMed] [Google Scholar]
- 8.Paulusma CC, van Geer MA, Evers R, Heijn M, Ottenhoff R, Borst P, Oude Elferink RP. Canalicular multispecific organic anion transporter/multidrug resistance protein 2 mediates low-affinity transport of reduced glutathione. Biochem J. 1999;338 ( Pt 2):393–401. [PMC free article] [PubMed] [Google Scholar]
- 9.Jansen PL, Peters WH, Lamers WH. Hereditary chronic conjugated hyperbilirubinemia in mutant rats caused by defective hepatic anion transport. Hepatology. 1985;5:573–579. doi: 10.1002/hep.1840050408. [DOI] [PubMed] [Google Scholar]
- 10.Kamisako T, Kobayashi Y, Takeuchi K, Ishihara T, Higuchi K, Tanaka Y, Gabazza EC, et al. Recent advances in bilirubin metabolism research: the molecular mechanism of hepatocyte bilirubin transport and its clinical relevance. J Gastroenterol. 2000;35:659–664. doi: 10.1007/s005350070044. [DOI] [PubMed] [Google Scholar]
- 11.Meyer zu Schwabedissen HE, Kroemer HK. In vitro and in vivo evidence for the importance of breast cancer resistance protein transporters (BCRP/MXR/ABCP/ABCG2) Handb Exp Pharmacol. 2011:325–371. doi: 10.1007/978-3-642-14541-4_9. [DOI] [PubMed] [Google Scholar]
- 12.Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A. 2005;102:17923–17928. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011:105–167. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 14.Scheffer GL, Kool M, de Haas M, de Vree JM, Pijnenborg AC, Bosman DK, Elferink RP, et al. Tissue distribution and induction of human multidrug resistant protein 3. Lab Invest. 2002;82:193–201. doi: 10.1038/labinvest.3780411. [DOI] [PubMed] [Google Scholar]
- 15.Lang T, Hitzl M, Burk O, Mornhinweg E, Keil A, Kerb R, Klein K, et al. Genetic polymorphisms in the multidrug resistance-associated protein 3 (ABCC3, MRP3) gene and relationship to its mRNA and protein expression in human liver. Pharmacogenetics. 2004;14:155–164. doi: 10.1097/00008571-200403000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Konig J, Rost D, Cui Y, Keppler D. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology. 1999;29:1156–1163. doi: 10.1002/hep.510290404. [DOI] [PubMed] [Google Scholar]
- 17.Chai J, He Y, Cai SY, Jiang Z, Wang H, Li Q, Chen L, et al. Elevated hepatic multidrug resistance-associated protein 3/ATP-binding cassette subfamily C 3 expression in human obstructive cholestasis is mediated through tumor necrosis factor alpha and c-Jun NH2-terminal kinase/stress-activated protein kinase-signaling pathway. Hepatology. 2012;55:1485–1494. doi: 10.1002/hep.24801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirohashi T, Suzuki H, Takikawa H, Sugiyama Y. ATP-dependent transport of bile salts by rat multidrug resistance-associated protein 3 (Mrp3) J Biol Chem. 2000;275:2905–2910. doi: 10.1074/jbc.275.4.2905. [DOI] [PubMed] [Google Scholar]
- 19.Akita H, Suzuki H, Hirohashi T, Takikawa H, Sugiyama Y. Transport activity of human MRP3 expressed in Sf9 cells: comparative studies with rat MRP3. Pharm Res. 2002;19:34–41. doi: 10.1023/a:1013699130991. [DOI] [PubMed] [Google Scholar]
- 20.Zelcer N, van de Wetering K, de Waart R, Scheffer GL, Marschall HU, Wielinga PR, Kuil A, et al. Mice lacking Mrp3 (Abcc3) have normal bile salt transport, but altered hepatic transport of endogenous glucuronides. J Hepatol. 2006;44:768–775. doi: 10.1016/j.jhep.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 21.Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011:299–323. doi: 10.1007/978-3-642-14541-4_8. [DOI] [PubMed] [Google Scholar]
- 22.Denk GU, Soroka CJ, Takeyama Y, Chen WS, Schuetz JD, Boyer JL. Multidrug resistance-associated protein 4 is up-regulated in liver but down-regulated in kidney in obstructive cholestasis in the rat. J Hepatol. 2004;40:585–591. doi: 10.1016/j.jhep.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Gradhand U, Lang T, Schaeffeler E, Glaeser H, Tegude H, Klein K, Fritz P, et al. Variability in human hepatic MRP4 expression: influence of cholestasis and genotype. Pharmacogenomics J. 2008;8:42–52. doi: 10.1038/sj.tpj.6500451. [DOI] [PubMed] [Google Scholar]
- 24.Mennone A, Soroka CJ, Cai SY, Harry K, Adachi M, Hagey L, Schuetz JD, et al. Mrp4−/− mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43:1013–1021. doi: 10.1002/hep.21158. [DOI] [PubMed] [Google Scholar]
- 25.Barnes SN, Aleksunes LM, Augustine L, Scheffer GL, Goedken MJ, Jakowski AB, Pruimboom-Brees IM, et al. Induction of hepatobiliary efflux transporters in acetaminophen-induced acute liver failure cases. Drug Metab Dispos. 2007;35:1963–1969. doi: 10.1124/dmd.107.016170. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Seward DJ, Li L, Boyer JL, Ballatori N. Expression cloning of two genes that together mediate organic solute and steroid transport in the liver of a marine vertebrate. Proc Natl Acad Sci U S A. 2001;98:9431–9436. doi: 10.1073/pnas.161099898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seward DJ, Koh AS, Boyer JL, Ballatori N. Functional complementation between a novel mammalian polygenic transport complex and an evolutionarily ancient organic solute transporter, OSTalpha-OSTbeta. J Biol Chem. 2003;278:27473–27482. doi: 10.1074/jbc.M301106200. [DOI] [PubMed] [Google Scholar]
- 28.Ballatori N, Li N, Fang F, Boyer JL, Christian WV, Hammond CL. OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front Biosci. 2009;14:2829–2844. doi: 10.2741/3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, Madejczyk MS, et al. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 30.Stieger B, O’Neill B, Meier PJ. ATP-dependent bile-salt transport in canalicular rat liver plasma-membrane vesicles. Biochem J. 1992;284 ( Pt 1):67–74. doi: 10.1042/bj2840067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishida T, Gatmaitan Z, Che M, Arias IM. Rat liver canalicular membrane vesicles contain an ATP-dependent bile acid transport system. Proc Natl Acad Sci U S A. 1991;88:6590–6594. doi: 10.1073/pnas.88.15.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Childs S, Yeh RL, Georges E, Ling V. Identification of a sister gene to P-glycoprotein. Cancer Res. 1995;55:2029–2034. [PubMed] [Google Scholar]
- 33.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046–10050. doi: 10.1074/jbc.273.16.10046. [DOI] [PubMed] [Google Scholar]
- 34.Strautnieks SS, Kagalwalla AF, Tanner MS, Knisely AS, Bull L, Freimer N, Kocoshis SA, et al. Identification of a locus for progressive familial intrahepatic cholestasis PFIC2 on chromosome 2q24. Am J Hum Genet. 1997;61:630–633. doi: 10.1086/515501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Woerd WL, van Mil SW, Stapelbroek JM, Klomp LW, van de Graaf SF, Houwen RH. Familial cholestasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy. Best Pract Res Clin Gastroenterol. 2010;24:541–553. doi: 10.1016/j.bpg.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 36.Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ, Koning JH, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 37.Jansen PL, Muller M. Genetic cholestasis: lessons from the molecular physiology of bile formation. Can J Gastroenterol. 2000;14:233–238. doi: 10.1155/2000/514172. [DOI] [PubMed] [Google Scholar]
- 38.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 39.Plass JR, Mol O, Heegsma J, Geuken M, de Bruin J, Elling G, Muller M, et al. A progressive familial intrahepatic cholestasis type 2 mutation causes an unstable, temperature-sensitive bile salt export pump. J Hepatol. 2004;40:24–30. doi: 10.1016/s0168-8278(03)00483-5. [DOI] [PubMed] [Google Scholar]
- 40.Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, Wanty C, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol. 2010;53:170–178. doi: 10.1016/j.jhep.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubin IN, Johnson FB. Chronic idiopathic jaundice with unidentified pigment in liver cells; a new clinicopathologic entity with a report of 12 cases. Medicine (Baltimore) 1954;33:155–197. doi: 10.1097/00005792-195409000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Paulusma CC, Kool M, Bosma PJ, Scheffer GL, ter Borg F, Scheper RJ, Tytgat GN, et al. A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology. 1997;25:1539–1542. doi: 10.1002/hep.510250635. [DOI] [PubMed] [Google Scholar]
- 43.Kartenbeck J, Leuschner U, Mayer R, Keppler D. Absence of the canalicular isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. Hepatology. 1996;23:1061–1066. doi: 10.1053/jhep.1996.v23.pm0008621134. [DOI] [PubMed] [Google Scholar]
- 44.Mikami T, Nozaki T, Tagaya O, Hosokawa S, Nakura T, Mori H, Kondou S. The characters of a new mutant in rats with hyperbilirubinuria syndrome. Congenital Anom. 1986;26:250–251. [Google Scholar]
- 45.Yue W, Lee JK, Abe K, Sugiyama Y, Brouwer KL. Decreased hepatic breast cancer resistance protein expression and function in multidrug resistance-associated protein 2-deficient (TR) rats. Drug Metab Dispos. 2011;39:441–447. doi: 10.1124/dmd.110.035188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huber M, Guhlmann A, Jansen PL, Keppler D. Hereditary defect of hepatobiliary cysteinyl leukotriene elimination in mutant rats with defective hepatic anion excretion. Hepatology. 1987;7:224–228. doi: 10.1002/hep.1840070204. [DOI] [PubMed] [Google Scholar]
- 47.Ishikawa T, Muller M, Klunemann C, Schaub T, Keppler D. ATP-dependent primary active transport of cysteinyl leukotrienes across liver canalicular membrane. Role of the ATP-dependent transport system for glutathione S-conjugates. J Biol Chem. 1990;265:19279–19286. [PubMed] [Google Scholar]
- 48.Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 49.Mayer R, Kartenbeck J, Buchler M, Jedlitschky G, Leier I, Keppler D. Expression of the MRP gene-encoded conjugate export pump in liver and its selective absence from the canalicular membrane in transport-deficient mutant hepatocytes. J Cell Biol. 1995;131:137–150. doi: 10.1083/jcb.131.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buchler M, Konig J, Brom M, Kartenbeck J, Spring H, Horie T, Keppler D. cDNA cloning of the hepatocyte canalicular isoform of the multidrug resistance protein, cMrp, reveals a novel conjugate export pump deficient in hyperbilirubinemic mutant rats. J Biol Chem. 1996;271:15091–15098. doi: 10.1074/jbc.271.25.15091. [DOI] [PubMed] [Google Scholar]
- 51.Taniguchi K, Wada M, Kohno K, Nakamura T, Kawabe T, Kawakami M, Kagotani K, et al. A human canalicular multispecific organic anion transporter (cMOAT) gene is overexpressed in cisplatin-resistant human cancer cell lines with decreased drug accumulation. Cancer Res. 1996;56:4124–4129. [PubMed] [Google Scholar]
- 52.Hirouchi M, Suzuki H, Sugiyama Y. Treatment of hyperbilirubinemia in Eisai hyperbilirubinemic rat by transfecting human MRP2/ABCC2 gene. Pharm Res. 2005;22:661–666. doi: 10.1007/s11095-005-2502-1. [DOI] [PubMed] [Google Scholar]
- 53.Fujimori S, Gudis K, Takahashi Y, Seo T, Yamada Y, Ehara A, Kobayashi T, et al. Distribution of small intestinal mucosal injuries as a result of NSAID administration. Eur J Clin Invest. 2010;40:504–510. doi: 10.1111/j.1365-2362.2010.02290.x. [DOI] [PubMed] [Google Scholar]
- 54.Endo H, Hosono K, Inamori M, Kato S, Nozaki Y, Yoneda K, Akiyama T, et al. Incidence of small bowel injury induced by low-dose aspirin: a crossover study using capsule endoscopy in healthy volunteers. Digestion. 2009;79:44–51. doi: 10.1159/000204465. [DOI] [PubMed] [Google Scholar]
- 55.Boelsterli UA, Ramirez-Alcantara V. NSAID acyl glucuronides and enteropathy. Curr Drug Metab. 2011;12:245–252. doi: 10.2174/138920011795101877. [DOI] [PubMed] [Google Scholar]
- 56.Lanas A, Scarpignato C. Microbial flora in NSAID-induced intestinal damage: a role for antibiotics? Digestion. 2006;73 (Suppl 1):136–150. doi: 10.1159/000089789. [DOI] [PubMed] [Google Scholar]
- 57.Wallace JL, Syer S, Denou E, De Palma G, Vong L, McKnight W, Jury J, et al. Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.06.075. [DOI] [PubMed] [Google Scholar]
- 58.Wax J, Clinger WA, Varner P, Bass P, Winder CV. Relationship of the enterohepatic cycle to ulcerogenesis in the rat small bowel with flufenamic acid. Gastroenterology. 1970;58:772–780. [PubMed] [Google Scholar]
- 59.Duggan DE, Hooke KF, Noll RM, Kwan KC. Enterohepatic circulation of indomethacin and its role in intestinal irritation. Biochem Pharmacol. 1975;24:1749–1754. doi: 10.1016/0006-2952(75)90450-5. [DOI] [PubMed] [Google Scholar]
- 60.Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug enteropathy in rats: role of permeability, bacteria, and enterohepatic circulation. Gastroenterology. 1997;112:109–117. doi: 10.1016/s0016-5085(97)70225-7. [DOI] [PubMed] [Google Scholar]
- 61.Seitz S, Kretz-Rommel A, Oude Elferink RP, Boelsterli UA. Selective protein adduct formation of diclofenac glucuronide is critically dependent on the rat canalicular conjugate export pump (Mrp2) Chem Res Toxicol. 1998;11:513–519. doi: 10.1021/tx970203+. [DOI] [PubMed] [Google Scholar]
- 62.Hodin RA, Shei A, Meng S. Transcriptional activation of the human villin gene during enterocyte differentiation. J Gastrointest Surg. 1997;1:433–438. doi: 10.1016/s1091-255x(97)80130-8. discussion 438. [DOI] [PubMed] [Google Scholar]
- 63.Oostendorp LJ, Stalmeier PF, Pasker-de Jong PC, Van der Graaf WT, Ottevanger PB. Systematic review of benefits and risks of second-line irinotecan monotherapy for advanced colorectal cancer. Anticancer Drugs. 2010;21:749–758. doi: 10.1097/CAD.0b013e32833c57cf. [DOI] [PubMed] [Google Scholar]
- 64.Sugiyama Y, Kato Y, Chu X. Multiplicity of biliary excretion mechanisms for the camptothecin derivative irinotecan (CPT-11), its metabolite SN-38, and its glucuronide: role of canalicular multispecific organic anion transporter and P-glycoprotein. Cancer Chemother Pharmacol. 1998;42 (Suppl):S44–49. doi: 10.1007/s002800051078. [DOI] [PubMed] [Google Scholar]
- 65.Chu XY, Kato Y, Ueda K, Suzuki H, Niinuma K, Tyson CA, Weizer V, et al. Biliary excretion mechanism of CPT-11 and its metabolites in humans: involvement of primary active transporters. Cancer Res. 1998;58:5137–5143. [PubMed] [Google Scholar]
- 66.Chabot GG. Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet. 1997;33:245–259. doi: 10.2165/00003088-199733040-00001. [DOI] [PubMed] [Google Scholar]
- 67.Schulz C, Boeck S, Heinemann V, Stemmler HJ. UGT1A1 genotyping: a predictor of irinotecan-associated side effects and drug efficacy? Anticancer Drugs. 2009;20:867–879. doi: 10.1097/CAD.0b013e328330c7d2. [DOI] [PubMed] [Google Scholar]
- 68.de Jong FA, Kehrer DF, Mathijssen RH, Creemers GJ, de Bruijn P, van Schaik RH, Planting AS, et al. Prophylaxis of irinotecan-induced diarrhea with neomycin and potential role for UGT1A1*28 genotype screening: a double-blind, randomized, placebo-controlled study. Oncologist. 2006;11:944–954. doi: 10.1634/theoncologist.11-8-944. [DOI] [PubMed] [Google Scholar]
- 69.Nozawa T, Minami H, Sugiura S, Tsuji A, Tamai I. Role of organic anion transporter OATP1B1 (OATP-C) in hepatic uptake of irinotecan and its active metabolite, 7-ethyl-10-hydroxycamptothecin: in vitro evidence and effect of single nucleotide polymorphisms. Drug Metab Dispos. 2005;33:434–439. doi: 10.1124/dmd.104.001909. [DOI] [PubMed] [Google Scholar]
- 70.Di Paolo A, Bocci G, Polillo M, Del Re M, Di Desidero T, Lastella M, Danesi R. Pharmacokinetic and pharmacogenetic predictive markers of irinotecan activity and toxicity. Curr Drug Metab. 2011;12:932–943. doi: 10.2174/138920011798062283. [DOI] [PubMed] [Google Scholar]
- 71.Lara PN, Jr, Natale R, Crowley J, Lenz HJ, Redman MW, Carleton JE, Jett J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–2535. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY, Lee JE, et al. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer. 2007;110:138–147. doi: 10.1002/cncr.22760. [DOI] [PubMed] [Google Scholar]
- 73.de Jong FA, Scott-Horton TJ, Kroetz DL, McLeod HL, Friberg LE, Mathijssen RH, Verweij J, et al. Irinotecan-induced diarrhea: functional significance of the polymorphic ABCC2 transporter protein. Clin Pharmacol Ther. 2007;81:42–49. doi: 10.1038/sj.clpt.6100019. [DOI] [PubMed] [Google Scholar]
- 74.Bowalgaha K, Miners JO. The glucuronidation of mycophenolic acid by human liver, kidney and jejunum microsomes. Br J Clin Pharmacol. 2001;52:605–609. doi: 10.1046/j.0306-5251.2001.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shipkova M, Armstrong VW, Wieland E, Niedmann PD, Schutz E, Brenner-Weiss G, Voihsel M, et al. Identification of glucoside and carboxyl-linked glucuronide conjugates of mycophenolic acid in plasma of transplant recipients treated with mycophenolate mofetil. Br J Pharmacol. 1999;126:1075–1082. doi: 10.1038/sj.bjp.0702399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shaw LM, Korecka M, Venkataramanan R, Goldberg L, Bloom R, Brayman KL. Mycophenolic acid pharmacodynamics and pharmacokinetics provide a basis for rational monitoring strategies. Am J Transplant. 2003;3:534–542. doi: 10.1034/j.1600-6143.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 77.Bullingham R, Monroe S, Nicholls A, Hale M. Pharmacokinetics and bioavailability of mycophenolate mofetil in healthy subjects after single-dose oral and intravenous administration. J Clin Pharmacol. 1996;36:315–324. doi: 10.1002/j.1552-4604.1996.tb04207.x. [DOI] [PubMed] [Google Scholar]
- 78.Xia ZW, Jun CY, Hao C, Bing C, Min SM, Jie XJ. The occurrence of diarrhea not related to the pharmacokinetics of MPA and its metabolites in liver transplant patients. Eur J Clin Pharmacol. 2010;66:671–679. doi: 10.1007/s00228-010-0833-2. [DOI] [PubMed] [Google Scholar]
- 79.Kobayashi M, Saitoh H, Tadano K, Takahashi Y, Hirano T. Cyclosporin A, but not tacrolimus, inhibits the biliary excretion of mycophenolic acid glucuronide possibly mediated by multidrug resistance-associated protein 2 in rats. J Pharmacol Exp Ther. 2004;309:1029–1035. doi: 10.1124/jpet.103.063073. [DOI] [PubMed] [Google Scholar]
- 80.Hesselink DA, van Hest RM, Mathot RA, Bonthuis F, Weimar W, de Bruin RW, van Gelder T. Cyclosporine interacts with mycophenolic acid by inhibiting the multidrug resistance-associated protein 2. Am J Transplant. 2005;5:987–994. doi: 10.1046/j.1600-6143.2005.00779.x. [DOI] [PubMed] [Google Scholar]
- 81.Haenisch S, Zimmermann U, Dazert E, Wruck CJ, Dazert P, Siegmund W, Kroemer HK, et al. Influence of polymorphisms of ABCB1 and ABCC2 on mRNA and protein expression in normal and cancerous kidney cortex. Pharmacogenomics J. 2007;7:56–65. doi: 10.1038/sj.tpj.6500403. [DOI] [PubMed] [Google Scholar]
- 82.Yang JW, Lee PH, Hutchinson IV, Pravica V, Shah T, Min DI. Genetic polymorphisms of MRP2 and UGT2B7 and gastrointestinal symptoms in renal transplant recipients taking mycophenolic acid. Ther Drug Monit. 2009;31:542–548. doi: 10.1097/FTD.0b013e3181b1dd5e. [DOI] [PubMed] [Google Scholar]
- 83.Ohmann EL, Burckart GJ, Brooks MM, Chen Y, Pravica V, Girnita DM, Zeevi A, et al. Genetic polymorphisms influence mycophenolate mofetil-related adverse events in pediatric heart transplant patients. J Heart Lung Transplant. 2010;29:509–516. doi: 10.1016/j.healun.2009.11.602. [DOI] [PubMed] [Google Scholar]
- 84.Naesens M, Kuypers DR, Verbeke K, Vanrenterghem Y. Multidrug resistance protein 2 genetic polymorphisms influence mycophenolic acid exposure in renal allograft recipients. Transplantation. 2006;82:1074–1084. doi: 10.1097/01.tp.0000235533.29300.e7. [DOI] [PubMed] [Google Scholar]
- 85.Miura M, Satoh S, Inoue K, Kagaya H, Saito M, Inoue T, Suzuki T, et al. Influence of SLCO1B1, 1B3, 2B1 and ABCC2 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Eur J Clin Pharmacol. 2007;63:1161–1169. doi: 10.1007/s00228-007-0380-7. [DOI] [PubMed] [Google Scholar]
- 86.Woillard JB, Rerolle JP, Picard N, Rousseau A, Drouet M, Munteanu E, Essig M, et al. Risk of diarrhoea in a long-term cohort of renal transplant patients given mycophenolate mofetil: the significant role of the UGT1A8 2 variant allele. Br J Clin Pharmacol. 2010;69:675–683. doi: 10.1111/j.1365-2125.2010.03625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Stieger B, Meier PJ. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–231. doi: 10.1067/mcp.2001.114667. [DOI] [PubMed] [Google Scholar]
- 88.Funk C, Pantze M, Jehle L, Ponelle C, Scheuermann G, Lazendic M, Gasser R. Troglitazone-induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male and female rats. Correlation with the gender difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology. 2001;167:83–98. doi: 10.1016/s0300-483x(01)00460-7. [DOI] [PubMed] [Google Scholar]
- 89.Lee JK, Marion TL, Abe K, Lim C, Pollack GM, Brouwer KL. Hepatobiliary disposition of troglitazone and metabolites in rat and human sandwich-cultured hepatocytes: use of Monte Carlo simulations to assess the impact of changes in biliary excretion on troglitazone sulfate accumulation. J Pharmacol Exp Ther. 332:26–34. doi: 10.1124/jpet.109.156653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, Afshari CA, et al. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci. 2010;118:485–500. doi: 10.1093/toxsci/kfq269. [DOI] [PubMed] [Google Scholar]
- 91.Daly AK, Aithal GP, Leathart JB, Swainsbury RA, Dang TS, Day CP. Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology. 2007;132:272–281. doi: 10.1053/j.gastro.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 92.Lee JK, Leslie EM, Zamek-Gliszczynski MJ, Brouwer KL. Modulation of trabectedin (ET-743) hepatobiliary disposition by multidrug resistance-associated proteins (Mrps) may prevent hepatotoxicity. Toxicol Appl Pharmacol. 2008;228:17–23. doi: 10.1016/j.taap.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Waterschoot RA, Eman RM, Wagenaar E, van der Kruijssen CM, Rosing H, Beijnen JH, Schinkel AH. ABCC2, ABCC3, and ABCB1, but not CYP3A, protect against trabectedin-mediated hepatotoxicity. Clin Cancer Res. 2009;15:7616–7623. doi: 10.1158/1078-0432.CCR-09-2127. [DOI] [PubMed] [Google Scholar]
- 94.Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- 95.Kondo C, Suzuki H, Itoda M, Ozawa S, Sawada J, Kobayashi D, Ieiri I, et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res. 2004;21:1895–1903. doi: 10.1023/b:pham.0000045245.21637.d4. [DOI] [PubMed] [Google Scholar]
- 96.Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, Miki Y, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–616. [PubMed] [Google Scholar]
- 97.Bailey KM, Romaine SP, Jackson BM, Farrin AJ, Efthymiou M, Barth JH, Copeland J, et al. Hepatic metabolism and transporter gene variants enhance response to rosuvastatin in patients with acute myocardial infarction: the GEOSTAT-1 Study. Circ Cardiovasc Genet. 2010;3:276–285. doi: 10.1161/CIRCGENETICS.109.898502. [DOI] [PubMed] [Google Scholar]
- 98.van Herwaarden AE, Schinkel AH. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol Sci. 2006;27:10–16. doi: 10.1016/j.tips.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 99.Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ, Wang A, et al. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006;373:99–103. doi: 10.1016/j.cca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 100.Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86:197–203. doi: 10.1038/clpt.2009.79. [DOI] [PubMed] [Google Scholar]
- 101.Coffman BL, Rios GR, King CD, Tephly TR. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos. 1997;25:1–4. [PubMed] [Google Scholar]
- 102.Paerregaard P. Excretion of morphine in the urine of non-addicts. Acta Pharmacol Toxicol (Copenh) 1957;14:53–57. doi: 10.1111/j.1600-0773.1957.tb01141.x. [DOI] [PubMed] [Google Scholar]
- 103.Moran TD, Smith PA. Morphine-3beta-D-glucuronide suppresses inhibitory synaptic transmission in rat substantia gelatinosa. J Pharmacol Exp Ther. 2002;302:568–576. doi: 10.1124/jpet.102.035626. [DOI] [PubMed] [Google Scholar]
- 104.Zelcer N, Saeki T, Reid G, Beijnen JH, Borst P. Characterization of drug transport by the human multidrug resistance protein 3 (ABCC3) J Biol Chem. 2001;276:46400–46407. doi: 10.1074/jbc.M107041200. [DOI] [PubMed] [Google Scholar]
- 105.Zelcer N, van de Wetering K, Hillebrand M, Sarton E, Kuil A, Wielinga PR, Tephly T, et al. Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception. Proc Natl Acad Sci U S A. 2005;102:7274–7279. doi: 10.1073/pnas.0502530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sasaki T, Hirota T, Ryokai Y, Kobayashi D, Kimura M, Irie S, Higuchi S, et al. Systematic screening of human ABCC3 polymorphisms and their effects on MRP3 expression and function. Drug Metab Pharmacokinet. 2011;26:374–386. doi: 10.2133/dmpk.dmpk-10-rg-103. [DOI] [PubMed] [Google Scholar]
- 107.Hitzl M, Klein K, Zanger UM, Fritz P, Nussler AK, Neuhaus P, Fromm MF. Influence of omeprazole on multidrug resistance protein 3 expression in human liver. J Pharmacol Exp Ther. 2003;304:524–530. doi: 10.1124/jpet.102.043547. [DOI] [PubMed] [Google Scholar]
- 108.Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther. 2009;328:652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 109.Ghibellini G, Leslie EM, Pollack GM, Brouwer KL. Use of tc-99m mebrofenin as a clinical probe to assess altered hepatobiliary transport: integration of in vitro, pharmacokinetic modeling, and simulation studies. Pharm Res. 2008;25:1851–1860. doi: 10.1007/s11095-008-9597-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sharma V, Beatty A, Wey SP, Dahlheimer J, Pica CM, Crankshaw CL, Bass L, et al. Novel gallium(III) complexes transported by MDR1 P-glycoprotein: potential PET imaging agents for probing P-glycoprotein-mediated transport activity in vivo. Chem Biol. 2000;7:335–343. doi: 10.1016/s1074-5521(00)00111-3. [DOI] [PubMed] [Google Scholar]
- 111.Brune K, Nuernberg B, Schneider HT. Biliary elimination of aspirin after oral and intravenous administration in patients. Agents Actions Suppl. 1993;44:51–57. [PubMed] [Google Scholar]
- 112.Ghibellini G, Leslie EM, Brouwer KL. Methods to evaluate biliary excretion of drugs in humans: an updated review. Mol Pharm. 2006;3:198–211. doi: 10.1021/mp060011k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Groothuis GM, Hulstaert CE, Kalicharan D, Hardonk MJ. Plasma membrane specialization and intracellular polarity of freshly isolated rat hepatocytes. Eur J Cell Biol. 1981;26:43–51. [PubMed] [Google Scholar]
- 114.Swift B, Pfeifer ND, Brouwer KL. Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev. 2010;42:446–471. doi: 10.3109/03602530903491881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sasaki M, Suzuki H, Ito K, Abe T, Sugiyama Y. Transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II cell monolayer expressing both human organic anion-transporting polypeptide (OATP2/SLC21A6) and Multidrug resistance-associated protein 2 (MRP2/ABCC2) J Biol Chem. 2002;277:6497–6503. doi: 10.1074/jbc.M109081200. [DOI] [PubMed] [Google Scholar]
- 116.Cui Y, Konig J, Keppler D. Vectorial transport by double-transfected cells expressing the human uptake transporter SLC21A8 and the apical export pump ABCC2. Mol Pharmacol. 2001;60:934–943. doi: 10.1124/mol.60.5.934. [DOI] [PubMed] [Google Scholar]
- 117.Kopplow K, Letschert K, Konig J, Walter B, Keppler D. Human hepatobiliary transport of organic anions analyzed by quadruple-transfected cells. Mol Pharmacol. 2005;68:1031–1038. doi: 10.1124/mol.105.014605. [DOI] [PubMed] [Google Scholar]
- 118.Hirouchi M, Kusuhara H, Onuki R, Ogilvie BW, Parkinson A, Sugiyama Y. Construction of triple-transfected cells [organic anion-transporting polypeptide (OATP) 1B1/multidrug resistance-associated protein (MRP) 2/MRP3 and OATP1B1/MRP2/MRP4] for analysis of the sinusoidal function of MRP3 and MRP4. Drug Metab Dispos. 2009;37:2103–2111. doi: 10.1124/dmd.109.027193. [DOI] [PubMed] [Google Scholar]
- 119.Fahrmayr C, Konig J, Auge D, Mieth M, Fromm MF. Identification of drugs and drug metabolites as substrates of multidrug resistance protein 2 (MRP2) using triple-transfected MDCK-OATP1B1-UGT1A1-MRP2 cells. Br J Pharmacol. 2012;165:1836–1847. doi: 10.1111/j.1476-5381.2011.01672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuteykin-Teplyakov K, Luna-Tortos C, Ambroziak K, Loscher W. Differences in the expression of endogenous efflux transporters in MDR1-transfected versus wildtype cell lines affect P-glycoprotein mediated drug transport. Br J Pharmacol. 2010;160:1453–1463. doi: 10.1111/j.1476-5381.2010.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 122.Kouzuki H, Suzuki H, Ito K, Ohashi R, Sugiyama Y. Contribution of organic anion transporting polypeptide to uptake of its possible substrates into rat hepatocytes. J Pharmacol Exp Ther. 1999;288:627–634. [PubMed] [Google Scholar]
- 123.Pang KS, Maeng HJ, Fan J. Interplay of transporters and enzymes in drug and metabolite processing. Mol Pharm. 2009;6:1734–1755. doi: 10.1021/mp900258z. [DOI] [PubMed] [Google Scholar]
- 124.Kanebratt KP, Andersson TB. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos. 2008;36:1444–1452. doi: 10.1124/dmd.107.020016. [DOI] [PubMed] [Google Scholar]
- 125.Chistiakov DA, Chistiakov PA. Strategies to produce hepatocytes and hepatocyte-like cells from pluripotent stem cells. Hepatol Res. 2011 doi: 10.1111/j.1872-034X.2011.00896.x. [DOI] [PubMed] [Google Scholar]
- 126.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 127.Liu Y, Chen HC, Yang SM, Sun TL, Lo W, Chiou LL, Huang GT, et al. Visualization of hepatobiliary excretory function by intravital multiphoton microscopy. J Biomed Opt. 2007;12:014014. doi: 10.1117/1.2710237. [DOI] [PubMed] [Google Scholar]
- 128.Castellino S, Groseclose MR, Wagner D. MALDI imaging mass spectrometry: bridging biology and chemistry in drug development. Bioanalysis. 2011;3:2427–2441. doi: 10.4155/bio.11.232. [DOI] [PubMed] [Google Scholar]
- 129.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]