

Abstract
GPR119 is a G protein-coupled receptor expressed on enteroendocrine L-cells that synthesize and secrete the incretin hormone glucagon-like peptide-1 (GLP-1). Although GPR119 agonists stimulate L-cell GLP-1 secretion, there is uncertainty concerning whether GLP-1 biosynthesis is under the control of GPR119. Here we report that GPR119 is functionally coupled to increased proglucagon (PG) gene expression that constitutes an essential first step in GLP-1 biosynthesis. Using a mouse L-cell line (GLUTag) that expresses endogenous GPR119, we demonstrate that PG gene promoter activity is stimulated by GPR119 agonist AS1269574. Surprisingly, transfection of GLUTag cells with recombinant human GPR119 (hGPR119) results in a constitutive and apparently ligand-independent increase of PG gene promoter activity and PG mRNA content. These constitutive actions of hGPR119 are mediated by cAMP-dependent protein kinase (PKA) but not cAMP sensor Epac2. Thus, the constitutive action of hGPR119 to stimulate PG gene promoter activity is diminished by: 1) a dominant-negative Gαs protein, 2) a dominant-negative PKA regulatory subunit, and 3) a dominant-negative A-CREB. Interestingly, PG gene promoter activity is stimulated by 6-Bn-cAMP-AM, a cAMP analog that selectively activates α and β isoforms of type II, but not type I PKA regulatory subunits expressed in GLUTag cells. Finally, our analysis reveals that a specific inhibitor of Epac2 activation (ESI-05) fails to block the stimulatory action of 6-Bn-cAMP-AM at the PG gene promoter, nor is PG gene promoter activity stimulated by: 1) a constitutively active Epac2, or 2) cAMP analogs that selectively activate Epac proteins. Such findings are discussed within the context of ongoing controversies concerning the relative contributions of PKA and Epac2 to the control of PG gene expression.
GPR119 is a class I GTP-binding protein-coupled receptor (GPCR) expressed on intestinal enteroendocrine cells (L-cells) that synthesize and secrete the incretin hormone glucagon-like peptide-1 (GLP-1) (1, 2). GPR119 is activated by synthetic small molecule agonists such as AR231453 (3), by monoacylglycerols such as 2-oleoyl glycerol derived from dietary fat hydrolysis (4), and by fatty acid amides such as oleoylethanolamide derived from plasma membrane phospholipid hydrolysis (5). By activating the L-cell GPR119, dietary nutrients stimulate GLP-1 secretion so that circulating GLP-1 is free to exert its actions to lower levels of blood glucose, slow gastric emptying, and suppress appetite (6). Because GPR119 is also expressed on pancreatic β-cells (7, 8), and because β-cell GPR119 activation promotes insulin secretion (7, 8), it is possible that the L-cell and β-cell GPR119 receptors constitute new molecular targets for pharmacological intervention in the treatment of type 2 diabetes and obesity (9, 10).
In the present study we sought to determine whether GPR119 also plays an important role in the control of L-cell GLP-1 biosynthesis by virtue of its putative action to stimulate proglucagon (PG) gene expression. This possibility is suggested by the prior report that GPR119 agonist AR231453 raised levels of cAMP in mouse L-cell line GLUTag (2). Because GLP-1 is derived from PG (11), and because PG gene transcription in the intestine and in GLUTag cells is stimulated by various cAMP-elevating agents (12, 13), there is good reason to predict that GPR119 agonists should enhance GLP-1 biosynthesis as a consequence of their as-yet-to-be established abilities to stimulate PG gene expression.
As is the case for certain types of GPCRs (14), GPR119 can exert a constitutive and apparently ligand-independent action to raise levels of cAMP in GLUTag cells and β-cell lines (2, 8). Thus, it may be hypothesized that a constitutive and ligand-independent action of GPR119 might also exist in L-cells to stimulate PG gene expression. If so, this constitutive signaling property of GPR119 could be exploited to identify small molecules that bind to GPR119 with high affinity and that act as inverse agonists (15). By identifying the structures of these inverse agonists, it might then be possible to identify GPR119 agonists that are stimulators of PG gene expression.
We now report that PG gene expression is stimulated by GPR119 agonist AS1269574 acting via endogenous GPR119 in GLUTag cells. However, transfection of GLUTag cells with human GPR119 also leads to an increase of PG gene promoter activity and PG mRNA content. This constitutive action of GPR119 is observed in the absence of added agonist, and it is mediated by cAMP-dependent protein kinase (PKA). Of particular interest is our finding that a stimulation of PG gene promoter activity can be achieved using N6-benzyladenosine-3′,5′-cyclic monophosphate acetoxymethyl ester (6-Bn-cAMP-AM). This “prodrug” is converted to bioactive 6-Bn-cAMP that selectively activates the RIIα and RIIβ PKA regulatory subunit isoforms we report to be expressed in GLUTag cells. We also find that the action of 6-Bn-cAMP-AM to stimulate PG gene promoter activity is unaffected by a specific inhibitor of cAMP sensor Epac2 activation (ESI-05), whereas it is blocked by a cAMP antagonist that is a competitive inhibitor of PKA activation (Rp-8-Br-cAMPS-pAB). Thus, findings presented here indicate that the RIIα and RIIβ subunits of PKA are of particular importance to cAMP-regulated PG gene expression that is under the control of GPR119 in GLUTag cells.
Materials and Methods
Cell culture
GLUTag cells provided by Dr Daniel J. Drucker (University of Toronto, Toronto, Canada) were cultured in DMEM containing 5.6 mM glucose as described previously (13). HIT-T15 cells from the American Type Culture Collection (ATCC, Manassas, Virginia) were cultured in Ham's F-12K medium containing 7 mM glucose, as described previously (16). INS-1 cells provided by Dr Maryam Asfari (Université Paris, Paris, France) were cultured in RPMI 1640 containing 11.1 mM glucose as described previously (17). HEK293 cells obtained from the ATCC, or HEK293-GLP-1R cells obtained from Novo Nordisk A/S (Bagsvaerd, Denmark), were cultured in DMEM containing 25 mM glucose as described previously (18). Cultures were passaged once a week while maintained at 37°C in a humidified incubator gassed with 5% CO2. All culture media and additives were from Invitrogen Life Technologies (Grand Island, New York).
Cloning of human GPR119
GPR119 cDNA derived by reverse transcription of human islet total RNA was amplified using an AccessQuick RT-PCR System (Promega, Madison, Wisconsin). PCR primers for amplification of the cDNA were based on the published human GPR119 mRNA sequence (GenBank NM_178471.2). The forward primer was 5′-GCCGCGAATTCACGAGACATGGAATCATCTTTC-3′ and the reverse primer was 5′-GGATCCTCTAGAGTCTTAGCCATCAAACTCTGA-3′. The forward and reverse primers contained EcoRI and XbaI cut sites, respectively (underlined), and they also incorporated start and stop codons, respectively (bold type). PCR was performed using an MJ MiniOpticon thermal cycler (Bio-Rad, Hercules, California). Samples containing the reaction mixtures were heated to 45°C for 45 minutes to initiate reverse transcription, after which the samples were denatured at 95°C for 2 minutes. PCR was then performed using 34 cycles in which the parameters for each cycle were 95°C for 30 seconds, 67°C for 60 seconds, and 72°C for 65 seconds, followed by an extension step of 72°C for 5 minutes. Amplified hGPR119 cDNA digested with EcoRI and XbaI was subcloned in-frame with a FLAG-epitope coding sequence in pFLAG-CMV4 mammalian expression vector (Sigma, St Louis, Missouri) after linearization of the vector with EcoRI and XbaI. DNA sequencing and restriction enzyme analysis confirmed the insert's orientation and identity. JM109 competent Escherichia coli cells (Promega) were used for transformation and antibiotic resistance selection to obtain a single colony expressing the pFLAG-CMV4-hGPR119 plasmid DNA. This plasmid was isolated from bacteria using a HiSpeed Midi Kit (Qiagen, Valencia, California).
Additional sources of plasmid DNA and methods of transfection
Glu-Luc luciferase reporter plasmids in which luciferase activity was placed under the control of −2400 bp, −1100 bp, or −302 bp of the rat PG gene promoter were provided by Dr Tianru Jin (University of Toronto, Toronto, Canada) (19). The RIP1-CRE-Luc reporter generated in our laboratory (20) consists of 4 multimerized nonpalindromic cAMP response elements (CREs) found within the rat insulin I gene promoter (RIP1) fused to the coding sequence of firefly luciferase in pLuc-MCS. The A-CREB expression plasmid (21) was obtained from Dr Charles Vinson (National Institutes of Health [NIH], Bethesda, Maryland). Dominant-negative (DN) “triple mutant” Gαs expression plasmid (22) was obtained from Dr Catherine Berlot (Geisinger Clinic, Danville, Pennsylvania). DN PKA regulatory subunit RIα plasmid containing a [Gly 200 to Glu] substitution in cAMP-binding site A, and dual [Gly 324 to Asp] and [Arg 332 to His] substitutions in cAMP-binding site B, was obtained from Dr G. S. McKnight (University of Washington, Seattle, WA) (23). The full-length mouse PKA Cα1 catalytic subunit in pFA-PKAcat plasmid was from Stratagene (La Jolla, California) (24). The Epac2 fluorescence resonance energy transfer (FRET) reporter was from Dr Jin Zhang (Johns Hopkins University, Baltimore, Maryland) (25). The mouse GPR119 cDNA (GenBank NM_181751) in pCMV6-Entry vector was from OriGene Tech (Rockville, Maryland). Transient transfections with these plasmids were performed using Lipofectamine and Plus reagent according to the manufacturer's protocol (Invitrogen Life Technologies). Experiments were performed 48 hours after transfection.
Adenoviral transduction of GLUTag cells
Mouse wild-type (WT) Epac2 (GenBank AB021132) and DN Epac2 (G114E, G422D) in pSRα vector were provided by Dr Susumu Seino (Kobe University, Kobe, Japan) (26) and subcloned into pFLAG-CMV4 vector (Sigma). Constitutively active (CA) mouse Epac2 with deletion of the N-terminal cAMP-binding region was provided by Dr Lawrence A. Quilliam (Indiana University, Indianapolis, Indiana) (27) and subcloned into pFLAG-CMV4 vector. Each of these Epac2 plasmids was used for the synthesis of corresponding bicistronic adenoviral constructs that contained the coding sequences for Epac2 and red fluorescent protein (RFP), as generated by Vector Biolabs (Philadelphia, Pennsylvania).
Luciferase reporter assay
The luciferase assay was performed as described in greater detail in the Supplemental Material, Part B, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org. Briefly, cells were exposed for 4 hours to serum-free medium containing 0.1% BSA and test substances. Cells were lysed in passive lysis buffer (Promega), and lysates were assayed in triplicate for photoemissions using a luciferase assay kit (Promega) and a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, California).
Bioassay of GLUTag cell conditioned medium
HEK293 cells were cotransfected with pFLAG-CMV4-hGPR119 and the RIP1-CRE-Luc reporter. After 48 hours, one set of cells was exposed for 4 hours to conditioned DMEM that was previously used to culture GLUTag cells for 24 hours. A second set of cells was exposed under identical conditions to unconditioned DMEM that was not exposed to GLUTag cells. This same protocol was also used for INS-1 cells expressing human GPR119, and for HEK293 cells expressing human GLP-1R. A bioassay was also performed using HIT-T15 cells transfected only with RIP1-CRE-Luc because these cells express endogenous GPR119 (8).
Quantitative PCR (QPCR) for PG
Total RNA was isolated from GLUTag cells using an RNEasy kit (Qiagen). RNA concentration and purity was assessed using a NanoDrop ND-1000 spectrofluorimeter (NanoDrop Technologies, Wilmington, Delaware). QPCR reactions were performed using QuantiTect SYBR Green one-step kits (Qiagen) with ∼100 ng of template RNA. Thermal cycling was performed using a MJ MiniOpticon cycler (Bio-Rad) using 34 cycles in which the parameters for each cycle were as follows: 94°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, followed by a melting curve analysis from 60 to 94°C. PCR products were then run on 2% agarose gels; resolved products in gels were extracted using QIAquick kits (Qiagen), and product identity was confirmed by sequencing. PCR primers were based on the published sequence for mouse PG (GenBank NM_008100.3). The forward primer was 5′-CAATGTTGTTCCGGTTCCTC-3′ and the reverse primer was 5′-CCCTGATGAGATGAATGAAGACA-3′. A mouse HPRT QuantiTest Primer Assay (Qiagen) primer pair was used to generate the reference RNA PCR product. All primers were tested using different starting template RNA concentrations to validate their equal efficiencies in the amplification reaction. Threshold crossing (Ct) values were set manually, and the difference (ΔCt) between the Ct value for the HPRT and PG mRNAs was calculated for each reaction. ΔCt values were entered into Origin 8.0 software (OriginLab, Northampton, Massachusetts) for statistical analyses using ANOVA. To confirm expression of enodgenous GPR119 in GLUTag cells, RT-PCR was performed using PCR primers based on the published sequence for mouse GPR119 (GenBank NM_181751.2). The forward primer for mouse GPR119 was 5′-GCCGCGAATTCCGAGACATGGAATCATCTTTC-3′ and the reverse primer was 5′-GATCCTCTAGACCGTCTTAGCCATCAAACTCTG-3′.
Western blot detection of Epac2 and FLAG-GPR119
Cells were harvested, lysed in Laemmli sample buffer, sonicated, and subjected to a determination of protein concentration, as measured using a BCA Protein Assay Kit (Thermo Scientific, Rockford, Illinois). Whole-cell lysates were electrophoresed on 4% to 20% gradient Mini-PROTEAN TGX Precast Gels (Bio-Rad) and the resolved proteins were transferred to polyvinyldifluoride (PVDF) membranes (EMD Millipore, Billerica, Massachusetts). The PVDF membranes were blocked for 1 hour at room temperature in Tris-buffered saline containing 0.1% Tween-20 (TBST) and 5% nonfat dry milk. For immunodetection of endogenous Epac2 in GLUTag cells, the primary monoclonal antibody (MAB) was a 1:100-fold dilution of anti-Epac2 5B1 (Cell Signaling, Danvers, Massachusetts) and the secondary antiserum was a 1:5000-fold dilution of goat antimouse horseradish peroxidase (HRP) –conjugated IgG (Sigma). For immunodetection of FLAG epitope-tagged mouse or human GPR119 in transfected HEK293 cells, we used a 1:4000-fold dilution of anti-FLAG M2 HRP-conjugated MAB (Sigma). PVDF membranes were initially incubated in TBST contained 10% goat serum with primary antibodies for 16 hours at 4°C. Where applicable, incubation with a secondary antiserum was performed for 1 hour at room temperature. The immunoreactive proteins were detected by enhanced chemiluminescence using a SuperSignal West Pico Kit (Thermo Scientific) and the immunoreactivity was quantified using a ChemiDoc XRS digital imaging system and Quantity One software (Bio-Rad).
Western blot detection of PKA regulatory subunit isoforms
GLUTag cell lysates containing approximately 15 μg total protein were solubilized in sample buffer and resolved by 12% SDS-PAGE for transfer to PVDF membranes. After blocking in TBST supplemented with 5% nonfat milk (1 hour, room temperature), the membranes were incubated overnight at 4°C with TBST containing the following PKA regulatory subunit isoform-specific primary antibodies: rabbit anti-RIα (Cell Signaling), affinity-purified sheep anti-RIβ (R&D Systems, Minneapolis, Minnesota), mouse anti-RIIα (BD Transduction Laboratories, San Jose, California), mouse anti-RIIβ (BD Transduction Laboratories). The HRP-conjugated secondary antisera were donkey antirabbit IgG (GE Healthcare, Waukesha, Wisconsin), rabbit antimouse IgG, (Sigma), and donkey antisheep IgG (Abcam, Cambridge, Massachusetts). PVDF membranes were incubated with these secondary antisera for 1 hour at room temperature and the immune complex was detected by digital imaging using an enhanced chemiluminescence kit (Perkin Elmer, Waltham, Massachusetts).
Rap1 activation assays
A pull-down assay was used to detect active Rap1 in lysates of GLUTag cells transfected with FLAG-tagged Rap1 and Epac2 (28). Activated Rap1-GTP and total Rap1 were detected using an anti-FLAG M2 MAB conjugated to HRP (Sigma). The in vitro Rap1 activation assay using mant-GDP is described in the Supplemental Material, Part D, Legend for Supplemental Figure 5.
FRET assay for Epac2 activation
The Epac2-C1 clone of HEK293 cells stably expressing a full-length Epac2 FRET reporter was described previously (29). These cells were plated at 80% confluence on 96-well clear bottom assay plates (Costar 3904) and maintained in culture medium overnight before their use. Assays were performed using a FlexStation 3 microplate reader, as described previously (29). After replacement of the culture medium with 170 μL per well of a standard extracellular saline solution, the excitation light was delivered at 435/9 nm (455 nm cutoff), and the emitted light was detected at 485/15 nm (cyan fluorescent protein) or 535/15 nm (yellow fluorescent protein). The excitation light source was a Xenon flash lamp, and the emission intensities were the average of 12 excitation flashes for each time point. Test solutions comprising cAMP analogs dissolved in standard extracellular saline solution containing 0.1% dimethylsulfoxide were placed in V-bottom 96-well plates (Greiner Bio-One, Monroe, North Carolina) and an automated pipetting procedure was used to transfer 30 μL of each test solution to the assay plate containing cells. The test solutions were injected into each well at a pipette height that corresponded to a fluid level of 150 μL, and the rate of injection was 31 μL/s. The cyan fluorescent protein/yellow fluorescent protein emission ratio was calculated for each well, and values for 8 to 12 wells were averaged. The time course of the change of FRET ratio was plotted after exporting these data to Origin 8.0 (OriginLab, Northhampton, Massachusetts).
Bioluminescence resonance energy transfer (BRET) assay for PKA activation
To perform time-resolved BRET measurements of PKA activation, the BRET2 system was optimized to eBRET by fusing the novel luciferase Rluc8 developed by the laboratory of S. Gambhir (30), to the C-terminus of human PKA regulatory subunits. In this BRET assay (31), the PKA regulatory subunit Rluc8 constructs are cotransfected with a GFP2-hCα construct in which a human PKA catalytic subunit is fused to a variant of green fluorescent protein. Resultant reconstitution of the PKA holoenzyme occurs in living cells, and BRET is measurable due to energy transfer between Rluc8 and GFP2. When levels of cytosolic cAMP rise, the resultant activation of PKA leads to holoenzyme dissociation that is measurable as a decrease of BRET. For the studies described here, GLUTag cells seeded on white 96-well plates were cotransfected with pGFP2-hCα and the desired human PKA regulatory subunits fused to Rluc8 (RIα, RIβ, RIIα, RIIβ). Experiments were also performed using GLUTag cells transfected with a unimolecular hEpac1ΔDEP-CDC25-HD(T781A/F782A) BRET sensor incorporating an engineered Epac1, as described previously (32). BRET measurements were performed 48 hours after transfection, at which time GLUTag cells were exposed to cAMP analogs under conditions of equilibration in saline containing a luciferase substrate (coelenterazine; 5 μM) that emits photons after its luciferase-catalyzed oxidation. Emitted light corresponding to Rluc8 luminescence or pGFP2-hCα fluorescence was detected at room temperature using a POLARstar Omega microplate reader (BMG Labtech, Cary, North Carolina), and the BRET ratio (pGFP2/Rluc8) was determined in real-time at 2-minute intervals. Additional details are provided in the Supplemental Material, Part C.
In vitro PKA activation assay
Recombinant human PKA Cα1 catalytic subunit was expressed and purified as described previously (33). Recombinant human PKA regulatory subunits (hRIα, hRIβ, hRIIα, hRIIβ) were expressed and purified according to the procedure of Bertinetti et al (34) using Sp-8-AEA-cAMPS agarose. SDS-PAGE was used to monitor protein expression and purification. Typically, the recombinant proteins were purified to ≥95% homogeneity. PKA activity was assayed in vitro using the coupled spectrophotometric assay described by Cook et al (35) in which 260 μM Kemptide (LRRASLG; Biosyntan, Berlin, Germany) served as a PKA substrate. PKA holoenzyme formation was carried out for 3 minutes at room temperature in a pH 7.0 assay mixture comprising 100 mM 3[N-morholino]propanesulfonic acid, 10 mM MgCl2, 100 μM ATP, 1 mM phosphoenolpyruvate, 15 U/mL lactate dehydrogenase, 70 U/mL pyruvate kinase, 200 mM reduced nicotinamide adenine dinucleotide, 5 mM β-mercaptoethanol with 15 to 50 nM human PKA Cα1 subunit and a 1.2-fold molar excess of cAMP-free RIα, RIβ, RIIα, or RIIβ subunits. Kinase activity was determined after preincubating the holoenzyme for 3 minutes with increasing amounts of cAMP or 6-Bn-cAMP (100 fM to 500 μM) before starting the reaction with substrate Kemptide. Enzymatic activities were quantified as U/mg, defined as μmol mg−1min−1. Data points were determined in duplicate and analyzed using GraphPad Prism 5.01 (GraphPad Software, Inc, La Jolla, California) by plotting the normalized enzymatic activities against the logarithm of the cyclic nucleotide concentration.
Analysis of luciferase assay data
The repeatability of findings concerning luciferase assays was confirmed by performing each experiment a minimum of 3 times. Statistical analyses of these data were performed using the Student's paired t test and SigmaStat software (Systat Software, Inc, Chicago, Illinois). A P value of < .05 was considered to be statistically significant and is indicated by an asterisk.
Sources of additional reagents
cAMP analogs (see Supplemental Material, Part A, for a complete list with abbreviations) were from the BIOLOG Life Science Institute (Bremen, Germany). Exendin-4, forskolin, IBMX, and H-89 were from Sigma. AR231453 was from Enzo Life Sciences (Farmingdale, New York). AS1269574 was from EMD Millipore.
Results
RT-PCR cloning and expression of functional human GPR119
Human GPR119 (hGPR119) cDNA was derived by RT-PCR using human islet mRNA from a nondiabetic anonymous donor (Supplemental Figure 1A). The hGPR119 cDNA was fully sequenced to confirm its identity and was ligated into pCMV4 to allow for the expression of FLAG epitope-tagged hGPR119 in transfected HEK293 cells and also GLUTag cells. Two days after transfection, the expression of hGPR119 was confirmed by Western blot analysis using an anti-FLAG MAB (Supplemental Figure 1, B and C). Luciferase-based assays were then performed using a cAMP-responsive reporter (RIP1-CRE-Luc) (20) to demonstrate that hGPR119 was activated by GPR119 agonist AR231453 in HEK293 cells transfected with hGPR119 (Supplemental Figure 2A). Control experiments using an empty vector demonstrated that AR231453 exerted no stimulatory action at RIP1-CRE-Luc under conditions in which HEK293 cells were not transfected with hGPR119 (Supplemental Figure 2A). This negative finding is consistent with one prior report that HEK293 cells do not express endogenous GPR119 (8). As expected, AR231453 stimulated RIP1-CRE-Luc activity in hamster HIT-T15 insulin-secreting cells that express endogenous GPR119 (Supplemental Figure 2B) (8), and also in rat INS-1 cells transfected with hGPR119 (Supplemental Figure 2B). In summary, these findings obtained using multiple cell lines demonstrated the functionality of hGPR119 derived by RT-PCR.
GPR119 agonist AS1269574 stimulates PG gene promoter activity
We developed a luciferase-based assay for assessment of PG gene promoter activity in GLUTag cells (Supplemental Material, Part B). In this assay, GLUTag cells were transfected with a Glu-Luc construct in which luciferase activity was placed under the control of −2400 bp of the PG gene promoter (19, 36). The activity of this reporter was stimulated after a 4-hour exposure of GLUTag cells to the cAMP-elevating agents forskolin and IBMX (Figure 1A), and this stimulation was suppressed under conditions in which GLUTag cells were treated with PKA inhibitor H-89 (37) (Figure 1A). By screening commercially available GPR119 agonists, we found that −2400 Glu-Luc activity in GLUTag cells was stimulated most strongly by GPR119 agonist AS1269574 (38), an effect reduced by H-89 (Figure 1B). This action of AS1269574 was expected because GLUTag cells express endogenous GPR119 (see Figure 6A) (2). Because the basal and agonist-stimulated activities of −2400 Glu-Luc were reduced by H-89, these initial findings provided evidence for a PKA-mediated action of GPR119 to stimulate PG gene expression.
Figure 1.
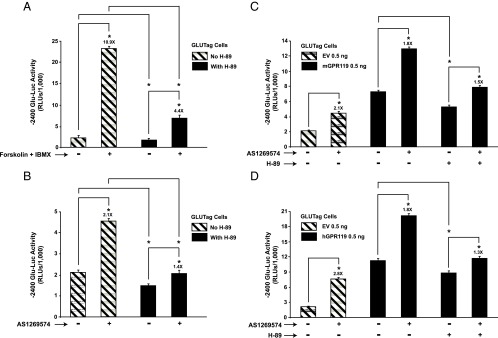
H-89-sensitive stimulation of −2400 Glu-Luc activity by forskolin and AS1269574 in GLUTag cells. A, treatment of GLUTag cells for 4 hours with forskolin (Fsk, 2 μM) and IBMX (100 μM) led to increased −2400 Glu-Luc activity, an effect reduced by H-89 (10 μM). B, a 4-hour exposure to GPR119 agonist AS1269574 (1 μM) stimulated −2400 Glu-Luc activity in an H-89-sensitive manner. C, transfection of GLUTag cells with mouse GPR119 (mGPR119) increased basal −2400 Glu-Luc activity in the absence of AS1269574, an effect reduced by H-89. Addition of AS12369574 (3 μM) to cells transfected with mGPR119 led to an additional increase of −2400 Glu-Luc activity, and this action of AS12369574 was also H-89 sensitive. D, same as for (C) except that GLUTag cells were transfected with human GPR119 (hGPR119). Abbreviations: EV, empty vector; RLUs, relative light units. *, P < .05 here and in subsequent figures. Values are 0.5 ng/well for mGPR119 and hGPR119 plasmids.
Figure 6.
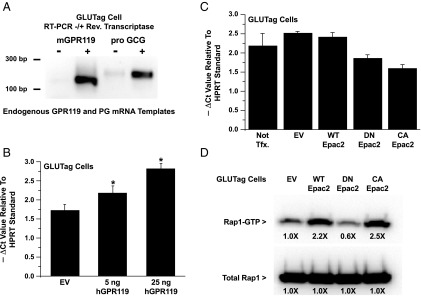
Human GPR119 (hGPR119) but not CA Epac2 increases levels of PG mRNA. A, RT-PCR for endogenous GPR119 and PG (pro GCG) using total RNA from GLUTag cells. B, QPCR analysis revealed that levels of PG mRNA were increased in GLUTag cells transfected with human GPR119. C, QPCR analysis demonstrated that levels of PG mRNA were not increased after transduction of GLUTag cells with a CA Epac2. For comparison, GLUTag cells were also transduced with WT or DN Epac2. D, a control Rap1 activation assay using transduced GLUTag cells expressing FLAG-Rap1 confirmed that CA Epac2 increased levels of active Rap1-GTP, whereas DN Epac2 exerted an opposite effect. For (B) and (C), the y-axes indicate the −ΔCt values in the QPCR reactions using HPRT mRNA as a reference standard. A more negative ΔCt value signifies higher levels of PG mRNA.
Constitutive stimulation of PG gene promoter activity by GPR119
Transfection of GLUTag cells with mouse GPR119 (mGPR119) or hGPR119 increased basal −2400 Glu-Luc activity, and under these conditions, AS1269574 produced an additional stimulatory effect that was H-89 sensitive (Figure 1, C and D). Such findings indicated that GPR119 signaled in a constitutive and ligand-independent manner to stimulate PG gene promoter activity. This possibility was evaluated in greater detail using GLUTag cells transfected with increasing amounts of mGPR119 or hGPR119 plasmid DNA. This analysis revealed a “dose-dependent” action of mGPR119 and hGPR119 to increase the activities of Glu-Luc reporters incorporating −1100 bp (Figure 2, A and C) or −2400 bp (Figure 2, B and D) of the PG gene promoter. When comparing mGPR119 and hGPR119 in this assay, a stronger constitutive stimulation was measurable with mGPR119 (Figure 2, A–D). Importantly, GPR119 acted in a PG gene promoter-specific manner because hGPR119 failed to elevate basal levels of luciferase in GLUTag cells transfected with a pSK-Luc reporter in which the cDNA for luciferase was not fused to the PG gene promoter.
Figure 2.
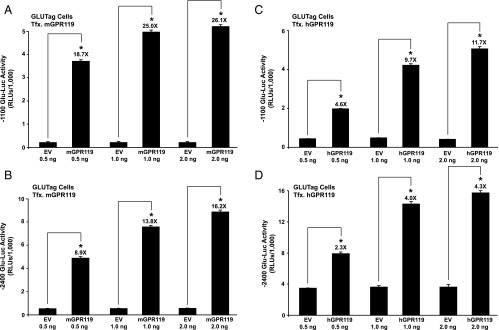
Constitutive ligand-independent stimulation of Glu-Luc activity by mouse GPR119 and human GPR119 in GLUTag cells. (A, B) GLUTag cells were cotransfected with mouse GPR119 (mGPR119) and either −1100 Glu-Luc (A) or −2400 Glu-Luc (B). The amount of mouse GPR119 plasmid or control empty vector (EV) added to the transfection mixture within each well of a 96-well plate is indicated on the x-axis (ng/well). (C, D) The same experiment as for (A) and (B) except that GLUTag cells were instead transfected with human GPR119 (hGPR119).
Because GPR119 stimulated Glu-Luc activity in the absence of added agonist, it was of interest to ascertain whether GLUTag cells secreted an autocrine factor that activated GPR119, thereby up-regulating PG gene promoter activity. This possibility was tested using a bioassay in which conditioned culture medium from GLUTag cells was screened for the presence of a GPR119 agonist. To this end, HEK293 cells were treated with conditioned medium after cotransfection with hGPR119 and RIP1-CRE-Luc. This analysis demonstrated that for HEK293 cells expressing hGPR119, the activity of RIP1-CRE-Luc was unaltered after treatment of cells with conditioned medium obtained from GLUTag cells (Figure 3A). Similar findings were obtained using INS-1 cells transfected with hGPR119 and RIP1-CRE-Luc (Figure 3B), or HIT-T15 cells transfected with RIP1-CRE-Luc only (Figure 3C). Such findings indicated that an autocrine GPR119 agonist was not secreted from GLUTag cells under the experimental conditions described here.
Figure 3.
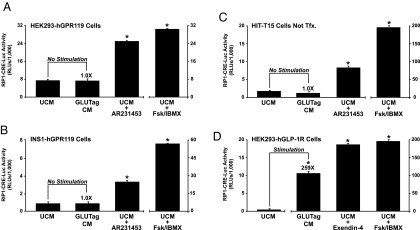
Conditioned DMEM from GLUTag cells does not contain a GPR119 agonist. A, HEK293 cells transfected with human GPR119 (hGPR119) and RIP1-CRE-Luc were exposed for 4 hours to unconditioned medium (UCM) or conditioned medium (CM) obtained from GLUTag cell cultures. As a positive control, the responsiveness of RIP1-CRE-Luc was validated under conditions in which these cells were treated for 4 hours with AR231453 (300 nM) or forskolin (Fsk, 2 μM) and IBMX (100 μM). B, same as (A) except that these assays were performed using INS-1 cells transfected with human GPR119 and RIP1-CRE-Luc. C, same as (A) except that these assays were performed using HIT-T15 cells that express endogenous GPR119, and that were transfected with RIP1-CRE-Luc only. D, same as (A) except that these assays were performed using GLP-1 receptor (GLP-1R) agonist Exendin-4 (10 nM) and HEK293 cells transfected with the human GLP-1R and RIP1-CRE-Luc. Note that (D) demonstrates the presence of GLP-1 in the conditioned DMEM of GLUTag cells.
Despite such findings, it might be argued that our bioassay using conditioned medium lacked appropriate sensitivity. This is unlikely to be the case because GPR119 agonist AR231453 stimulated an increase of RIP1-CRE-Luc activity in the HEK293, INS-1, and HIT-T15 cells described above (Figure 3, A–C). Furthermore, the sensitivity of this bioassay was validated by demonstrating the expected finding that conditioned medium of GLUTag cells contained secreted GLP-1, as verified in assays of HEK293 cells transfected with the human GLP-1 receptor (Figure 3D). Thus, under conditions in which GLUTag cells were transfected with hGPR119 (Figure 2, C and D), there existed a constitutive and apparently ligand-independent action of hGPR119 to stimulate −1100 and −2400 Glu-Luc activity. This constitutive action of hGPR119 was independent of the extracellular glucose concentration when comparing 5 mM vs 25 mM glucose (Supplemental Figure 3).
PKA links GPR119 to PG gene promoter activity
Because GPR119 agonists are cAMP-elevating agents in GLUTag cells (8), we evaluated whether human GPR119 acted via PKA to stimulate Glu-Luc activity in the absence of an added receptor agonist. It was demonstrated that the constitutive action of human GPR119 to stimulate −2400 Glu-Luc activity was reduced after cotransfection of GLUTag cells with a DN “triple mutant” Gαs protein (22) that uncouples GPCRs from cAMP production (Figure 4A). Moreover, the stimulatory action of hGPR119 at −2400 Glu-Luc was reduced (Figure 4B) after cotransfection with a DN PKA regulatory subunit that prevents activation of endogenous PKA by cAMP (23). Conversely, for cells not expressing hGPR119, −2400 Glu-Luc activity was stimulated after overexpression of a PKA catalytic subunit (Figure 4C).
Figure 4.
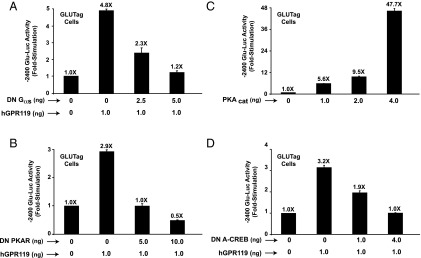
Molecular analysis of human GPR119 (hGPR119) signal transduction in GLUTag cells. (A, B) The constitutive action of human GPR119 to activate −2400 Glu-Luc was dose-dependently blocked when GLUTag cells were transfected with a DN Gαs protein (A) or a DN PKA regulatory subunit (DN PKAR) (B). C, −2400 Glu-Luc was activated in GLUTag cells transfected with a PKA catalytic subunit (PKAcat). D, the constitutive action of human GPR119 at −2400 Glu-Luc was blocked in GLUTag cells transfected with a DN A-CREB.
It was also possible to demonstrate that for GLUTag cells expressing hGPR119, the activity of −2400 Glu-Luc was reduced after cotransfection with a DN A-CREB that blocks CREB-dependent activation of gene transcription (Figure 4D) (21). Furthermore, the stimulatory action of hGPR119 at −2400 Glu-Luc was reduced but not eliminated when the CRE of the PG gene promoter was mutated to reduce its affinity for CREB. Thus, for a −302-bp PG gene promoter luciferase construct incorporating a WT CRE (19), the fold-stimulation in response to hGPR119 was 7.9 ± 0.2 (mean ± SD, n = 3 assays). If the CRE was mutated to impair the binding of CREB (19), the fold-stimulation was reduced to 5.7 ± 0.3. Thus, the proglucagonotropic action of hGPR119 was mediated, at least in part, by a cAMP signal transduction mechanism comprising Gαs, PKA, CREB, and the CRE of the PG promoter.
PG gene promoter activity is unaffected by selective Epac activators
Prior studies of GLUTag cells provided evidence for a PKA-independent cAMP signaling mechanism, one that uses Epac2 to stimulate PG gene expression (39–41). Epac2 is expressed in GLUTag cells (Figure 5A), but our luciferase assays revealed that −2400 Glu-Luc activity was not stimulated by cAMP analogs that incorporate a 2′-O-methyl substitution on the ribose moiety, a modification that allows selective activation of Epac proteins (42, 43). For example, 8-pMeOPT-2′-O-Me-cAMP (30–100 μM) was without effect at −2400 Glu-Luc (Figure 5B), as was also the case for a phosphodiesterase-resistant Sp-8-pCPT-2′-O-Me-cAMPS analog that has improved stability against hydrolysis (Figure 5B). In marked contrast, cAMP analogs that activate both PKA and Epac did stimulate −2400 Glu-Luc activity. Such analogs included 8-pCPT-2′-OH-cAMP and Sp-8-pCPT-2′-OH-cAMPS (Figure 5B). In summary, our findings do not support prior studies in which it was proposed that Epac2 mediates the stimulatory action of cAMP at the PG gene promoter (39–41, 44, 45).
Figure 5.
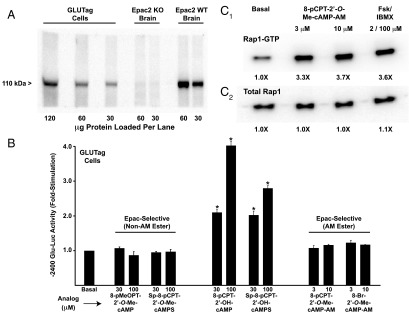
Epac-selective cAMP analogs fail to activate the PG gene promoter. A, expression of endogenous 110 kDa Epac2 in GLUTag cells was confirmed by Western blot analysis using an anti-Epac2 5B1 MAB. Epac2 immunoreactivity was also detected in a lysate from WT mouse brain but not a negative control lysate from an Epac2 knockout (KO) mouse brain (72). B, −2400 Glu-Luc activity was not stimulated after a 4-hour exposure of GLUTag cells to the non-AM esters (left) or AM-esters (right) of Epac-selective cAMP analogs. As a positive control, the responsiveness of −2400 Glu-Luc was confirmed using GLUTag cells treated with non-AM esters of cAMP analogs that activate PKA (middle). C, the efficacy of Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP-AM was validated in a pull-down Rap1 activation assay using lysates of GLUTag cells transfected with FLAG-Rap1. Levels of active Rap1-GTP (C1) and total Rap1 (C2) were determined by Western blot analysis using an anti-FLAG MAB.
One potential criticism of our analysis using Epac-selective cAMP analogs is that these compounds might not enter GLUTag cells. Therefore, we tested Epac-selective cAMP analogs that are highly membrane permeable by virtue of their substitution with an acetoxymethyl ester (AM-ester) moiety. Such analogs include 8-pCPT-2′-O-Me-cAMP-AM and 8-Br-2′-O-Me-cAMP-AM, each of which is a “prodrug” activated by cytosolic esterases that remove the AM-ester moiety (46). In assays using GLUTag cells transfected with −2400 Glu-Luc, these Epac-selective AM-esters also failed to exert stimulatory effects when tested at concentrations of 3 or 10 μM (Figure 5B). In contrast, control Rap1 activation assays demonstrated the expected finding that the Epac2 effector Rap1 GTPase was activated by 8-pCPT-2′-O-Me-cAMP-AM in GLUTag cells (Figure 5C).
PG mRNA content is increased by hGPR119 but not CA Epac2
Because luciferase-based measurements of PG gene promoter activity do not necessarily reflect corresponding changes in levels of PG mRNA, real-time QPCR was performed to determine the PG mRNA content of GLUTag cells transfected with hGPR119. Using a primer set that amplified endogenous PG mRNA (Figure 6A), this analysis revealed that levels of PG mRNA were elevated in GLUTag cells transfected with hGPR119 (Figure 6B). This action of hGPR119 was proportional to the amount of receptor plasmid present in the transfection reagent, and it did not require the addition of a GPR119 agonist. Thus, these findings obtained in the QPCR assay paralleled findings obtained using the −1100 and −2400 Glu-Luc reporters in which a constitutive and apparently ligand-independent action of hGPR119 to stimulate PG gene promoter activity was observed.
Next, we evaluated a potential role for Epac2 in the control of PG mRNA content. Experiments were performed in which the PG mRNA content was measured after adenoviral transduction of GLUTag cells with WT, DN, or CA Epac2. This analysis revealed that basal levels of PG mRNA were not significantly altered in cells transduced with WT Epac2, whereas for DN Epac2 a small reduction was measured (Figure 6C). However, CA Epac2 also reduced levels of PG mRNA (Figure 6C). In marked contrast, control Rap1 activation assays using GLUTag cells demonstrated that levels of active Rap1 were increased by CA Epac2, whereas they were decreased by DN Epac2 (Figure 6D). Thus, the Rap1 activation assay faithfully reported the catalytic activities of DN and CA Epac2, whereas the QPCR assay for PG mRNA did not. Such findings seem to indicate that the overexpression of mutant forms of Epac2 leads to nonphysiological “squelching” of PG gene expression.
6-Bn-cAMP-AM is a powerful stimulator of PG gene expression
We confirmed a prior finding (39) that the Epac-selective cAMP analog 8-pMeOPT-2′-O-Me-cAMP (100 μM) stimulated a small but statistically significant increase of PG mRNA content in GLUTag cells (Figure 7A). However, levels of PG mRNA were increased to a greater extent after treatment of GLUTag cells with the AM-ester of a cAMP analog designated as 6-Bn-cAMP-AM (Figure 7A). Similarly, 6-Bn-cAMP-AM was the strongest stimulator of PG gene promoter activity when comparing the efficacies of various cAMP analogs that activate PKA (Figure 7B). Because N6-substituted cAMP analogs are commonly used as selective activators of PKA (43), we evaluated the properties of the N6-benzyladenine-containing analog in greater detail. By performing in vitro PKA activation assays to determine cyclic nucleotide activation constants (Table 1; Supplemental Figure 4, A–D), it was found that 6-Bn-cAMP had a higher selectivity for the RIIα and RIIβ isoforms of PKA regulatory subunits, as compared with the RIα and RIβ isoforms. Collectively, these findings provided the first indication that 6-Bn-cAMP-AM might constitute a novel pharmacological tool with which to explore the putative roles of RIIα and RIIβ in the control of PG gene expression.
Figure 7.
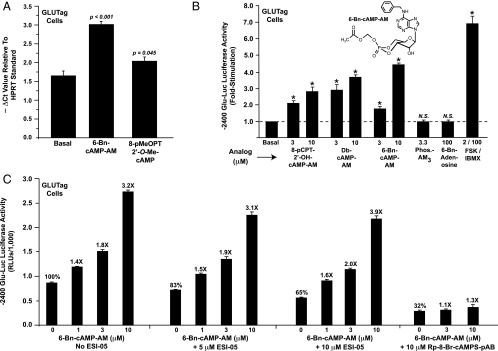
6-Bn-cAMP-AM stimulates PG gene expression. A, levels of PG mRNA were increased in GLUTag cells treated for 4 hours with 100 μM of the Epac-selective cAMP analog 8-pMeOPT-2′-O-Me-cAMP. A stronger stimulation was measured when cells were treated with 6-Bn-cAMP-AM (10 μM). B, the activity of −2400 Glu-Luc was stimulated by AM-esters of cAMP analogs that have the capacity to activate PKA. These included Db-cAMP-AM and 8-pCPT-2′-OH-cAMP-AM. However, no such stimulation was measured using a negative control phosphate-acetoxymethyl ester (Phos.-AM3) or a negative control 6-Bn-adenosine that is a metabolite of 6-Bn-cAMP-AM. C, the action of 6-Bn-cAMP-AM to stimulate −2400 Glu-Luc was blocked by Rp-8-Br-cAMPS-pAB, a competitive antagonist of PKA activation. In contrast, a specific inhibitor of Epac2 activation (ESI-05) failed to block the action of 6-Bn-cAMP-AM.
Table 1.
PKA Holoenzyme Activation Constants for cAMP and 6-Bn-cAMP
6-Bn-cAMP-AM acts independently of Epac2 to stimulate PG gene promoter activity
Because 6-Bn-cAMP-AM was a powerful stimulator of PG gene promoter activity, and because PG gene promoter activity was not stimulated by selective activators of Epac proteins, it might be concluded that 6-Bn-cAMP-AM acted at PKA and not at Epac2 to exert its effects in the assays reported here. However, this conclusion does not take into account the possibility that dual activation of PKA and Epac2 by 6-Bn-cAMP-AM might synergistically up-regulate PG gene promoter activity. Thus, we evaluated the capacity of 6-Bn-cAMP-AM to activate Epac2. Surprisingly, we found that the non-AM ester of 6-Bn-cAMP activated recombinant Epac2, as measured in a solution-based in vitro Rap1 activation assay using fluorescent mant-GDP (Supplemental Figure 5A). By performing FRET assays using HEK293 cells transfected with biosensors that report Epac2 or PKA activation (29), we independently confirmed that 6-Bn-cAMP-AM activated both Epac2 and PKA (Supplemental Figure 5, B and C). With these findings in mind, we evaluated whether the action of 6-Bn-cAMP-AM at −2400 Glu-Luc might be blocked by a pharmacological inhibitor of Epac2 activation. To this end, we used ESI-05, a molecule that inhibits Epac2 activation while having no effect at Epac1 or PKA (29). Our analysis revealed that ESI-05 (10 μM) reduced basal −2400 Glu-Luc activity, yet when findings were expressed as the fold-stimulation of luciferase activity, ESI-05 failed to inhibit the stimulatory action of 6-Bn-cAMP-AM (Figure 7C). Importantly, the efficacy of ESI-05 was confirmed in a control experiment, whereby it was demonstrated that 10 μM ESI-05 blocked activation of the Epac2 FRET reporter by 6-Bn-cAMP-AM (Supplemental Figure 6, A and B).
Next, it was demonstrated that the stimulatory action of 6-Bn-cAMP-AM at −2400 Glu-Luc was blocked by Rp-8-Br-cAMPS-pAB (Figure 7C). This highly membrane-permeable “prodrug” derivative of Rp-8-Br-cAMPS acts as a cAMP antagonist to competitively inhibit PKA activation by endogenous cAMP (47, 48). Thus, unlike findings obtained with ESI-05, Rp-8-Br-cAMPS-pAB nearly abrogated the action of 6-Bn-cAMP-AM at −2400 Glu-Luc (Figure 7C). It may be concluded that 6-Bn-cAMP-AM up-regulated PG gene promoter activity in a PKA-mediated manner, and that this action of 6-Bn-cAMP-AM was not contingent on concomitant Epac2 activation. Finally, we noted that basal −2400 Glu-Luc activity was reduced not only by H-89 (Figure 1, A and B), but also by Rp-8-Br-cAMPS-pAB (Figure 7C) and ESI-05 (Figure 7C). Such findings are expected if basal PG gene promoter activity is dually regulated by PKA and Epac2.
6-Bn-cAMP-AM activates RIIα and RIIβ PKA regulatory subunits in GLUTag cells
Taken as a whole, these findings obtained using −2400 Glu-Luc and 6-Bn-cAMP-AM indicated that it was PKA and not Epac2 that participated in the stimulatory control of PG gene promoter activity by cAMP-elevating agents. However, these findings did not reveal which PKA regulatory subunit isoform mediated the action of cAMP, nor did they rule out a role for Epac1 in the process. To address these issues, GLUTag cells were transfected with BRET biosensors based on PKA regulatory and catalytic subunits, as well as Epac1. In these live-cell, multiwell, plate-reader assays, a reduction of the BRET signal reflects: 1) activation of the respective PKA holoenzyme due to cAMP-dependent dissociation of the catalytic and regulatory subunits (31), or 2) a conformational change in Epac1 due to binding of cAMP (32) (for additional details, see Supplemental Material, Part C and also Materials and Methods).
Our BRET analysis revealed that 6-Bn-cAMP-AM was a selective activator of PKA RIIα and RIIβ isoforms expressed by transient transfection in GLUTag cells (Figure 8, A and B). In fact, when tested at a concentration of 10 μM, 6-Bn-cAMP-AM had a greatly reduced capacity to activate the RIα and RIβ isoforms of PKA regulatory subunits (Figure 8, C and D). Moreover, 6-Bn-cAMP-AM did not activate the Epac1 BRET sensor (data not shown). Thus, these findings obtained using the BRET assay indicated that endogenous RIIα and/or RIIβ regulatory subunits of PKA played an especially important role in the control of PG gene promoter activity in GLUTag cells. Using antisera that were confirmed to be selective for recombinant PKA regulatory subunit isoforms (Figure 8E), we performed Western blot analyses using GLUTag cell lysates to demonstrate that these cells expressed endogenous RIIα and RIIβ in addition to RIα and RIβ (Figure 8F).
Figure 8.
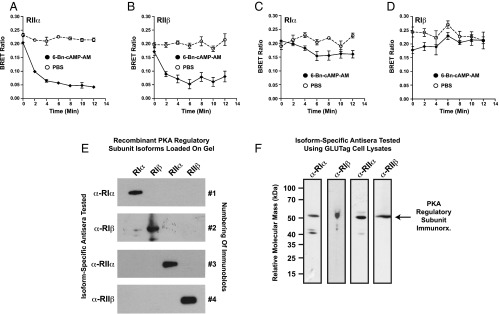
6-Bn-cAMP-AM is a selective activator of RIIα and RIIβ PKA regulatory subunits. A–D, GLUTag cells were transfected with BRET sensors that report cAMP-dependent activation of reconstituted PKA holoenzymes in situ (see Supplemental Material, Part C). The BRET sensors contained RIIα (A), RIIβ (B), RIα (C), or RIβ (D) regulatory subunit isoforms of PKA. 6-Bn-cAMP-AM (10 μM) added at time 0 selectively activated RIIα (A) and RIIβ (B), but PBS was without significant effect. Note that no significant stimulation of RIα or RIβ was measured in response to 6-Bn-cAMP-AM (C, D). E, Purified recombinant PKA regulatory subunits (25 ng/lane) were subjected to Western blot analysis to validate the specificity of isoform-specific antiregulatory subunit antisera (α-RIα, α-RIβ, α-RIIα, α-RIIβ). For this analysis, each immunoblot contained RIα, RIβ, RIIα, and RIIβ PKA regulatory subunit isoforms resolved by SDS-PAGE and transferred to the blot in the indicated lanes. For each blot, a single antiregulatory subunit antiserum was tested. F, for untransfected GLUTag cells, Western blot analysis revealed that the isoform-specific PKA regulatory subunit antisera detected endogenous RIα, RIβ, RIIα, and RIIβ immunoreactivity (arrow) in whole-cell lysates.
Discussion
PG gene expression is under the control of GPR119
This study is the first to report that a GPR119 agonist has the capacity to stimulate PG gene expression, as demonstrated using GLUTag cells treated with AS1269574. Surprisingly, we also report a constitutive and apparently ligand-independent action of GPR119 to stimulate PG gene expression. Thus, in the absence of added GPR119 agonist, PG gene promoter activity and PG mRNA content are found to be increased in GLUTag cells transfected with GPR119. Collectively, these findings are consistent with recent receptor mutagenesis studies that report key structural features of GPR119 that allow it to be activated by GPR119 agonists, while also stimulating cAMP production independently of agonist binding (49, 50). Because a knockout of GPR119 expression in mice leads to a reduction in the ability of orally administered glucose to stimulate L-cell GLP-1 secretion (51, 52), these agonist-induced and constitutive actions of GPR119 might play a role in determining how much GLP-1 is available for release. Thus, a rationale exists for future studies that will determine whether or not an up- or down-regulation of GPR119 expression influences intestinal GLP-1 production in the L-cells.
GPR119 signals via PKA and bZIP transcription factors to stimulate PG gene expression
We find that PKA inhibitor H-89 reduces the action of GPR119 agonist AS1269574 to stimulate PG gene promoter activity, whereas constitutive up-regulation of PG gene promoter activity by GPR119 is reduced by 1) a DN Gαs that uncouples GPCRs from cAMP production (22), or 2) a DN PKA regulatory subunit that fails to bind cAMP and that traps PKA catalytic subunits in holoenzyme complexes that are not cAMP responsive (23). We also find that a DN A-CREB completely blocks the constitutive action of GPR119 to stimulate PG gene promoter activity. Because A-CREB sequesters basic region-leucine zipper (bZIP) transcription factors by dimerizing with them to prevent their binding to DNA promoter elements (21), and because bZIPs are PKA substrates, it could be that GPR119 signals exclusively through a bZIP such as CREB to transactivate PG gene expression. This outcome is surprising given that evidence exists for PKA-mediated but bZIP-independent actions of cAMP to up-regulate PG gene expression in GLUTag cells. For example, PKA signals through β-catenin and TCF7L2 to up-regulate PG gene promoter activity (45). To what extent A-CREB antagonizes such alternative bZIP-independent signaling pathways remains to be determined. However, a CREB-independent action of GPR119 might exist because inactivating mutations introduced into the CRE of the PG gene promoter only modestly reduce the constitutive action of GPR119 to up-regulate Glu-Luc activity.
PG gene expression is under the control of RIIα and RIIβ PKA subunits
PG gene promoter activity in GLUTag cells is stimulated by 6-Bn-cAMP-AM, a cAMP analog that preferentially activates RIIα/β but not RIα/β isoforms of PKA regulatory subunits expressed in GLUTag cells. Because this action of 6-Bn-cAMP-AM is not reproduced by cAMP analogs that are selective activators of Epac proteins, and because this action of 6-Bn-cAMP-AM is blocked by an inhibitor of PKA activation (Rp-8-Br-cAMPS-pAB), the available evidence indicates that GPR119 signals through RIIα and/or RIIβ to up-regulate PG gene promoter activity. Proof that this is the case will require additional studies using knockdowns or knockouts of each of the 4 PKA regulatory subunits. Furthermore, the possible involvement of A-kinase anchoring proteins as RII interacting proteins should be explored in view of the fact that A-kinase anchoring proteins participate in the control of endocrine cell functions (53, 54).
Does Epac2 participate in the control of PG gene expression?
An unanticipated outcome of the present study is that Epac-selective cAMP analogs failed to stimulate PG gene promoter activity in GLUTag cells. This finding is surprising in view of prior reports that Epac2 participates in the stimulatory control of PG gene expression by cAMP in GLUTag cells (39–41). However, these prior studies are based on the use of forskolin and IBMX, rather than Epac-selective cAMP analogs such as 8-pCPT-2′-O-Me-cAMP-AM. Although one prior study of GLUTag cells documents an ability of DN Epac2 to diminish the stimulatory actions of forskolin and IBMX at the PG gene promoter (40), this effect of DN Epac2 is not dose-dependent nor is it reported that a CA Epac2 exerts an opposite effect (40). Because our analysis using DN and CA Epac2 indicates that these mutant forms of Epac2 act nonphysiologically to squelch PG gene promoter activity in GLUTag cells (Figure 6C), the findings of this prior study are drawn into question.
Another prior study reports an increase of PG mRNA content in GLUTag cells treated with Epac activator 8-pMeOPT-2′-O-Me-cAMP (39). Although we confirm this finding (Figure 7A), we find that PG gene promoter activity is not stimulated by 8-pMeOPT-2′-O-Me-cAMP (Figure 5B). Thus, 8-pMeOPT-2′-O-Me-cAMP might raise levels of PG mRNA by slowing mRNA degradation rather than by stimulating PG gene transcription. Future studies in which PG mRNA turnover is quantified under conditions of Epac2 activation seem warranted. Finally, we report that Epac2 might participate in the control of basal PG gene promoter activity since −2400 Glu-Luc activity is dose-dependently reduced under conditions in which GLUTag cells are treated with the specific Epac2 inhibitor ESI-05 (Figure 7C). However, because the pharmacological properties of ESI-05 are not yet fully defined (29), this effect of ESI-05 needs to be investigated in greater detail.
Conclusion
Recent advances in cyclic nucleotide research were exploited in the present study so as to reveal an unlikely role for Epac2 in the control of PG gene promoter activity by hGPR119. New pharmacological tools for this analysis included ESI-05, Rp-8-Br-cAMPS-pAB, and 6-Bn-cAMP-AM. Through the use of live-cell BRET assays in combination with biosensors that report PKA activation, we came to the conclusion that it is the RIIα and RIIβ subunits of PKA that mediate the action of cAMP to stimulate PG gene promoter activity. Although our QPCR assays also revealed a potential role for Epac2 in the control of PG mRNA stability, this concept remains to be substantiated. An interesting parallel exists when considering findings presented here as they relate to prior studies of GLP-1 secretion. In such studies it was demonstrated that GPR119 agonists stimulate GLUTag cell GLP-1 secretion (55), and that this occurs in a glucose-independent manner (56). Remarkably, we found that human GPR119 also acted in a glucose-independent manner to constitutively stimulate PG gene promoter activity in GLUTag cells. Thus, unlike the glucose-dependent action of GPR119 agonists to stimulate pancreatic β-cell insulin secretion (56), it could be that L-cell glucose metabolism is not a prerequisite for proglucagonotropic actions of GPR119. Future challenges are to more clearly define the signaling properties of GPR119 that are under agonist control rather than constitutive control. In particular, it remains to be established whether the G protein coupling of GPR119 is identical when assessing its ability to signal in an agonist-stimulated vs constitutive manner, or in a manner that targets GLP-1 secretion as opposed to PG gene expression. These matters are topics of interest because new evidence indicates that GPR119 signals not only through Gαs proteins, but also through the G protein α-gustducin (57–60).
Supplementary Material
Acknowledgments
We thank M. Kriebel and E. Franz for technical assistance. Plasmids for expression of PKA subunits were a kind gift of S.S. Taylor (University of California at San Diego, California). The Epac1 BRET sensor hybrid cDNA was supplied by M. Zaccolo and G. Hamilton (Balliol College, University of Oxford, United Kingdom) and was used to generate the Epac1 Rluc8 BRET sensor by A. Meyer. We thank Dr Tianru Jin (University of Toronto, Canada) for providing the Glu-Luc reporters. G.G.H. acknowledges the support of the National Institute of Health (NIH) (5R01-DK069575) and the American Diabetes Association (Basic Science Award). F.W.H. acknowledges the support of the Federal Ministry of Education and Research Project 0315449D “Modeling pain switches (MoPS)” and the European Union (EU) FP7 collaborative project Affinomics (Contract No. 241481). X.C. acknowledges the support of the NIH (5R01-GM066170). C.A.L. acknowledges the support of the American Diabetes Association (Basic Science Award).
Disclosure Summary: O.G.C., D.B., M.D., C.A.L., P.A., T.T., X.C., F.W.H., and G.G.H. have nothing to disclose. F.S. and H-G.G. are employed by the BIOLOG Life Science Institute that sells the cAMP analogs that were used in this study.
Footnotes
- AM
- acetoxymethyl
- 6-Bn-cAMP-AM
- N6-benzyladenosine-3′,5′-cyclic monophosphate acetoxymethyl ester
- BRET
- bioluminescence resonance energy transfer
- bZIP
- basic region-leucine zipper
- CA
- constitutively active
- CRE
- cAMP response element
- CREB
- cAMP response element binding protein
- Ct
- threshold crossing
- DN
- dominant-negative
- Epac2
- type-2 isoform of guanyl nucleotide exchange factor activated by cAMP
- ESI-05
- specific inhibitor of Epac2 activation
- FRET
- fluorescence resonance energy transfer
- GLP-1
- glucagon-like peptide-1
- GPCR
- G protein-coupled receptor
- HRP
- horseradish peroxidase
- MAB
- monoclonal antibody
- PG
- proglucagon
- PKA
- cAMP-dependent protein kinase
- PVDF
- polyvinyldifluoride
- QPCR
- quantitative PCR
- RFP
- red fluorescent protein
- RIP
- rat insulin I gene promoter
- TBST
- Tris-buffered saline containing 0.1% Tween-20
- WT
- wild-type.
References
- 1. Fredriksson R, Höglund PJ, Gloriam DE, Lagerström MC, Schiöth HB. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003;554:381–388 [DOI] [PubMed] [Google Scholar]
- 2. Chu ZL, Carroll C, Alfonso J, et al. A role for intestinal endocrine cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucagon-like peptide-1 and glucose-dependent insulinotropic peptide release. Endocrinology. 2008;149:2038–2047 [DOI] [PubMed] [Google Scholar]
- 3. Jones RM, Leonard JN, Buzard DJ, Lehmann J. GPR119 agonists for the treatment of type 2 diabetes. Expert Opin Ther Pat. 2009;19:1339–1359 [DOI] [PubMed] [Google Scholar]
- 4. Hansen KB, Rosenkilde MM, Knop FK, et al. 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J Clin Endocrinol Metab. 2011;96:E1409–E1417 [DOI] [PubMed] [Google Scholar]
- 5. Overton HA, Babbs AJ, Doel SM, et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167–175 [DOI] [PubMed] [Google Scholar]
- 6. Shah U, Kowalski TJ. GPR119 agonists for the potential treatment of type 2 diabetes and related metabolic disorders. Vitam Horm. 2010;84:415–448 [DOI] [PubMed] [Google Scholar]
- 7. Soga T, Ohishi T, Matsui T, et al. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem Biophys Res Commun. 2005;326:744–751 [DOI] [PubMed] [Google Scholar]
- 8. Chu ZL, Jones RM, He H, et al. A role for β-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology. 2007;148:2601–2609 [DOI] [PubMed] [Google Scholar]
- 9. Overton HA, Fyfe MC, Reynet C. GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br J Pharmacol. 2008;153(Suppl 1):S76–S81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8:369–385 [DOI] [PubMed] [Google Scholar]
- 11. Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem. 1986;261:11880–11889 [PubMed] [Google Scholar]
- 12. Drucker DJ, Brubaker PL. Proglucagon gene expression is regulated by a cyclic AMP-dependent pathway in rat intestine. Proc Natl Acad Sci USA. 1989;86:3953–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drucker DJ, Jin T, Asa SL, Young TA, Brubaker PL. Activation of proglucagon gene transcription by protein kinase-A in a novel mouse enteroendocrine cell line. Mol Endocrinol. 1994;8:1646–1655 [DOI] [PubMed] [Google Scholar]
- 14. Smit MJ, Vischer HF, Bakker RA, et al. Pharmacogenomic and structural analysis of constitutive G protein-coupled receptor activity. Annu Rev Pharmacol Toxicol. 2007;47:53–87 [DOI] [PubMed] [Google Scholar]
- 15. Chalmers DT, Behan D. The use of constitutively active GPCRs in drug discovery and functional genomics. Nat Rev Drug Discov. 2002;1:599–608 [DOI] [PubMed] [Google Scholar]
- 16. Santerre RF, Cook RA, Crisel RM, et al. Insulin synthesis in a clonal cell line of simian virus 40-transformed hamster pancreatic β cells. Proc Natl Acad Sci USA. 1981;78:4339–4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178 [DOI] [PubMed] [Google Scholar]
- 18. Gromada J, Rorsman P, Dissing S, Wulff BS. Stimulation of cloned human glucagon-like peptide 1 receptor expressed in HEK 293 cells induces cAMP-dependent activation of calcium-induced calcium release. FEBS Lett. 1995;373:182–186 [DOI] [PubMed] [Google Scholar]
- 19. Lü F, Jin T, Drucker DJ. Proglucagon gene expression is induced by gastrin-releasing peptide in a mouse enteroendocrine cell line. Endocrinology. 1996;137:3710–3716 [DOI] [PubMed] [Google Scholar]
- 20. Chepurny OG, Holz GG. A novel cyclic adenosine monophosphate responsive luciferase reporter incorporating a nonpalindromic cyclic adenosine monophosphate response element provides optimal performance for use in G protein coupled receptor drug discovery efforts. J Biomol Screen. 2007;12:740–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berlot CH. A highly effective dominant negative αs construct containing mutations that affect distinct functions inhibits multiple Gs-coupled receptor signaling pathways. J Biol Chem. 2002;277:21080–21085 [DOI] [PubMed] [Google Scholar]
- 23. Rogers KV, Goldman PS, Frizzell RA, McKnight GS. Regulation of Cl− transport in T84 cell clones expressing a mutant regulatory subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1990;87:8975–8979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uhler MD, Carmichael DF, Lee DC, Chrivia JC, Krebs EG, McKnight GS. Isolation of cDNA clones coding for the catalytic subunit of mouse cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1986;83:1300–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herbst KJ, Coltharp C, Amzel LM, Zhang J. Direct activation of Epac by sulfonylurea is isoform selective. Chem Biol. 2011;18:243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ozaki N, Shibasaki T, Kashima Y, et al. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811 [DOI] [PubMed] [Google Scholar]
- 27. Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem. 2006;281:2506–2514 [DOI] [PubMed] [Google Scholar]
- 28. Chepurny OG, Leech CA, Kelley GG, et al. Enhanced Rap1 activation and insulin secretagogue properties of an acetoxymethyl ester of an Epac-selective cyclic AMP analog in rat INS-1 cells: studies with 8-pCPT-2′-O-Me-cAMP-AM. J Biol Chem. 2009;284:10728–10736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsalkova T, Mei FC, Li S, et al. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci USA. 2012;109:18613–18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loening AM, Fenn TD, Wu AM, Gambhir SS. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel. 2006;19:391–400 [DOI] [PubMed] [Google Scholar]
- 31. Prinz A, Diskar M, Erlbruch A, Herberg FW. Novel, isotype-specific sensors for protein kinase A subunit interaction based on bioluminescence resonance energy transfer (BRET). Cell Signal. 2006;18:1616–1625 [DOI] [PubMed] [Google Scholar]
- 32. Ponsioen B, Zhao J, Riedl J, et al. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olsen SR, Uhler MD. Affinity purification of the Cα and Cβ isoforms of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1989;264:18662–18666 [PubMed] [Google Scholar]
- 34. Bertinetti D, Schweinsberg S, Hanke SE, et al. Chemical tools selectively target components of the PKA system. BMC Chem Biol. 2009;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook PF, Neville ME, Jr, Vrana KE, Hartl FT, Roskoski R., Jr Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry. 1982;21:5794–5799 [DOI] [PubMed] [Google Scholar]
- 36. Jin T, Drucker DJ. Activation of proglucagon gene transcription through a novel promoter element by the caudal-related homeodomain protein cdx-2/3. Mol Cell Biol. 1996;16:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chijiwa T, Mishima A, Hagiwara M, et al. Inhibition of foskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272 [PubMed] [Google Scholar]
- 38. Yoshida S, Ohishi T, Matsui T, Shibasaki M. Identification of a novel GPR119 agonist, AS1269574, with in vitro and in vivo glucose-stimulated insulin secretion. Biochem Biophys Res Commun. 2010;400:437–441 [DOI] [PubMed] [Google Scholar]
- 39. Lotfi S, Li Z, Sun J, et al. Role of the exchange protein directly activated by cyclic adenosine 5′-monophosphate (Epac) pathway in regulating proglucagon gene expression in intestinal endocrine L cells. Endocrinology. 2006;147:3727–3736 [DOI] [PubMed] [Google Scholar]
- 40. Islam D, Zhang N, Wang P, et al. Epac is involved in cAMP-stimulated proglucagon expression and hormone production but not hormone secretion in pancreatic α- and intestinal L-cell lines. Am J Physiol Endocrinol Metab. 2009;296:E174–E181 [DOI] [PubMed] [Google Scholar]
- 41. Wang P, Wang Q, Sun J, et al. POU homeodomain protein Oct-1 functions as a sensor for cyclic AMP. J Biol Chem. 2009;284:26456–26465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Enserink JM, Christensen AE, de Rooij J, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906 [DOI] [PubMed] [Google Scholar]
- 43. Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jin T. Mechanisms underlying proglucagon gene expression. J Endocrinol. 2008;198:17–28 [DOI] [PubMed] [Google Scholar]
- 45. Yu Z, Jin T. New insights into the role of cAMP in the production and function of the incretin hormone glucagon-like peptide-1 (GLP-1). Cell Signal. 2010;22:1–8 [DOI] [PubMed] [Google Scholar]
- 46. Vliem MJ, Ponsioen B, Schwede F, et al. 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem. 2008;9:2052–2054 [DOI] [PubMed] [Google Scholar]
- 47. Gjertsen BT, Mellgren G, Otten A, et al. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1β action. J Biol Chem. 1995;270:20599–20607 [DOI] [PubMed] [Google Scholar]
- 48. Jessen HJ, Schulz T, Balzarini J, Meier C. Bioreversible protection of nucleoside diphosphates. Angew Chem Int Ed Engl. 2008;47:8719–8722 [DOI] [PubMed] [Google Scholar]
- 49. Holst B, Nygaard R, Valentin-Hansen L, et al. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285:3973–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Valentin-Hansen L, Holst B, Frimurer TM, Schwartz TW. PheVI:09 (Phe6.44) as a sliding microswitch in seven-transmembrane (7TM) G protein-coupled receptor activation. J Biol Chem. 2012;287:43516–43526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lan H, Vassileva G, Corona A, et al. GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J Endocrinol. 2009;201:219–223 [DOI] [PubMed] [Google Scholar]
- 52. Hansen HS, Rosenkilde MM, Holst JJ, Schwartz TW. GPR119 as a fat sensor. Trends Pharmacol Sci. 2012;33:374–381 [DOI] [PubMed] [Google Scholar]
- 53. Lester LB, Langeberg LK, Scott JD. Anchoring of protein kinase A facilitates hormone-mediated insulin secretion. Proc Natl Acad Sci USA. 1997;94:14942–14947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fraser ID, Tavalin SJ, Lester LB,. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes. 2009;58:1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lan H, Lin HV, Wang CF. Agonists at GPR119 mediate secretion of GLP-1 from mouse enteroendocrine cells through glucose-independent pathways. Br J Pharmacol. 2012;165:2799–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rozengurt N, Wu SV, Chen MC, Huang C, Sternini C, Rozengurt E. Colocalization of the α-subunit of gustducin with PYY and GLP-1 in L cells of human colon. Am J Physiol Gastrointest Liver Physiol. 2006;291:G792–G802 [DOI] [PubMed] [Google Scholar]
- 58. Jang HJ, Kokrashvili Z, Theodorakis MJ, et al. Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci USA. 2007;104:15069–15074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kokrashvili Z, Mosinger B, Margolskee RF. Taste signaling elements expressed in gut enteroendocine cells regulate nutrient-responsive secretion of gut hormones. Am J Clin Nutr. 2009;90:822S–825S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Y, Kokrashvili Z, Mosinger B, Margolskee RF. Gustducin couples fatty acid receptors to GLP-1 release in colon. Am J Physiol Endocrinol Metab. 2013;304:E651–E660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Herberg FW, Dostmann WR, Zorn M, Davis SJ, Taylor SS. Crosstalk between domains in the regulatory subunit of cAMP-dependent protein kinase: influence of amino terminus on cAMP binding and holoenzyme formation. Biochemistry. 1994;33:7485–7494 [DOI] [PubMed] [Google Scholar]
- 62. Herberg FW, Taylor SS, Dostmann WR. Active site mutations define the pathway for the cooperative activation of cAMP-dependent protein kinase. Biochemistry. 1996;35:2934–2942 [DOI] [PubMed] [Google Scholar]
- 63. Herberg FW, Doyle ML, Cox S, Taylor SS. Dissection of the nucleotide and metal-phosphate binding sites in cAMP-dependent protein kinase. Biochemistry. 1999;38:6352–6360 [DOI] [PubMed] [Google Scholar]
- 64. Gibson RM, Ji-Buechler Y, Taylor SS. Interaction of the regulatory and catalytic subunits of cAMP-dependent protein kinase. Electrostatic sites on the type Iα regulatory subunit. J Biol Chem. 1997;272:16343–16350 [DOI] [PubMed] [Google Scholar]
- 65. Ringheim GE, Taylor SS. Dissecting the domain structure of the regulatory subunit of cAMP-dependent protein kinase I and elucidating the role of MgATP. J Biol Chem. 1990;265:4800–4808 [PubMed] [Google Scholar]
- 66. Diskar M, Zenn H-M, Kaupisch A, et al. Regulation of cAMP-dependent protein kinases: the human protein kinase X (PrKX) reveals the role of the catalytic subunit αH-αI loop. J Biol Chem. 2010;285:35910–35918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moll D, Prinz A, Brendel CM, et al. Biochemical characterization and cellular imaging of a novel, membrane permeable fluorescent cAMP analog. BMC Biochem. 2008;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cadd GG, Uhler MD, McKnight GS. Holoenzymes of cAMP-dependent protein kinase containing the neural form of type I regulatory subunit have an increased sensitivity to cyclic nucleotides. J Biol Chem. 1990;265:19502–19506 [PubMed] [Google Scholar]
- 69. Diskar M, Zenn HM, Kaupisch A, Prinz A, Herberg FW. Molecular basis for isoform-specific autoregulation of protein kinase A. Cell Signal. 2007;19:2024–2034 [DOI] [PubMed] [Google Scholar]
- 70. Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, Taylor SS. Structure and allostery of the PKA RIIβ tetrameric holoenzyme. Science. 2012;335:712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vetter MM, Zenn HM, Méndez E, van den Boom H, Herberg FW, Skålhegg BS. The testis-specific Cα2 subunit of PKA is kinetically indistinguishable from the common Cα1 subunit of PKA. BMC Biochem. 2011;12:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dzhura I, Chepurny OG, Kelley GG, et al. Epac2-dependent mobilization of intracellular Ca2+ by glucagon-like peptide-1 receptor agonist exendin-4 is disrupted in β-cells of phospholipase C-ϵ knockout mice. J Physiol. 2010;588:4871–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.