

Abstract
The mechanism of Rh-catalyzed (5+2) cycloadditions of 3-acyloxy-1,4-enyne (ACE) and alkynes is investigated using density functional theory calculations. The catalytic cycle involves 1,2-acyloxy migration, alkyne insertion, and reductive elimination to form the cycloheptatriene product. In contrast to the (5+2) cycloadditions with vinylcyclopropanes (VCP), in which alkyne inserts into a rhodium-allyl bond, alkyne insertion into a Rh–C(sp2) bond is preferred. The 1,2-acyloxy migration is found to be the rate-determining step of the catalytic cycle. The electron-rich p-dimethylaminobenzoate substrate promotes 1,2-acyloxy migration and significantly increases the reactivity. In the regioselectivity-determining alkyne insertion step, the alkyne substituent prefers to be distal to the forming C–C bond and thus distal to the OAc group in the product.
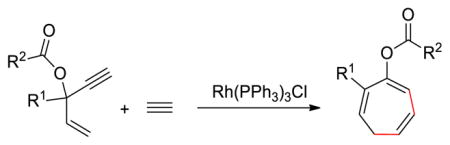
Seven-membered carbocycles have attracted increasing interests in the fields of natural product synthesis and pharmaceutical chemistry.1 In contrast to the facile synthetic routes to five- and six-membered rings, efficient cycloaddition strategies to form seven-membered rings, such as (5+2) and (4+3) reactions, are still limited.2 In 1995, Wender group reported the first transition metal-catalyzed (5+2) cycloadditions employing vinylcyclopropanes (VCPs) as the five-carbon synthon. This methodology has evolved to a general and effective route to seven-membered rings.3 A variety of (5+2) cycloadditions of VCPs and 2π systems with different transition metal catalysts were developed by Wender,4 Trost,5 Louie,6 Fürstner,7 Yu,8 Murai,9 and others.10 Stryker11 and Tanino12 also reported different types of (5+2) cycloadditions using stoichiometric amount of metals. The Houk group, along with Wender and Yu explored the detailed mechanism of the VCP cycloadditions with rhodium catalysts using computations.13 Recently, the Tang group reported a new class of (5+2) cycloadditions using 3-acyloxy-1,4-enyne (ACE) in place of VCP as five-carbon synthon.14 This methodology provides direct route to seven-membered rings with three C=C double bonds that could be selectively functionalized (Scheme 1). Both intra- and intermolecular cycloadditions have been achieved to produce various substituted cyclic compounds from ACEs and readily available terminal or internal alkynes.
Scheme 1.
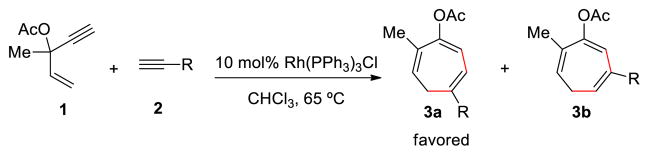
Rh-Catalyzed (5+2) Cycloadditions of 3-Acyloxy-1,4-Enyne (ACE) and Alkynes.
In contrast to the detailed computational and mechanistic studies on the (5+2) cycloadditions with VCPs,13 the mechanism of the reaction with ACE remains unclear. In previous experimental studies, we proposed a mechanism involving 1,2-acyloxy migration of ACE via possible intermediates 4–7 (Scheme 2a).15 Subsequent alkyne insertion into the rhodium–carbon bond and reductive elimination afford the cycloadduct. The (5+2) cycloadditions with VCPs involves the formation of rhodium-allyl complex 11, in close analogy to one of the possible intermediates (7) in the reaction with ACE, alkynes insertion, and reductive elimination (Scheme 2b). Although the formation of the intermediate 7 is much more complicated than that of rhodium-allyl complex 11, the latter half of the proposed mechanism in Scheme 2a appears to be similar to the (5+2) cycloaddition with VCP in Scheme 2b. The alkyne insertion into complex 11 takes place at the rhodium-allyl bond (C1), and forms the first C–C bond with the terminal alkenyl carbon of VCP. The Xu, Houk and Tang groups have now worked together to explore the mechanisms of the (5+2) cycloadditions involving the new 5-carbon building block—ACE. The DFT investigations described here reveal several fundamental differences in the nature of rate- and regioselectivity-determining steps, and the order of formation of the two new C–C bonds, when employing ACE in place of VCP as the five-carbon synthon. The change of mechanism results in different regiochemical control as well as unique substituent effects on reactivity. Here we report the first computational investigation on the mechanism and the origins of reactivities and regioselectivities of Rh-catalyzed (5+2) cycloadditions of ACE and alkynes.
Scheme 2.
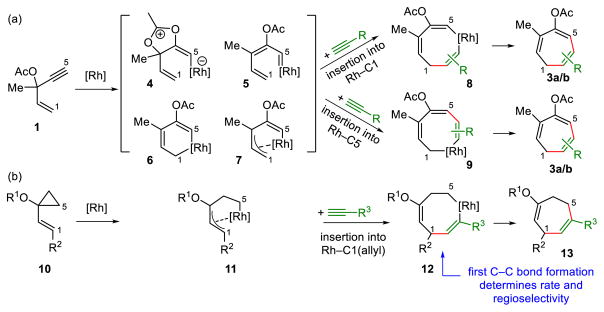
Proposed mechanisms of Rh-catalyzed (5+2) cycloadditions of (a) ACE and alkynes; (b) VCP and alkynes.
The computed Gibbs free energy profiles of several possible reaction pathways of Rh(PPh3)3Cl-catalyzed reaction of ACE and acetylene are shown in Figure 1. Geometry optimization and frequency analysis were performed in CHCl3 solvent with the SMD solvation model using B3LYP and a mixed basis set of SDD for Rh and 6-31G(d) for other atoms. Single point energies were calculated at the M06/6-311+G(d,p) level (SDD for Rh) with the SMD model (CHCl3 solvent) on B3LYP-optimized geometries. All calculations were carried out with Gaussian 09.16 Entropic contributions to the reported free energies were calculated from partition functions evaluated using Truhlar’s quasiharmonic approximation.17
Figure 1.

Gibbs free energy profile of the Rh(PPh3)3Cl-catalyzed (5+2) cycloaddition of ACE and acetylene. Energies are in kcal/mol and calculated using M06/SDD-6-311+G(d,p)/SMD(CHCl3)//B3LYP/SDD-6-31G(d)/SMD(CHCl3).
In the presence of π-acidic transition metal catalysts, terminal propargylic esters undergo 1,2-acyloxy migration, while internal propargylic esters undergo 1,3-migration.18 Previous computational studies indicated gold- and platinum-catalysts promote the 1,2-acyloxy migration of ACE via a stepwise mechanism involving intermediates similar to 4.19 We investigated the 1,2- and 1,3-acyloxy migration pathways with ACE. The reaction initiates from the ACE-Rh(PPh3)Cl π complex (14).20 With the rhodium catalyst, 1,2-acyloxy migration is a concerted process, requiring an activation free energy of 17.7 kcal/mol (TS1). In TS1, the rhodium prefers to be anti to the acyloxy group on ACE (Figure 2). The syn pathway, in which the rhodium is syn to the acyloxy, is stepwise and disfavored by 5.4 kcal/mol (TS1′). 1,3-Acyloxy migration requires a much higher activation barrier of 23.5 kcal/mol (TS2). This is consistent with the experimental observation where 1,2-acyloxy migration product is formed exclusively with ACE 1.14 The preference of 1,2-migration is also consistent with the reaction of terminal propargylic esters with other transition metal catalysts.
Figure 2.
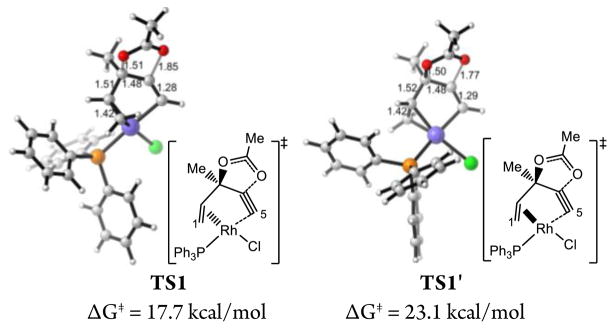
Transition states in two possible 1,2-acyloxy migration pathways.
The 1,2-acyloxy migration leads directly to a rhodium-allyl complex 15, in which PPh3 is trans to the alkenyl group (C5). Complex 15 isomerizes to a more stable rhodium-allyl intermediate 16, in which the Cl is trans to the alkenyl group. We also considered other possible intermediates proposed by Rautenstrauch and others (4–6, Scheme 1). They are all higher in energy than 15 and 16 (see SI for details).
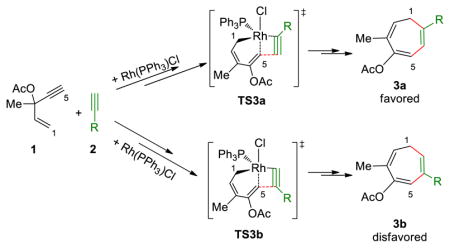
Subsequent alkyne insertion takes place from the Rh-allyl intermediate 16 via two distinct pathways: the alkyne may insert into the Rh–C5(sp2) bond (TS3) to form rhodacyclooctatriene 20, in which the three double bonds are all conjugated (pathway I), or insert into the Rh–C1(allyl) bond (TS5) to form an isomeric metallacycle 23 (pathway II). The computed activation barrier of the alkyne insertion into the Rh–C5(sp2) bond (TS3) is 7.4 kcal/mol lower than the insertion into the Rh–C1(allyl) bond (TS5, see Figure 3 for structures of the transition states). The higher activity of alkyne insertion into the Rh–C(sp2) bond is attributed to greater orbital overlap of the HOMO of the rhodium-allyl intermediate 16 and LUMO of the alkyne. As illustrated in Figure 4, the HOMO of 16 is mainly localized at the π orbital of the conjugated diene. Therefore, attack of the alkyne at the sp2 carbon results in favorable orbital overlap between the diene π orbital and alkyne π* orbital. Thus, in the (5+2) cycloaddition of ACE, the first C–C bond is formed with the alkynyl carbon of ACE and the 2 new C–C bond with the alkenyl carbon is formed later in the reductive elimination. In contrast, in the (5+2) reaction with VCPs, the alkyne preferentially inserts into the Rh-allyl bond and forms the first C–C bond with the alkenyl carbon of VCP. In the reaction with VCP, the C4-C5 bond is saturated, and the alkyne insertion into the rhodium–C(allyl) bond is favorable. The different order of bond formation in the ACE reaction suggests a different regiochemical preference for the (5+2) cycloadduct that may lead to complementary products to the reactions of VCPs. The regioselectivity will be discussed in more details later. The overall barrier of alkyne insertion via TS3 is 12.2 kcal/mol with respect to intermediate 16. This suggests alkyne insertion is faster than corresponding alkyne insertion step in the reaction with VCP. It also has lower barrier than the previous 1,2-acyloxy migration step.
Figure 3.

Optimized geometries of the alkyne insertion and reductive elimination transition states in pathway I (TS3 and TS4) and pathway II (TS5 and TS6).
Figure 4.
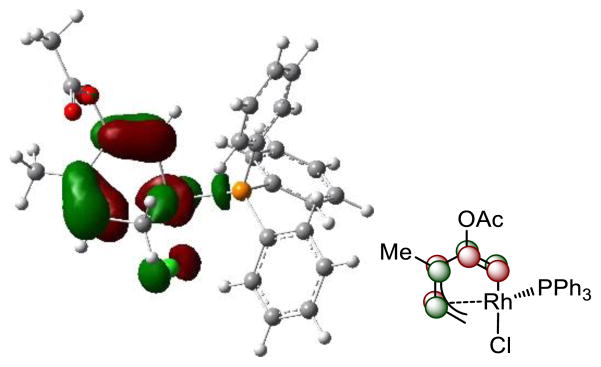
HOMO of rhodacyclohexadiene intermediate 16.
The rhodacyclooctatriene intermediate 20 subsequently undergoes C(sp2)–C(sp3) reductive elimination to form the second new C–C bond via transition state TS4, leading to the product π complex 21. This step has a free energy barrier of 12.8 kcal/mol, comparable to the alkyne insertion step. Finally, 21 liberates the product and coordinates with another reactant molecule to regenerate 14 to complete the catalytic cycle. Although pathway II requires a low barrier for reductive elimination between two sp2 carbons (23 → TS6, Δ G‡ = 3.2 kcal/mol), the high barrier of alkyne insertion (16 → TS5, Δ G‡ = 19.6 kcal/mol) ruled out this pathway in the preferred catalytic cycle.
In summary, the preferred catalytic cycle of Rh-catalyzed (5+2) cycloadditions of ACE and alkynes involves three key steps: 1,2-acyloxy migration, alkyne insertion into the Rh–C(sp2) bond in the Rh–allyl complex, and finally the C(sp2)–C(sp3) reductive elimination (pathway I, shown in black in Figure 1). The rate-determining step in the catalytic cycle is 1,2-acyloxy migration.
We then investigated the effects of substituents on the rate of the reaction, which is determined by the barrier of 1,2-acyloxy migration. Experimentally, electron-rich esters dramatically increase the reactivity of ACEs in the (5+2) cycloaddition.21 The computed free energy barriers provide good agreement with the experimental reactivities of different esters (Table 1). Apparently, the electron-donating group stabilizes the positive charge building up in the oxocyclic transition state (TS1, see SI for NPA charge analysis). The ACE without methyl substitution at C3 (entry 1, Table 1) requires significantly higher 1,2-migration barrier than other ACE substrates, in agreement with the low reactivity observed in experiment.22
Table 1.
Computed Relative Reaction Rates for (5+2) Cycloadditions of ACE Derivatives and Acetylene.a
| |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | ΔG‡ (TS1–14)b | predicted relative ratec | exp. relative ratec,d |
| 1 | H | Me | 20.0 | 0.01 | no reaction |
| 2 | Me | Me | 17.7 | 0.36 | 0.68 |
| 3 | Me | Ph | 17.1 | 1.00 | 1.00 |
| 4 | Me | p-Me2NC6H4 | 14.5 | 80.2 | 46.3 |
See SI for computed rates for more substrates.
The activation free energy (in kcal/mol) of the rate-determining 1,2-acyloxy migration step.
Rates are relative to the rate of entry 3.
Experimental relative rates from Table 1 of ref. 21.
The computed potential energy profile indicated the formation of the first C–C bond to form the rhodacyclooctatriene intermediate 20 is irreversible, and thus is the regioselectivity-determining step.23 Cycloadditions of ACE with unsymmetrically substituted alkynes may lead to two regioisomeric products due to the different orientations of alkyne during the alkyne insertion step (Table 2). We computed the differences of activation barriers of the regioisomeric alkyne insertion transition states for the reactions with propargylic alcohol 24 and tri-methylsilylacetylene 25. The predicted regioselectivities are summarized in Table 2. The regioselectivity is dominated by steric effects. In both reactions, the terminal alkyne substituent prefers to be adjacent to the metal to avoid steric repulsions around the forming C–C bond (Figure 5). This leads to the major regioisomeric product 3a, in which the alkyne substituent R is distal to the first forming C–C bond, i.e. the former terminal alkynyl carbon (C5). The agreement of predicted regioselectivities with experiment24 further validated the computed mechanism that alkyne insertion into the Rh–C(sp2) bond is preferred.
Table 2.
Computed Regioselectivities for (5+2) Cycloadditions of ACE and Substituted Alkynes.
| ||||
|---|---|---|---|---|
| entry | R | ΔΔG‡ (TS3a–TS3b)a | predicted ratio b | exp. ratiob |
| 1 | CH2OH 24 | −4.1 | >20:1 | >20:1 |
| 2 | TMS 25 | −2.4 | >20:1 | 10:1 |
Gibbs free energy differences between TS3a and TS3b in kcal/mol.
Ratio of 3a : 3b; exp. ratio from Table 7 of ref. 14b.
Figure 5.
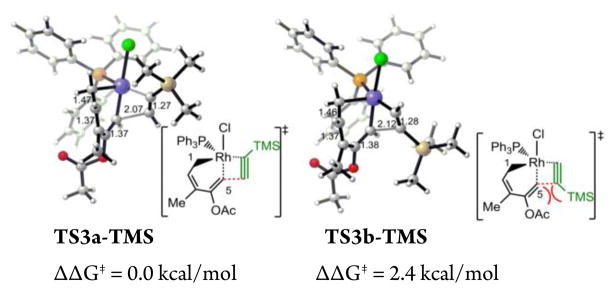
Regioisomeric alkyne insertion transition states for intermolecular (5+2) cycloaddition of ACE with TMS acetylene.
In summary, we performed DFT calculations to explore the mechanism, reactivity, and regioselectivity in Rh(PPh3)3Cl-catalyzed intermolecular (5+2) cycloaddition of ACE and alkynes. The catalytic cycle involves an unprecedented concerted 1,2-acyloxy migration to form a rhodium-allyl intermediate, alkyne insertion into the Rh–C(sp2) bond, and reductive elimination. The product regioselectivity is determined during formation of the first C–C bond in the alkyne insertion step. Bulkier alkyne substituent prefers to be distal to the first formed C–C bond. Our calculation predicts that the 1,2-acyloxy migration is the rate-determining step, which is consistent with higher reactivity experimentally observed for ACEs bearing an electron-rich benzoate. The significant differences between Rh-catalyzed cycloadditions of ACE and VCP disclosed here may have broad implications in understanding and development of transition metal-catalyzed reactions.
Supplementary Material
Acknowledgments
We are grateful to the NSF (K.N.H., CHE-1059084), NIH (W.T., R01GM088285), and the Natural Science Foundation of China (X.X., 21103094) for financial support of this research. Calculations were performed on the Hoffman2 cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF.
Footnotes
Complete reference of Gaussian 09, optimized geometries and energies of all computed species. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Battiste MA, Pelphrey PM, Wright DL. Chem Eur J. 2006;12:3438. doi: 10.1002/chem.200501083. [DOI] [PubMed] [Google Scholar]; (b) Butenschön H. Angew Chem Int Ed. 2008;47:5287. doi: 10.1002/anie.200801738. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ylijoki KEO, Stryker JM. Chem Rev. 2013;113:2244. doi: 10.1021/cr300087g. [DOI] [PubMed] [Google Scholar]; (b) Pellissier H. Adv Synth Catal. 2011;353:189. [Google Scholar]; (c) Harmata M. Chem Commun. 2010;46:8886. doi: 10.1039/c0cc03620j. [DOI] [PubMed] [Google Scholar]; (d) Harmata M. Chem Commun. 2010;46:8904. doi: 10.1039/c0cc03621h. [DOI] [PubMed] [Google Scholar]; (e) Lohse AG, Hsung RP. Chem-Eur J. 2011;17:3812. doi: 10.1002/chem.201100260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wender PA, Takahashi H, Witulski B. J Am Chem Soc. 1995;117:4720. [Google Scholar]
- 4.(a) Wender PA, Husfeld CO, Langkopf E, Love JA. J Am Chem Soc. 1998;120:1940. [Google Scholar]; (b) Wender PA, Glorius F, Husfeld CO, Langkopf E, Love JA. J Am Chem Soc. 1999;121:5348. [Google Scholar]; (c) Wender PA, Barzilay CM, Dyckman AJ. J Am Chem Soc. 2001;123:179. doi: 10.1021/ja0021159. [DOI] [PubMed] [Google Scholar]; (d) Wegner HA, De Meijere A, Wender PA. J Am Chem Soc. 2005;127:6530. doi: 10.1021/ja043671w. [DOI] [PubMed] [Google Scholar]
- 5.(a) Trost BM, Toste FD, Shen H. J Am Chem Soc. 2000;122:2379. [Google Scholar]; (b) Trost BM, Shen HC. Angew Chem Int Ed. 2001;40:2313. [PubMed] [Google Scholar]
- 6.Zuo G, Louie J. J Am Chem Soc. 2005;127:5798. doi: 10.1021/ja043253r. [DOI] [PubMed] [Google Scholar]
- 7.Fürstner A, Majima K, Martín R, Krause H, Kattnig E, Goddard R, Lehmann CW. J Am Chem Soc. 2008;130:1992. doi: 10.1021/ja0777180. [DOI] [PubMed] [Google Scholar]
- 8.(a) Jiao L, Ye S, Yu Z. J Am Chem Soc. 2008;130:7178. doi: 10.1021/ja8008715. [DOI] [PubMed] [Google Scholar]; (b) Li Q, Jiang G, Jiao L, Yu Z. Org Lett. 2010;12:1332. doi: 10.1021/ol100237h. [DOI] [PubMed] [Google Scholar]
- 9.Inagaki F, Sugikubo K, Miyashita Y, Murai C. Angew Chem Int Ed. 2010;49:2206. doi: 10.1002/anie.200906994. [DOI] [PubMed] [Google Scholar]
- 10.(a) Ashfeld BL, Miller KA, Smith AJ, Tran K, Martin SF. Org Lett. 2005;7:1661. doi: 10.1021/ol0504300. [DOI] [PubMed] [Google Scholar]; (b) Ashfeld BL, Miller KA, Smith AJ, Tran K, Martin SF. J Org Chem. 2007;72:9018. doi: 10.1021/jo701290b. [DOI] [PubMed] [Google Scholar]
- 11.(a) Dzwiniel TL, Stryker JM. J Am Chem Soc. 2004;126:9184. doi: 10.1021/ja047852+. [DOI] [PubMed] [Google Scholar]; (b) Witherell RD, Ylijoki KEO, Stryker JM. J Am Chem Soc. 2008;130:2176. doi: 10.1021/ja710568d. [DOI] [PubMed] [Google Scholar]
- 12.Tanino K, Shimizu T, Miyama M, Kuwajima I. J Am Chem Soc. 2000;122:6116. [Google Scholar]
- 13.(a) Yu Z, Wender PA, Houk KN. J Am Chem Soc. 2004;126:9154. doi: 10.1021/ja048739m. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Wang J, Su JC, Huang F, Jiao L, Liang Y, Yang D, Zhang S, Wender PA, Yu Z. J Am Chem Soc. 2007;129:10060. doi: 10.1021/ja072505w. [DOI] [PubMed] [Google Scholar]; (c) Yu Z, Cheong PHY, Liu P, Legault CY, Wender PA, Houk KN. J Am Chem Soc. 2008;130:2378. doi: 10.1021/ja076444d. [DOI] [PubMed] [Google Scholar]; (d) Liu P, Cheong PHY, Yu Z, Wender PA, Houk KN. Angew Chem Int Ed. 2008;47:3939. doi: 10.1002/anie.200800420. [DOI] [PubMed] [Google Scholar]; (e) Liu P, Sirois LE, Cheong PHY, Yu Z, Hartung IV, Rieck H, Wender PA, Houk KN. J Am Chem Soc. 2010;132:10127. doi: 10.1021/ja103253d. [DOI] [PubMed] [Google Scholar]; (f) Xu X, Liu P, Lesser A, Sirois LE, Wender PA, Houk KN. J Am Chem Soc. 2012;134:11012. doi: 10.1021/ja3041724. [DOI] [PubMed] [Google Scholar]; (g) Hong X, Liu P, Houk KN. J Am Chem Soc. 2013;135:1456–1462. doi: 10.1021/ja309873z. [DOI] [PubMed] [Google Scholar]; (h) Hong X, Trost BM, Houk KN. J Am Chem Soc. 2013 doi: 10.1021/ja4012657. [DOI] [PubMed] [Google Scholar]
- 14.(a) Shu XZ, Huang S, Shu D, Guzei IA, Tang W. Angew Chem Int Ed. 2011;50:8153. doi: 10.1002/anie.201103136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shu XZ, Li X, Shu D, Huang S, Schienebeck CM, Zhou X, Robichaux PJ, Tang W. J Am Chem Soc. 2012;134:5211. doi: 10.1021/ja2109097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Rautenstrauch V. J Org Chem. 1984;49:950. [Google Scholar]; (b) Zheng H, Zheng J, Yu B, Chen Q, Wang X, He Y, Yang Z, She X. J Am Chem Soc. 2010;132:1788. doi: 10.1021/ja910346m. [DOI] [PubMed] [Google Scholar]; (c) Garayalde D, Gómez-Bengoa E, Huang X, Goeke A, Nevado C. J Am Chem Soc. 2010;132:4720. doi: 10.1021/ja909013j. [DOI] [PubMed] [Google Scholar]
- 16.Frisch MJ, et al. Gaussian 09, revision B.01. Gaussian, Inc; Wallingford, CT: 2010. [Google Scholar]
- 17.Ribeiro RF, Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2011;115:14556. doi: 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- 18.(a) Marion N, Nolan SP. Angew Chem, Int Ed. 2007;46:2750. doi: 10.1002/anie.200604773. [DOI] [PubMed] [Google Scholar]; (b) Shu XZ, Shu D, Schienebeck CM, Tang W. Chem Soc Rev. 2012;41:7698. doi: 10.1039/c2cs35235d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Faza ON, Lopez CS, Alvarez R, de Lera AR. J Am Chem Soc. 2006;128:2434. doi: 10.1021/ja057127e. [DOI] [PubMed] [Google Scholar]; (b) Soriano E, Marco-Contelles J. Chem–Eur J. 2008;14:6771. doi: 10.1002/chem.200800305. [DOI] [PubMed] [Google Scholar]; (c) Soriano E, Marco-Contelles J. Acc Chem Res. 2009;42:1026. doi: 10.1021/ar800200m. [DOI] [PubMed] [Google Scholar]
- 20.The Rh(PPh3)3Cl catalyst first undergoes ligand exchange to eliminate two PPh3 molecules and bind to the π bonds of ACE to form 14. This step is highly exergonic by 22.1 kcal/mol. This indicates 14 is the most stable pre-reaction complex.
- 21.Schienebeck CM, Robichaux PJ, Li X, Chen L, Tang W. Chem Commun. 2013;49:2616. doi: 10.1039/c3cc40634b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R1 substitution stabilizes the Rh-allyl intermediate (15) and thus lowers the barrier of acyloxy migration. See SI for more details about the effects of R1.
- 23.After formation of 20, the forward reaction barrier to form the product (20 → TS4) is much lower than the reverse reaction to regenerate the alkyne (20 → TS3).
- 24.Computations predicted the correct major regioisomeric products for the reactions with alkynes 24 and 25. However, the regioselectivity with 25 is slightly overestimated.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.