

Abstract
Formaldehyde is produced in most living systems and is present in the environment. Evidence that formaldehyde causes cancer in experimental animals infers that it may be a carcinogenic hazard to humans. Formaldehyde reacts with the exocyclic amino group of deoxyguanosine, resulting in the formation of N2-methyl-2′-deoxyguanosine (N2-Me-dG) via reduction of the Schiff base. The same reaction is likely to occur in living cells, because cells contain endogenous reductants such as ascorbic acid and gluthathione. To explore the miscoding properties of formaldehyde-derived DNA adducts a site-specifically modified oligodeoxynucleotide containing a N2-Me-dG was prepared and used as the template in primer extension reactions catalyzed by the Klenow fragment of Escherichia coli DNA polymerase I. The primer extension reaction was slightly stalled one base before the N2-Me-dG lesion, but DNA synthesis past this lesion was readily completed. The fully extended products were analyzed to quantify the miscoding specificities of N2-Me-dG. Preferential incorporation of dCMP, the correct base, opposite the lesion was observed, along with small amounts of misincorporation of dTMP (9.4%). No deletions were detected. Steady-state kinetic studies indicated that the frequency of nucleotide insertion for dTMP was only 1.2 times lower than for dCMP and the frequency of chain extension from the 3′-terminus of a dT:N2-Me-dG pair was only 2.1 times lower than from a dC:N2-Me-dG pair. We conclude that N2-Me-dG is a miscoding lesion capable of generating G→A transition mutations.
INTRODUCTION
N2-methyl-2′-deoxyguanosine (N2-Me-dG) is one of the formaldehyde-derived DNA adducts. Formaldehyde is abundantly produced in industry and is used as a fixative and preservative for medical and biological specimens. The annual world production is apparently 12 000 000 tons. Therefore, humans are occupationally exposed to formaldehyde. Formaldehyde is also produced in most living systems, where it is derived from the metabolism of serine, choline and methionine (1). Common non-occupational sources of exposure include vehicle emissions, some building materials, food, tobacco smoke and its use as a disinfectant. Levels of formaldehyde in the indoor air of houses are in the range 0.02–0.06 mg/m3 (2,3).
Evidence that formaldehyde causes cancer in experimental animals infers that it may be a carcinogenic hazard to humans. Several studies in which formaldehyde was administered to rats by inhalation showed carcinogenicity, particularly induction of squamous cell carcinomas of the nasal cavities. In rats, administered formaldehyde in the drinking water increased incidences of forestomach papillomas, leukemias and gastrointestinal tract tumors were reported (4).
Aldehydes are known to produce DNA–protein and/or DNA–DNA crosslinks (5–8) and mainly react with dG in the exocyclic amino group producing N-alkyl-dG via an unstable Schiff base, which can be stabilized by reduction. For instance, acetaldehyde is known to form stable N2-ethyl-2′-deoxyguanosine (N2-Et-dG) (9,10). This adduct has been detected in granulocyte and lymphocyte DNA of alcoholic patients and in human buccal cells exposed to acetaldehyde (11,12). In addition, N2-Et-dG can be detected in human urine (9). Due to the structural similarities of acetaldehyde and formaldehyde, formaldehyde should produce the N2-Me-dG adduct in cells.
In this study to explore the miscoding properties of formaldehyde-derived DNA adducts site-specifically modified oligodeoxynucleotides containing a N2-Me-dG were prepared and used as DNA templates in primer extension reactions catalyzed by the Klenow fragment of Escherichia coli DNA polymerase I. Using these templates, miscoding specificities of N2-Me-dG were investigated with an in vitro experimental system that can quantify base substitutions and deletions (13) and determine steady-state kinetics. The results suggest that the N2-Me-dG adduct is a miscoding lesion capable of generating G→A transition mutations.
MATERIALS AND METHODS
Materials
[γ-32P]ATP (sp. act. >6000 Ci/mmol) was obtained from Amersham Corp. The Klenow fragment of E.coli DNA polymerase I (exo+), T4 polynucleotide kinase, T4 DNA ligase and EcoRI restriction endonuclease were purchased from Takara Shuzo (Kyoto, Japan). Micrococcal nuclease and spleen phoshodiesterase were obtained from Worthington Biochemical Corp. Alkaline phosphatase was purchased from Sigma Chemical Corp. A Waters 996 HPLC instrument equipped with a photodiode array detector was used for separation and purification of modified and unmodified oligodeoxynucleotides. NMR spectroscopy was performed on a Bruker 600 MHz instrument. Mass spectroscopy was performed on a TSQ 7000 Triple-Stage Quadrupole (Finnigan MAT, San Jose).
Synthesis of N2-Me-dGTP and N2-Me-dG
N2-Me-dGTP was purchased from Trilink Biochemical Corp. (San Diego, CA) or synthesized by ourselves using dGTP and formalin (37% formaldehyde and 10–15% methanol) as the starting materials. dGTP (3 mg/reaction) was reacted overnight with 3.7% (v/v) formalin in 1 ml of 0.2 mM sodium acetate buffer (pH 4.5) and reduced by adding 1 mg NaBH3CN (Fig. 1A). Five batches of the reactant were gathered and passed through a Sep-pak C18 cartridge (Waters). The cartridge was washed with 5 ml of 50 mM triethylamine acetate (pH 7.0) and eluted with 5 ml of methanol. The extract was evaporated to dryness and redissolved in 100 µl of pure water. The sample containing N2-Me-dGTP was fractionated by HPLC on a reversed phase Wakosil 5C18-200T column (0.46 × 25 cm; Wako), eluted over 40 min at a flow rate of 1.0 ml/min with a linear gradient of 50 mM triethylamine acetate (pH 7.0) containing 0–40% acetonitrile. The yield of N2-Me-dGTP was ∼15% of the starting material dGTP. About 10 mg of the N2-Me-dGTP was prepared and identified using 31P-NMR, 1H-NMR and ESI LC/MS. 31P-NMR and 1H-NMR spectra were recorded in D2O using orthophosphoric acid and 3-(trimethylsilyl)propionic acid-d4 sodium salt as the internal standards, respectively. N2-Me-dGTP: 31P-NMR (D2O) δ (p.p.m.), 4.50 (d, J = 18.3 Hz), 10.05 (d, J = 18.2 Hz), 19.66 (t); 1H-NMR (D2O) δ (p.p.m.), 2.51 (m, 1H, 2′-Ha), 2.86 (m, 1H, 2′-Hb), 2.97 (s, 3H, NHCH3), 4.20 (m, 2H, 5′-H), 4.26 (m, 1H, 4′-H), 4.80 (overlapping with DOH, 3′-H), 6.41 (t, 1H, J = 6.8 Hz, 1′-H), 8.08 (s, 1H, 8-H). Using negative ion ESI LC/MS/MS the base peak was at m/z 520 (M–1).
Figure 1.

(A) Formation of N2-Me-dGTP via the Schiff base. dGTP was reacted with formaldehyde in sodium acetate buffer, pH 4.5, and reduced by NaBH3CN as described in Materials and Methods. (B) HPLC separation of N2-Me-dGTP. N2-Me-dGTP was isolated from dGTP on a reversed phase Wakopak Wakosil 5C18-200T column (0.46 × 25 cm) using a linear gradient of 50 mM triethylamine acetate, pH 7.0, containing 0–40% acetonitrile over 40 min and a flow rate of 1.0 ml/min.
N2-Me-dG was prepared essentially by the same method as described above except that dG was the starting material instead of dGTP. About 1 mg of N2-Me-dG was prepared and identified using 1H-NMR and ESI LC/MS/MS. N2-Me-dG: 1H-NMR, δ 10.49 (brs, NH), 7.89 (s, H8), 6.30 (brs, 1H, J = 5.2 Hz, N2H), 6.17 (t, J = 7.2 Hz, 1′-H), 5.26 (d, J = 3.6 Hz, 3′-OH), 4.85 (t, J = 6.0 Hz, 5′-OH), 4.36 (m, 3′-H), 3.81 (m, 4′-H), 3.55 and 3.50 (m, 5′-H), 3.30 (d, -CH3), 2.20 and 2.60 (m, 2′-H). Me2SO-d6 solution was measured with tetramethylsilane as the internal standard. Using positive ion ESI LC/MS/MS the parent ion was at m/z 282 (M+1). This molecular ion gave a daughter ion at m/z 166 (base+1).
Synthesis and purification of oligodeoxynucleotides
DNA template, primers and standard markers, listed in Table 1, were purchased from Amersham Corp. These were purified on a reversed phase Wakopak WS-DNA column (0.46 × 25 cm; Wako) eluted over 60 min at a flow rate of 1.0 ml/min with a linear gradient of 50 mM triethylamine acetate (pH 7.0) containing 10–20% acetonitrile. DNA templates and primers were further purified by electrophoresis on a 20% polyacrylamide gel (20 × 40 × 0.035 cm) in the presence of 7 M urea. The oligomers recovered from PAGE were again subjected to HPLC to remove urea. Oligomers were labeled at the 5′-terminus by treatment with T4 polynucleotide kinase in the presence of [γ-32P]ATP and subjected to electrophoresis to establish homogeneity.
Table 1. Sequences of oligodeoxynucletidesa.
| Number |
Sequence |
| 1 | 5′-CATGCTGATGAATTCCTTC-3′ |
| 2 | 5′-CATGCTGATGAATTCCTTCX-3′ |
| 3 | 5′-CTTCTTTCCTCTCCCTTT-3′ |
| 4 | 5′-CATGCTGATGAATTCCTTCXCTTCTTTCCTCTCCCTTT-3′ |
| 5 | 5′-AAAGGGAGAGGAAAGAAGCGAAGGAATTCATCAGCATG-3′ |
| 6 | 5′-AGGAAAGAAGCGAAGGAATTCATC-3′ |
| 7 | 5′-AGAGGAAAGA-3′ |
| 8 | 5′-AGAGGAAAGAAG-3′ |
| 9 | 5′-AGAGGAAAGAAGN-3′ |
| 10 | 5′-AGAGGAAAGAAGNGAAGG-3′ |
| 11 | 5′-AGAGGAAAGAAGGAAGG-3′ |
| 12 | 5′-AGAGGAAAGAAGAAGG-3′ |
| 13 | 5′-AGAGGAAAGAAGNGAAGGAATTCATCAGCATG-3′ |
aSequences of template, primers and standard markers. X = N2-Me-dG or dG; N = dC, dA, dG or dT.
The 38mer modified template (5′-CATGCTGATGAATTCCTTCGMeCTTCTTTCCTCTCCCTTT, GMe = N2-Me-dG) containing a single N2-Me-dG was prepared by enzymatic incorporation of N2-Me-dGTP using the 19mer oligodeoxynucleotide (5′-CATGCTGATGAATTCCTTC) and the Klenow fragment of E.coli DNA polymerase I following ligation to an 18mer (5′-CTTCTTTCCTCTCCCTTT) with T4 DNA ligase, as shown in Figure 2.
Figure 2.

Diagram of preparation of N2-Me-dG-modified 38mer template.
To synthesize a 20mer (sequence 2 in Table 1, X = N2-Me-dG) containing N2-Me-dG at the 3′-terminus, N2-Me-dGTP (2 µg) was mixed with a 19mer primer (1 µg, sequence 1 in Table 1) fully annealed to a 38mer template (10 µg, sequence 5 in Table 1). The mixture was incubated with Klenow fragment (0.1 U) at 25°C for 45 min in 50 µl of 10 mM Tris–HCl (pH 7.5) containing 7 mM MgCl2 and 0.1 mM dithiothreitol (DTT). The reaction was stopped by heating the sample to 95°C for 5 min. Samples were subjected to electrophoresis on a 20% polyacrylamide gel containing 7 M urea (20 × 40 × 0.035 cm). The N2-Me-dG-modified 20mer recovered from PAGE was again subjected to HPLC to remove urea. Four micrograms of 18mer (sequence 3 in Table 1) was phosphorylated at the 5′-terminus for 40 min at 37°C using T4 kinase (35 U) and 2 mM ATP in 50 µl of 50 mM Tris–HCl (pH 8.0) containing 10 mM MgCl2 and 5 mM DTT and subsequently ligated to N2-Me-dG-modified 20mer (2 µg, sequence 2 in Table 1) at 8°C overnight using T4 DNA ligase (1050 U), 0.3 mM ATP and a 24mer template (6 µg, sequence 6 in Table 1) in 50 µl of 66 mM Tris–HCl (pH 7.6) containing 6.6 mM MgCl2 and 10 mM DTT. The resultant N2-Me-dG 38mer was also isolated by HPLC and its length was confirmed by PAGE.
Enzymatic digestion
A 20mer oligodeoxynucleotide containing N2-Me-dG at the 3′-terminus (sequence 2 in Table 1, X = N2-Me-dG) was digested to deoxynucleoside 3′-monophosphates at 37°C for 3 h in 20 µl of 17 mM sodium succinate (pH 6.0) containing 8 mM CaCl2 using micrococcal nuclease (3 U) and spleen phoshodiesterase (0.1 U). The reaction mixture was further incubated at 37°C for 2 h with alkaline phosphatase (3 U) in 120 µl of 8 mM Tris–HCl (pH 8.5) containing 0.8 mM zinc sulfate. The mixture was evaporated to dryness and the resultant deoxynucleosides were extracted twice each with 300 µl of methanol. Methanol extracts were evaporated to dryness, dissolved in distilled water and analyzed by HPLC, using a Mightysil RP-18 GP Aqua column (0.46 × 25 cm; Kanto, Japan). Elution was carried out using a linear gradient of 50 mM sodium phosphate buffer (pH 6.0) containing 0–40% acetonitrile over 30 min at a flow rate of 1.0 ml/min.
Primer extension reactions
The primer (5′-AGAGGAAAGA) was labeled at the 5′-terminus by treatment with T4 polynucleotide kinase in the presence of [γ-32P]ATP. Using a 32P-labeled 10mer (0.5 pmol) primed with a 38mer template (0.75 pmol, 5′-CATGCTGATGAATTCCTTCXCTTCTTTCCTCTCCCTTT, X = dG or N2-Me-dG), primer extension reactions catalyzed by Klenow fragment (exo+) were carried out at 20°C for 1 h in 10 µl of buffer containing all four dNTPs (100 µM each). The reaction buffer consisted of 50 mM Tris–HCl (pH 8.0), 8 mM MgCl2 and 5 mM 2-mercaptoethanol. Reactions were stopped by adding formamide dye and heating to 95°C for 3 min. Samples were subjected to electrophoresis on a 20% polyacrylamide gel containing 7 M urea (20 × 40 × 0.035 cm). Bands were identified by autoradiography and the radioactivities were measured with a FLA-2000 instrument (Fuji Photo Film Corp., Japan).
Quantitation of miscoding specificity
Reaction of a 38mer template (0.75 pmol, sequence 4 in Table 1) and a 32P-labeled 10mer primer (0.5 pmol, sequence 7 in Table 1) was catalyzed by Klenow fragment (exo+) and carried out at 20°C for 1 h in the presence of all four dNTPs. Reactions were stopped by heating at 95°C for 3 min and then subjected to electrophoresis on a 20% polyacrylamide gel containing 7 M urea (20 × 40 × 0.035 cm). Fully extended products were recovered from the gel and then cleaved with EcoRI after annealing with an unmodified 38mer (sequence 4). Samples were incubated with EcoRI (100 U) for 1 h at 30°C and then for 1 h at 15°C to produce complete cleavage, as shown in Figure 3. Electrophoresis was carried out on two-phase 20% polyacrylamide gels (20 × 40 × 0.035 cm) containing 7 M urea in the upper phase and no urea in the lower phase (13). Standards representing products containing dC, dA, dG or dT opposite the lesion (sequence 10) or one and two base deletions (sequences 11 and 12) were completely resolved on the gel, based on their different migrations.
Figure 3.

Diagram of the primer extension methodology and analysis of the reaction products.
Kinetic studies of nucleotide insertion and chain extension
Kinetic parameters associated with nucleotide insertion opposite the lesion and chain extension from the primer 3′-terminus were determined in reactions containing a single dNTP (13–15). Reaction mixtures containing varying amounts (0.004–0.04 U) of Klenow fragment were incubated at 25°C for 1.0–10.0 min in 10 µl of Tris–HCl (pH 8.0) containing 1.0 pmol of 38mer template (sequence 4 in Table 1) primed with 0.5 pmol of 32P-labeled 12mer (5′-AGAGGAAAGAAG) to measure nucleotide insertion kinetics or with a 32P-labeled 13mer (5′-AGAGGAAAGAAGN, N = C, A, G or T) to measure chain extension kinetics. Base insertion was measured in reactions using 0.004 U Klenow fragment for 1 min (C:G, C:GMe and T:GMe), 0.01 U for 10 min (T:G) or 0.04 U for 10 min (other pairs).
Reaction mixtures were subjected to electrophoresis on 20% polyacrylamide gels (20 × 40 × 0.035 cm) in the presence of 7 M urea. The Michaelis constant (Km) and maximum rate of reaction (Vmax) were obtained from Hanes–Woolf plots of the kinetic data. Frequencies of insertion (Fins) and extension (Fext) were determined relative to the dC:dG base pair according to the equation developed by Mendelman et al. (14,15), where F = (Vmax/Km)[wrong pair]/(Vmax/Km)[correct pair = dG:dC] is defined for any base pair containing a single modified nucleoside. All reactions were linear over the course of the experiment. Data reported represent the averages of two to four separate experiments in which <20% of the primer was extended (14).
RESULTS
Preparation of site-specifically modified oligonucleotide containing a single N2-Me-dG
When dGTP was reacted with formalin and reduced by NaBH3CN, N2-Me-dGTP (tR = 14.8 min) could be completely isolated from dGTP (tR = 12.5 min) by HPLC (Fig. 1B). The peak (tR = 16.3 min) eluting after N2-Me-dGTP has not been identified.
N2-Me-dG-modified 20mer oligodeoxynucleotide (sequence 2 in Table 1, X = N2-Me-dG) was prepared by enzymatic incorporation of N2-Me-dGTP as described in Materials and Methods. The N2-Me-dG-modified 20mer was separable from unmodified 20mer by HPLC, and no contamination of the unmodified 20mer was observed. The N2-Me-dG-modified 20mer and unmodified 20mer (sequence 2 in Table 1, X = dG) were digested enzymatically and subjected to HPLC (Fig. 4A and B). The UV spectrum of the product detected at 19.2 min was identical to that of N2-Me-dG (Fig. 4C). The obtained molar ratio of dC:dG:dT:dA:dGMe estimated from UV absorbance (5.0:3.5:7.4:4.4:1.0) was almost consistent with the theoretical value (5:3:7:4:1) of the nucleoside composition of the 20mer. Moreover, the N2-Me-dG-modified 20mer was characterized by the use of negative ion ESI LC/MS/MS. As shown in Figure 5, the spectrum of the N2-Me-dG-modified 20mer (mol. wt 6097) exhibited multivalent ion peaks from 6 to 9 electric charges at m/z 1015.17, 870.09, 765.89 and 676.19, respectively (Fig. 5A). The observed molecular weight of the N2-Me-dG-modified 20mer was determined to be 6097.0 Da using deconvolution of the mass spectrum, which is also consistent with the theoretical value (Fig. 5B).
Figure 4.

HPLC elution profiles of enzyme digests of unmodified and N2-Me-dG-modified 20mer. Three micrograms of unmodified (A) or N2-Me-dG-modified (B) 20mer (5′-CATGCTGATGAATTCCTTCX, X = dG or N2-Me-dG) was digested to nucleosides as described in Materials and Methods. Methanol extracts of the digested samples were evaporated to dryness and placed on a Mightysil RP-18 GP Aqua column (0.46 × 25 cm; Kanto). Elution was carried out using a linear gradient of 50 mM sodium phosphate buffer, pH 6.0, containing 0–40% acetonitrile over 40 min at a flow rate of 1.0 ml/min. (C) UV spectrum of N2-Me-dG and dG.
Figure 5.

(A) Mass spectrum of N2-Me-dG-modified 20mer. A gas sheath flow of 60 p.s.i. (483 kPa) nitrogen, an electrospray voltage of –4.5 kV, a tubelens voltage of –114 V and a capillary temperature of 175°C were used. (B) Deconvolution spectrum of the N2-Me-dG-modified 20mer. Using BioWorks software (Finnigan MAT) the molecular weight of N2-Me-dG-modified 20mer was calculated from the mass spectrum data.
The characterized N2-Me-dG-modified 20mer was ligated to an 18mer (sequence 3 in Table 1), resulting in formation of a 38mer containing a single N2-Me-dG (Fig. 6A). The ligation product was subjected to HPLC (Fig. 6B). Fractions 1–3 were collected and subjected to PAGE containing 7 M urea. This revealed that fr-1 (tR = 28.1 min), fr-2 (tR = 30.9 min) and fr-3 (tR = 33.5 min) were 24mer, 18mer and N2-Me-dG-modified 38mer (ligation product), respectively. No peak of N2-Me-dG-modified 20mer was observed at its retention time (tR = 29.5 min, indicated by an arrow in Fig. 6B), indicating that most of the modified 20mer was ligated to the 18mer, resulting in formation of the N2-Me-dG-modified 38mer oligonucleotide. Since we used very pure oligonucleotides, in terms of both HPLC and PAGE analysis, and we used a ligation method to make the N2-Me-dG-modified 38mer, contamination with undesired sequences was unlikely to occur. Moreover, there was no contamination with unmodified 38mer when checked by HPLC (modified and unmodified 38mer are separable by HPLC; an example of this separation is shown in Fig. 7).
Figure 6.

(A) Diagram of the method of ligation of N2-Me-dG-modified 20mer with 18mer. (B) HPLC elution profile after ligation. fr-1 and fr-2 were 24mer and 18mer, respectively. The N2-Me-dG-modified 20mer (tR = 29.5 min) was completely consumed by the ligation reaction (no peak at 29.5 min). The desired N2-Me-dG-modified 38mer was fr-3.
Figure 7.

HPLC trace of N2-Me-dG-modified and unmodified oligomers. The two 38mers were separable on a reversed phase Wakopak WS-DNA column (0.46 × 25 cm) using a linear gradient of 50 mM triethylamine acetate, pH 7.0, containing 5–20% acetonitrile over 60 min and a flow rate of 1.0 ml/min.
Primer extension reactions
Primer extension reactions catalyzed by Klenow fragment (exo+) were carried out in the presence of four dNTPs using unmodified or N2-Me-dG-modified 38mer templates (sequence 4 in Table 1). As shown in Figure 8, primer extension on an unmodified template rapidly produced a single fully extended product (Fig. 8, lanes 1–4). Using N2-Me-dG-modified template the primer extension reaction was slightly stalled one and two bases before the lesion (Fig. 8, lanes 5–7). A small amount of 12mer (8.2% of the starting primer) and 11mer (9.3%) was observed after 60 min incubation (lane 7), but DNA synthesis past this lesion was readily completed.
Figure 8.

Primer extension reactions catalyzed by Klenow fragment (exo+). Using unmodified or N2-Me-dG-modified 38mer template (sequence 4 in Table 1) primed with a 5′-32P-labeled 10mer (sequence 7 in Table 1) primer extension reactions were conducted at 20°C using 0.004 U Klenow fragment as described in Materials and Methods. One-third of the reaction mixture was subjected to denaturing 20% PAGE. 13× shows the location opposite the N2-Me-dG lesion.
Quantitation of base substitutions
Fully extended reaction products were analyzed by two-phase gel electrophoresis (13) to determine the miscoding specificities of N2-Me-dG. As shown in Figure 9, a standard mixture of 32P-labeled oligodeoxynucleotides containing dC, dA, dG or dT at the position of the lesion and one or two base deletions served as standard markers for these experiments (Fig. 9, lane 2). DNA synthesis on an unmodified template led to the expected incorporation of dCMP opposite dG at position 13 (Fig. 9, lane 1). The full-length product represents 97.3% of the starting primer. When an N2-Me-dG-modified template was used (Fig. 9, lane 3) dCMP (84.0%) and dTMP (9.4%) were incorporated opposite the N2-Me-dG lesion. Incorporation of dAMP and dGMP was not observed. In addition, no deletions were detected.
Figure 9.
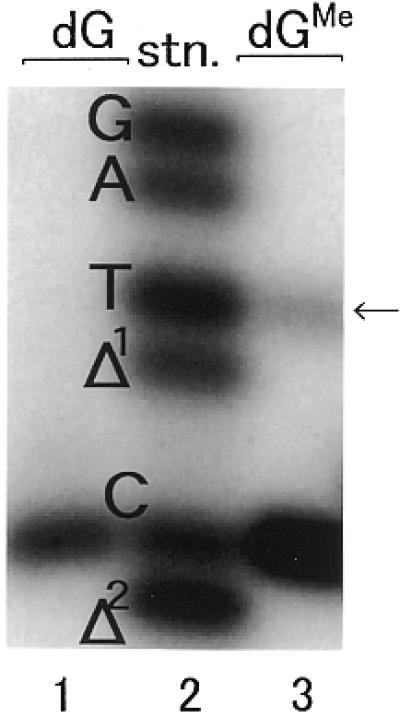
Quantitation of miscoding specificities induced by Klenow fragment (exo+). Using a 38mer template (sequence 4 in Table 1) primed with a 5′-32P-labeled 10mer (sequence 7) primer extension reactions were conducted at 20°C for 1 h using 0.004 U Klenow fragment for unmodified and N2-Me-dG-modified templates. The fully extended products recovered by PAGE were cleaved by EcoRI restriction enzyme at 30°C for 1 h and subsequently at 15°C for 1 h as described in Materials and Methods. The reaction samples were subjected to two-phase 20% PAGE. Mobilities of reaction products were compared with those of 18mer standards (sequences 10–12) containing dC, dA, dG or dT opposite the lesion and one base (Δ1) or two base (Δ2) deletions (lane 2).
Kinetic studies of nucleotide insertion and extension
Steady-state kinetic parameters were established for nucleotide insertion opposite N2-Me-dG and for chain extension from 3′-termini containing this lesion using Klenow fragment (exo+). The frequencies of nucleotide insertion (Fins) and extension (Fext) were defined as the ratio Vmax/Km relative to that of the correct base pair (dCTP opposite dG in the template). As shown in Table 2, Fins for dCTP opposite N2-Me-dG was 0.704 and for dTTP opposite N2-Me-dG was 0.582. Fins for dTTP opposite N2-Me-dG was 730 times higher than Fins for dTTP opposite unmodified dG. The frequency of chain extension (Fext) from 3′-termini containing a dC:N2-Me-dG pair was 0.795 and from those containing a dT:N2-Me-dG pair was 0.381. Fext from 3′-termini containing a dT:N2-Me-dG pair was 9.1 times higher than that from a dT:dG pair. The relative frequency of translesional synthesis (Fins × Fext) past N2-Me-dG in reactions involving dT was 0.222.
Table 2. Kinetic parameters for the nucleotide insertion and chain extension reactions catalyzed by Klenow fragmenta.
| N:X | Insertion | Extension | Fins×Fext | ||||
| dNTP | dGTP | ||||||
| ↓GAAGAAAGGAGA32P | ↓NGAAGAAAGGAGA32P | ||||||
| 5′-CCTTCXCTT
CTT TCC TCT CCCTTT |
|
|
5′-CCTTCXCTT
CTT TCC TCT CCCTTT |
|
|
||
| |
Km (µM) |
Vmax (% min–1) |
Fins |
Km (µM) |
Vmax (% min–1) |
Fext |
|
| C:dG | 1.76 ± 0.7b | 49.3 ± 10 | 1.0 | 5.31 ± 0.09 | 106 ± 5.4 | 1.0 | 1.0 |
| C:N2-Me-dG | 2.10 ± 0.3 | 41.3 ± 1.2 | 0.704 | 6.17 ± 0.7 | 98.1 ± 2.4 | 0.795 | 0.56 |
| T:dG | 51.3 ± 10.5 | 1.17 ± 0.3 | 7.96 × 10–4 | 18.5 ± 3.9 | 15.6 ± 2.1 | 4.20 × 10–2 | 3.34 × 10–5 |
| T:N2-Me-dG | 2.40 ± 0.4 | 39.1 ± 0.7 | 0.582 | 10.8 ± 2.7 | 82.6 ± 2.7 | 0.381 | 0.222 |
| A:dGc | 89.0 ± 37.3 | 0.82 ± 0.18 | 1.31 × 10–4 | 21.5 ± 9.6 | 0.10 ± 0.01 | 9.16 × 10–5 | 1.20 × 10–8 |
| A:N2-Me-dG | 71.9 ± 17.3 | 0.49 ± 0.20 | 2.44 × 10–4 | 96.7 ± 8.6 | 0.15 ± 0.08 | 7.85 × 10–5 | 1.92 × 10–8 |
| G:dGb | 25.1 ± 1.7 | 3.10 ± 0.42 | 1.53 × 10–3 | 8.64 ± 2.78 | 0.74 ± 0.02 | 1.42 × 10–3 | 2.17 × 10–6 |
| G:N2-Me-dG | 26.1 ± 1.4 | 2.68 ± 0.85 | 3.68 × 10–3 | 40.8±9.8 | 2.16 ± 0.17 | 2.65 × 10–3 | 9.75 × 10–6 |
aKinetics of nucleotide insertion and chain extension reactions were determined as described in Materials and Methods. Frequencies of nucleotide insertion (Fins) and chain extension (Fext) were estimated by the equation: F = (Vmax/Km)[wrong pair]/(Vmax/Km)[correct pair]. X = dG or N2-Me-dG lesion.
bData are expressed as means ± SD.
cData are taken from Shibutani et al. (16).
DISCUSSION
The site-specifically modified 38mer template containing a single N2-Me-dG can be prepared by the enzymatic incorporation of N2-Me-dGTP into the 3′-terminus of a 19mer oligomer following enzymatic ligation to another 18mer oligomer. Our preliminary experiments indicate that N2-Me-dGTP was well utilized as a substrate during DNA synthesis catalyzed by Klenow fragment. The insertion of N2-Me-dGTP opposite template dC with this enzyme was almost equal to that of dGTP. In addition, most of the N2-Me-dG-modified 20mer was ligated to an 18mer, resulting in formation of a 38mer containing a single N2-Me-dG. This shows that T4 DNA ligase was able to efficiently execute its function even at the N2-Me-dG site. This is probably because the N2-Me-dG:dC pair does not significantly change the conformation of the DNA duplex, although the guanine N2 position is responsible for base pairing via a hydrogen bond.
The miscoding properties of N2-Me-dG observed during translesional synthesis in reactions catalyzed by Klenow fragment were analyzed by two-phase gel electrophoresis. Preferential targeted incorporation of dCMP, the correct base, was observed. In addition, 9.4% misincorporation of dTMP was detected opposite the N2-Me-dG lesion, generating G→A transitions.
The miscoding specificities of methylated DNA adducts such as 8-methyl-2′-deoxyguanosine (8-Me-dG) (17) and O6-methyl-2′-deoxuguanosine (O6-Me-dG) (13) have been determined using the same in vitro experimental system. 8-Me-dG is a weak miscoding lesion, generating G→C (1.1%) or G→T (0.41%) transitions or deletions (1.2%). In contrast, O6-Me-dG is a strong miscoding lesion, generating primarily G→A transitions (75.5%). The position of methylation on dG adducts significantly influences the miscoding specificity and frequency.
Analysis of full-length reaction products should reflect relative rates of nucleotide insertion opposite the lesion. This assumption was confirmed by steady-state kinetic analyses, in which the frequency of nucleotide insertion opposite N2-Me-dG and chain extension from the primer 3′-terminus were measured in the presence of a single dNTP. With Klenow fragment Fins for dTTP was only 1.2 times lower than for dCTP and Fext from a dT:N2-Me-dG terminus was only 2.1 times lower than from dC:N2-Me-dG. The relative translesional synthesis (Fins × Fext) for the dT:N2-Me-dG pair is only 2.5-fold lower than that for the dC:N2-Me-dG pair.
It is instructive to compare the miscoding properties of N2-Me-dG with those of another endogenously generated DNA lesion, 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxo-dG) (18). As shown in Table 3, Fins × Fext for dCTP and dATP opposite 8-oxo-dG are 0.0116 and 0.025, respectively. Fins × Fext for dCTP and dTTP opposite N2-Me-dG are 0.56 and 0.222, respectively, which is much higher than that of 8-oxo-dG. This shows that Klenow fragment bypasses the N2-Me-dG lesion more easily than 8-oxo-dG. In addition, Fins × Fext for dTTP opposite N2-Me-dG is ∼9 times higher than that for dATP opposite 8-oxo-dG. Thus, N2-Me-dG is considered a more mutagenic lesion than 8-oxo-dG.
Table 3. Comparison of translesional synthesis of N2-Me-dG with 8-oxo-dG.
| N:X |
Fins |
Fext |
Fins × Fext |
| C:N2-Me-dG | 0.704 | 0.795 | 0.56 |
| T:N2-Me-dG | 0.582 | 0.381 | 0.222 |
| C:8-oxodGa | 0.35 | 0.033 | 0.0116 |
| A:8-oxodGa | 0.05 | 0.5 | 0.025 |
aData are taken from Shibutani et al. (18)
In a preliminary experiment utilization of N2-Me-dGTP opposite dN (dC, dA, dG or dT) embedded in templates during DNA synthesis catalyzed by Klenow fragment (exo+) was analyzed. Incorporation of N2-Me-dGTP opposite template dC and dT was observed (unpublished data), consistent with this study. Thus, N2-Me-dG tends to couple with dT. In chain extension from dT:dG, a mismatch base pair, most of the dT at the 3′-terminus was excised by Klenow fragment, but not from dT:N2-Me-dG. dT excision from dT:N2-Me-dG was markedly less than that from dT:dG. These results indicate that addition of a -CH3 moiety to the N2 position of dG promotes misincorporation of dTMP, resulting in dT:N2-Me-dG base pairing, and evasion of the 3′→5′ exonuclease function of Klenow fragment in chain extension from dT:N2-Me-dG.
Rife et al. showed that N2-methylguanosine can form base pairs with C, U and A in RNA molecules in vitro (19). However N2-Me-dG paired with dC and dT but not with dA in this study. The incorporation rate (Fins × Fext) of dATP opposite N2-Me-dG was very low (Table 2). Thus, although thermodynamically N2-methylguanine can pair with adenine, the enzymatic reaction data show that N2-Me-dG does not pair with dA during DNA synthesis.
In this study, using a N2-Me-dG-modified template, the miscoding specificities of N2-Me-dG were investigated using an in vitro experimental system that can quantify base substitutions and steady-state kinetics. The results suggest that the formaldehyde-derived DNA adduct N2-Me-dG is a miscoding lesion capable of generating G→A transition mutations.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in part by The Japanese Ministry of Education, Science, Sports and Culture (Grants in Aid for Scientific Research 09750636 and 12055101) and the National Institute of Environmental Health Sciences (Grants ES09418 and ES04068).
References
- 1. IPCS (1989) Formaldehyde. Environmental Health Criteria 89. World Health Organization, Geneva, pp. 11–176.
- 2.Spengler M.D. and Sefton,K. (1983) Indoor air pollutants: a public health perspective. Science, 221, 9–17. [DOI] [PubMed] [Google Scholar]
- 3. International Agency for Research on Cancer (1995) IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans: Wood Dust and Formaldehyde. IARC, Lyon, France.
- 4. International Agency for Research on Cancer (1986) IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans: Tobacco Smoking. IARC, Lyon, France.
- 5.Monticello T.M., Miller,F.J. and Morgan,K.T. (1991) Regional increases in rat nasal epithelial cell proliferation following acute and subchronic inhalation of formaldehyde. Toxicol. Appl. Pharmacol., 111, 409–421. [DOI] [PubMed] [Google Scholar]
- 6.Heck H.D.A., Casanova,M. and Starr,T.B. (1990) Formaldehyde toxicity—new understanding. Crit. Rev. Toxicol., 20, 397–426. [DOI] [PubMed] [Google Scholar]
- 7.Feron V.J., Til,H.P., de Vrijer,F., Woutersen,R.A., Cassee,F.R. and van Bladeren,P.J. (1991) Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat. Res., 259, 363–385. [DOI] [PubMed] [Google Scholar]
- 8.Chaw Y.F.M., Crane,L.E., Lange,P. and Shapiro,R. (1980) Isolation and identification of cross-links from formaldehyde-treated nucleic acids. Biochemistry, 19, 5525–5531. [DOI] [PubMed] [Google Scholar]
- 9.Matsuda T., Terashima,I., Matsumoto,Y., Yabushita,H., Matsui,S. and Shibutani,S. (1999) Effective utilization of N2-ethyl-2′-deoxyguanosine triphosphate during DNA synthesis catalyzed by mammalian replicative DNA polymerases. Biochemistry, 38, 929–935. [DOI] [PubMed] [Google Scholar]
- 10.Vaca C.E., Fang,J.L. and Schweda,E.K.H. (1995) Studies of the reaction of acetaldehyde with deoxynucleosides. Chem. Biol. Interact., 98, 51–67. [DOI] [PubMed] [Google Scholar]
- 11.Fang F.L. and Vaca,C.E. (1997) Detection of DNA adducts of acetaldehyde in peripheral white blood cells of alcohol abusers. Carcinogenesis, 18, 627–632. [DOI] [PubMed] [Google Scholar]
- 12.Vaca C.E., Nilsson,J.A., Fang,J.L. and Grafstrom,R.C. (1998) Formation of DNA adducts in human buccal epithetial cells exposed to acetaldehyde and methylglyoxal in vitro. Chem. Biol. Interact., 108, 197–208. [DOI] [PubMed] [Google Scholar]
- 13.Shibutani S. (1993) Quantitation of base substitutions and deletions induced by chemical mutagens during DNA synthesis in vitro. Chem. Res. Toxicol., 6, 625–629. [DOI] [PubMed] [Google Scholar]
- 14.Mendelman L.V., Boosalis,M.S., Petruska,J. and Goodman,M.F. (1989) Nearest neighbor influences on DNA polymerase insertion fidelity. J. Biol. Chem., 264, 14415–14423. [PubMed] [Google Scholar]
- 15.Mendelman L.V., Petruska,J. and Goodman,M.F. (1990) Base mispair extension kinetics. J. Biol. Chem., 265, 2338–2346. [PubMed] [Google Scholar]
- 16.Shibutani S., Takeshita,M. and Grollman,A.P. (1997) Translesional synthesis on DNA templates containing a single abasic site. J. Biol. Chem., 272, 13916–13922. [DOI] [PubMed] [Google Scholar]
- 17.Kohda K., Tsunomoto,H., Minoura,Y., Tanabe,K. and Shibutani,S. (1996) Synthesis, miscoding specificity and thermodynamic stability of oligodeoxynucleotide containing 8-methyl-2′-deoxyguanosine. Chem. Res. Toxicol., 9, 1278–1284. [DOI] [PubMed] [Google Scholar]
- 18.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 19.Rife J., Cheng,C., Moore,P.B. and Strobel,S.A. (1998) N2-methylguanosine is iso-energetic with guanosine in RNA duplexes and GNRA tetraloops. Nucleic Acids Res., 26, 3640–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]