

Abstract
A small number of proteins, called hubs, have high connectivity and are essential for interactome functionality and integrity. Keap1 is a crucial hub in the oxidative stress response and apoptosis. The Kelch domain of Keap1 preferentially binds to disordered regions of its partners, which share similar binding motifs, but have a wide range of binding affinities. Isothermal titration calorimetry (ITC) and multi-microsecond molecular dynamics (MD) simulations were used to determine the factors that govern the affinity of all currently known disordered binding partners to Kelch. Our results show that the affinities to this hub are largely determined by the extent of preformed bound state-like conformation in the free state structures of these disordered targets. Based on our findings, we have designed a high-affinity peptide that can specifically disrupt the Keap1-NRF2 interaction and has the potential for therapeutic applications.
Characterization of protein interaction networks is essential for understanding the functions of individual proteins, signaling pathways and their interconnections1,2. Some proteins have higher connectivity relative to others and they are commonly referred to as hubs3,4. Hub proteins are essential for interactome functionality and stability; therefore, it is not surprising that their disruptions in the human interactome are frequently associated with diseases4,5.
The structural plasticity of intrinsically disordered proteins (IDPs) allows them to interact with numerous different targets. It is also common for well-structured hub proteins to preferentially interact with disordered partners6. In contrast to protein-protein interactions between well-folded proteins, which usually involve larger interaction surfaces that are discontinuous in sequence, IDPs typically bind to targets using short (~6 residues) consecutive stretches of amino acid residues called linear motifs (LMs)7,8. These motifs often have distinct sequence characteristics with the primary difference being an increased hydrophobic content9, which may promote local structure formation10,11,12,13. Accurate detection of LMs and detailed characterization of their structures or conformational propensities would provide unique insight into the relationships between sequence and structure.
Here, we used a combined experimental and computational strategy to examine the interactions between the well-folded Kelch domain hub, from Keap1 (Kelch-like ECH-associated protein 1), and all of its identified (to date) disordered partners (Fig. 1a). NRF2 is arguably the most well known target of Keap1. The interaction of these proteins is critical for regulating the cellular anti-inflammatory oxidative stress responses14. The C-terminal Kelch domain of Keap1, which adopts a ~32 kDa β-propeller structure, is responsible for mediating the interaction with the Neh2 domain of NRF2. Structural studies have shown that Neh2 is intrinsically disordered15. It has high and low affinity ‘ETGE' and ‘DLG' containing Kelch domain binding regions, respectively, which will be referred to as sites 1 and 2, respectively hereafter15,16. These sites are located at separate ends of the ~100 residue Neh2 domain, connected by a segment with high helical propensity15. When both sites are bound to two separate Kelch domains of a Keap1 dimer, NRF2 is ubiquitinated, which targets it for proteasomal degradation. When only site 1 is bound, NRF2 avoids the degradation pathway and can promote expression of its target genes16. In addition to NRF2, at least 10 other proteins have been shown to interact with the Kelch domain of Keap1 to date. These include WTX17, p6218, PGAM519, PALB220, FAC121, PTMA22, IKKβ23 and BCL224. Several of these partners have been identified to disrupt the low affinity site 2-Kelch domain interaction, allowing NRF2 to promote cytoprotective gene expression. Most of the Kelch domain interacting proteins contain sequences resembling the site 1 motif of the Neh2 domain (Fig. 1a) and will be referred to as site 1-type proteins hereafter. The one exception is BCL2, which contains a site 2-type sequence and will be referred to as a site 2-type protein (Fig. 1a). Although not included in this study, as it does not appear to be directly linked to the oxidative stress response or apoptosis, myosin-VIIa has also been shown to interact with Kelch25. The region of human myosin-VIIa capable of binding Kelch includes a site 1-type sequence 1635LDHDTGE1641.
Figure 1. Sequence analysis of the Kelch domain interacting proteins.
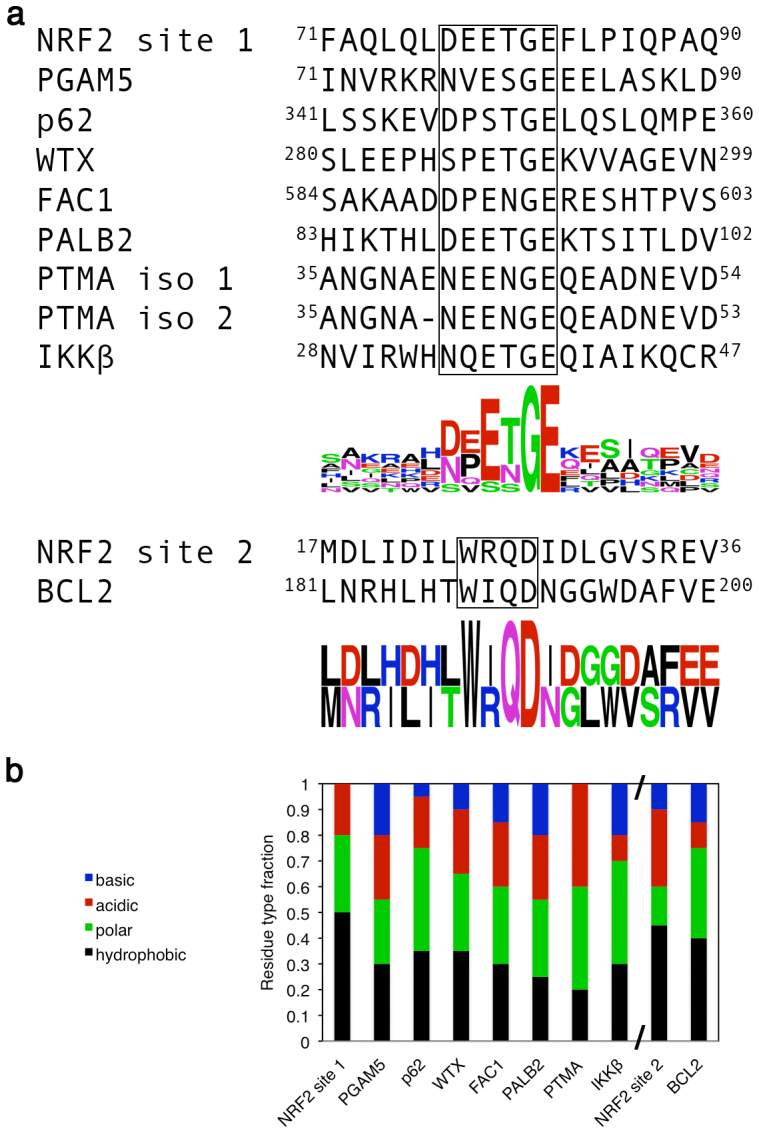
a) Manual sequence alignment and sequence WebLogos of the site 1- and site 2-type regions of the Kelch domain binding proteins60,61. B) Residue type fractions of the sequences. A, F, I, L, M, P, V, W and Y are hydrophobic; C, G, N, Q, S and T are polar; D and E are acidic; K, H and R are basic.
Through sequence analysis, disorder predictions, binding parameter measurements and MD simulations, the factors that govern the binding affinities and specificities of the different IDP-Kelch interactions were determined. Our findings were also used to design a new higher affinity Kelch-binding peptide.
Results
Kelch domain interacting proteins are predominantly disordered
Even though a number of targets of the Kelch domain of Keap1 have been identified, structural information of only a few of them is currently available. NMR studies show that the sites 1 and 2 Kelch binding regions of Neh2 are located in disordered regions15. Similarly, the site 1-type binding motif of PTMA have also been found to be largely unstructured in its free state26. Based on the amino acids sequences of the various proteins (Fig. 1a), intrinsic disorder in the Kelch domain binding regions of the site 1-type binders may be a common attribute. Predictions by PONDR-FIT27, a meta-predictor of intrinsically disordered amino acids, show that for NRF2, p62, WTX, FAC1, PALB2, and PTMA, their site 1-type Kelch binding motifs are all located in long stretches of disordered regions (Supplementary Fig. 1). PGAM5 and IKKβ were the only site 1-type proteins with disorder scores < 0.5. Based on the prediction, the Kelch binding region of PGAM5 is located on the border of the N-terminal disordered region and C-terminal structured domain. Meanwhile, the binding site of IKKβ is predicted to be in a structured part of the protein (Supplementary Fig. 1), which is consistent with the homology model28 that illustrates that IKKβ is a well-folded protein (Supplementary Fig. 2). For the site 2-type binders, NRF2 and BCL2 were predicted to have disorder tendencies of 0.40 and 0.07, respectively (Supplementary Fig. 1). The predicted low disorder tendency of BCL2 was not surprising because its binding region is found in a well-folded part of the protein29. Overall, the results suggest that while both intrinsically disordered and well-folded protein segments are able to bind to the Kelch domain, the majority of the partners (identified to date) are disordered, particularly around the site 1-type binding regions.
Sequence and structure comparison of the Kelch domain interacting proteins
Sequence comparison shows that the site 1-type proteins share high sequence similarity in a 6-residue stretch corresponding to the ‘DEETGE' of NRF2 site 1 (Fig. 1a). These residues comprise the Kelch domain binding interface30 and will be referred to as positions i – i + 5. Notably, G and E always occupy positions i + 4 and i + 5, respectively. E is found at i + 2 in all of the proteins, except p62, which contains an S at this position. The other positions are more variable. Outside of this 6-residue stretch, there are no clear sequence similarities between the different site 1-type proteins (Fig. 1a). On the other hand, the two site 2-type proteins have a short, 4-residue, ‘WXQD' consensus region. Like the site 1-type proteins, these two proteins do not share apparent sequence consensus outside of this short motif (Fig. 1a).
The hydrophobic content amongst the site 1-type binders varied considerably between 20–50%, with the NRF2 site 1 having the highest fraction of hydrophobic residues, followed by p62/WTX, PGAM5/FAC1/IKKβ, PALB2 and PTMA. Meanwhile, acidic content fell into a narrower range of 10–25% for all proteins, except PTMA, which had a considerably higher fraction of 40% (Fig. 1b). For the site 2-type sequences, NRF2 and BCL2 have similar amounts of hydrophobic content between 40–45%, whereas the latter has considerably higher polar and less acidic content (Fig. 1b).
Crystal structures of p62, PTMA and Neh2 peptides in complex with the Kelch domain are currently available. The structures show that the site 1-type regions of these three proteins all bind to the same site on Kelch18,22,31. Further, in their bound states, the PTMA and p62 peptides both adopt β-hairpin structures with low backbone rmsd (<0.3 Å Cα rmsd for 8 atom pairs) and similar sidechain conformations to the NRF2 site 1 peptide bound to the Kelch domain18,22,31 (Supplementary Fig. 2 and Supplementary Table 1). Notably, even though the structure of Kelch-IKKβ complex is currently not available, the free-state structure of IKKβ reveals that the Kelch domain binding region of this protein28 also forms a β-hairpin that has considerable resemblance (~0.5 Å Cα rmsd for 8 atom pairs) to the bound state structure of the NRF2 site 1 peptide (Supplementary Fig. 2). When comparing the site 2-type binders, it is clear that in its free state, the ‘WIQD' sequence of BCL229 has considerable structural resemblance (<0.5 Å Cα rmsd for 4 atom pairs) to the ‘WRQD' sequence of the NRF2 site 2 peptide bound to Kelch32. These residues appear to adopt a ‘turn' conformation and share similar backbone, but not χ1, dihedral angles (Supplementary Fig. 2 and Supplementary Table 1). Intriguingly, although the site 1- and 2-type Kelch domain interacting proteins do not have obvious sequence similarities, the residues that are largely buried in the Kelch domain binding interface (EE in NRF2 site 1 and PTMA, PS in p62, QE in IKKβ and QD in NRF2 site 2 and BCL2) all have similar ϕ and ψ angles (Supplementary Table 1).
Binding parameters of the Kelch domain interacting proteins
With the aim to identify the mechanisms by which the Kelch domain binds to different disordered targets, we have determined the thermodynamic parameters of binding (Table 1) for peptides encoding the binding regions of site 1-type Kelch domain interacting proteins with high disorder tendencies. For systematic comparisons, peptides of equal length (20 amino acids) were used and the experiments were performed using the same buffer conditions.
Table 1. Thermodynamic parameters for the binding of the peptides to the human Kelch domaina.
| Protein | nb | Kdc (10−6 M) | ΔHc (kcal/mol) | TΔSc (kcal/mol) | ΔGc (kcal/mol) |
|---|---|---|---|---|---|
| NRF2 site 1 | 1.08 | 0.023 ± 0.002 | −16.96 ± 0.05 | −6.56 | −10.40 ± 0.03 |
| NRF2 site 1 E78P | 0.99 | 0.007 ± 0.001 | −16.76 ± 0.05 | −5.64 | −11.12 ± 0.03 |
| PALB2 | 1.01 | 0.087 ± 0.007 | −19.29 ± 0.11 | −9.66 | −9.63 ± 0.05 |
| PGAM5 | 1.07 | 0.23 ± 0.01 | −19.12 ± 0.06 | −10.05 | −9.07 ± 0.03 |
| WTX | 1.04 | 0.25 ± 0.02 | −18.04 ± 0.09 | −9.03 | −9.01 ± 0.04 |
| FAC1 | 0.99 | 1.1 ± 0.05 | −15.11 ± 0.08 | −6.98 | −8.13 ± 0.04 |
| p62 | 0.97 | 1.3 ± 0.02 | −18.38 ± 0.07 | −10.34 | −8.04 ± 0.03 |
| WTX pS286 | 0.98 | 1.5 ± 0.14 | −10.83 ± 0.14 | −2.88 | −7.95 ± 0.10 |
| PTMA iso 2 | 1.05 | 2.62 ± 0.05 | −17.29 ± 0.06 | −9.69 | −7.60 ± 0.03 |
| PTMA iso 1 | 1.07 | 11.6 ± 0.23 | −14.74 ± 0.12 | −8.02 | −6.72 ± 0.05 |
aThe peptide sequences are shown in Fig. 1a.
bBinding stoichiometry.
cKd is the dissociation constant. ΔH, ΔS and ΔG are the change in enthalpy, entropy and Gibbs free energy upon binding at T = 298.15 K, respectively.
Despite having similar Kelch-binding motifs, a large variation in the binding affinity (Kd ranging from ~12 μM to 23 nM) was observed for different disordered Kelch domain interacting proteins (Table 1 and Supplementary Table 2 and Supplementary Fig. 3). Among these binders, the NRF2 site 1 peptide displays the highest affinity to Kelch, with a Kd of 23 ± 2 nM. The value agrees well with what has been measured for a 16-mer peptide (~20 nM)30. It is interesting that PALB2, which contains the same ‘LDEETGE' sequence as NRF2 site 1, was ~4-fold weaker. Our ITC data showed that while the interaction between the PALB2 peptide and Kelch domain is enthalpically more favorable than that for the NRF2 site 1 peptide, it lost considerable entropy upon binding. Compared to NRF2 site 1, PALB2 has less hydrophobic content in its binding region, which may hamper local folding and allow for more conformational freedom. This could possibly explain the more significant entropy loss of PALB2 upon binding, resulting in a weaker interaction. The unphosphorylated WTX peptide and PGAM5 bind to Kelch with similar affinity. However, upon phosphorylation of S286, the binding affinity of WTX was substantially decreased by ~6-fold (Table 1). The affinity of the p62 peptide for the Kelch domain was on par with the reported value of 1851 ± 103 nM for a (mouse) fragment containing residues 168–39118. The high similarity between these measurements indicates that regions distant from the binding regions may not contribute to the interaction. The ~5 fold lower affinity of PTMA isoform 1 compared to isoform 2 (Table 1) was intriguing. The two isoforms have nearly identical sequences, with the only difference being a deletion of E at position i − 1 in isoform 2 (Fig. 1a). Factors that contribute to the difference in binding affinity are not clear, but we speculate that having amino acids with the same charge (E40 and E48) in isoform 1 somewhat close to each other in a β-turn conformation is unfavorable. Similarly, the D589 and E597 pair in FAC1 would also be in close proximity, assuming a β-turn conformation is adopted.
Free state structures of Kelch domain interacting peptides
To help identify factors that cause the large variation in binding affinity, MD simulations were performed to assess the relationships between peptide conformation in the free and bound states of the various Kelch domain interacting peptides. The results show that most peptides had free state contacts that were generally suggestive of turn or hairpin formations, with the turns occurring at the Kelch binding site (Figs. 2,3,4 and Supplementary Figs. 4–6).
Figure 2. Cαi-Cαi+3 1/r3 averaged distances from the MD simulations.
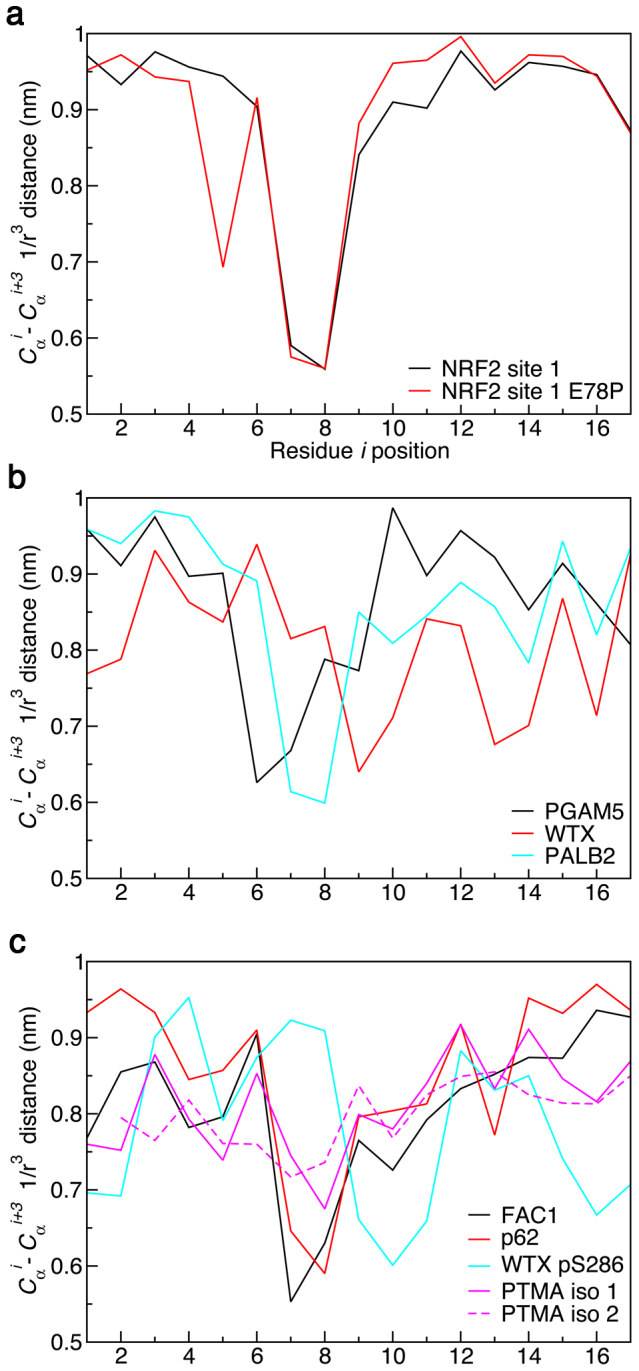
The 1/r3 averaged distances were calculated using the g_rmsdist tool in GROMACS 4.5 over the last 0.5 μs of the trajectories.
Figure 3. Cluster analysis of the peptide structures.

The center structures and percentages of structures in the highest population cluster are shown. Snapshots at 0.4 ns intervals from the 1 μs trajectories were clustered based on the rmsd of all Cα atoms using a 0.15 nm cutoff33,34. The Kelch binding ‘DEETGE' like regions (Fig. 1a) are indicated in atomistic detail. The mainchain (N, Cα, C′, and CO) rmsd of the atoms from the ‘DEETGE' like regions (Fig. 1a) were compared to the corresponding atoms of the bound state NRF2-Kelch structure (PDB id: 2FLU)30: NRF2, 0.04 nm; NRF2 E78P 0.08 nm; PALB2, 0.10 nm; PGAM5, 0.21 nm; WTX, 0.24 nm; p62, 0.19 nm; FAC1, 0.17 nm; WTX pS286, 0.24 nm; PTMA isoform 1, 0.12 nm; PTMA isoform 2, 0.23 nm.
Figure 4. Uncomplexed to complexed state comparison.

For each peptide, 6 residues, including 24 mainchain (N, Cα, C′, and CO) atoms from the ‘DEETGE' like regions (Fig. 1a) were compared to the corresponding atoms of the bound state NRF2-Kelch structure (PDB id: 2FLU)30. Snapshots of the peptide conformations were rendered with VMD62.
While most of the peptides had a region of compactness at their Kelch binding regions (Fig. 2), there were some variations. For instance, it was clear that the region of compactness in the WTX pS286 simulation was shifted from its expected location, towards the C-terminal end of the peptide, relative to the unphosphorylated version (Figs. 2b and c). The MD data indicates that interactions between the pS286 residue and a lysine on the opposite side of the turn could potentially be the cause of the turn distortion (Fig. 2 and Supplementary Fig. 5). In our previous MD simulations, we found that phosphorylation of T80 in an NRF2 peptide severely inhibited formation of the expected β-hairpin structure10. In the case of WTX, it appears that S286 phosphorylation may actually enhance free state structure formation. This is supported by our ITC data (Table 1), which revealed that the binding of WTX pS286 peptide to Kelch had the smallest entropy change. However, this peptide interacted with the Kelch domain with the least favorable enthalpy, which suggests that the peptide conformation induced by phosphorylation is not optimal for binding.
The conformational ensembles and bound state resemblance of the peptides was examined by cluster analysis and rmsd comparison to the NRF2 site 1 bound state33,34. It was apparent that NRF2 adopted a relatively stable bound-state-like conformation with the majority of structures occupying a single cluster (Fig. 3 and 4). Bound-state-like structures formation was more infrequent in the other peptides and their largest clusters were less populated, relative to NRF2 (Fig. 3 and 4). Secondary structure analysis by DSSP further illustrates that turn or hairpin formations at the Kelch binding regions were more transient for PALB2, WTX, FAC1 and PTMA (Supplementary Fig. 6).
The MD simulations also provide insights into factors that stabilize the binding regions of the peptides. For instance, in addition to cross-strand hydrophobic contacts, electrostatic interactions between oppositely charged residues and hydrogen bonding pairs in cross-strand locations were commonly observed in the simulations (Supplementary Figs. 4 and 5).
NMR experiments were also performed on the peptides to assess their free state structures. Small 1Hα chemical shifts deviations from the random coil values, and narrow range of amide 1H peak dispersions (~1 ppm) suggest that the peptides do not adopt stable structures in solution (Supplementary Table 3). NOESY cross-peaks between protons > 2 residues apart were evident in some of the peptides (Supplementary Fig. 4) and these contacts were consistent with turn formation at the Kelch binding motifs. The NRF2 site 1 peptide clearly had the largest number of NOESY cross-peaks of all the peptides. Several of these cross-peaks were between residues comprising the β-turn motif that forms the binding interface with the Kelch domain. The NOEs span about the same area as documented for the full-length mouse Neh2 domain15. NOESY cross-peaks between residues in i and i + 3 positions were also found in the PGAM5 and p62 peptides (Supplementary Fig. 4). Even though NOESY cross-peaks between residues > 2 residues apart were not clearly observed for the WTX, FAC1, PALB2 and PTMA peptides, the presence of such contacts cannot be ruled out. Incomplete resonance assignments and overcrowding made analysis of these spectra challenging. It is also important to note that the observed NOEs > 2 residues apart were weak-moderate for all peptides. This, along with near random coil chemical shifts for most protons suggest low folded populations in general. While the NMR results are consistent with the MD findings, further experimental work is necessary for a comprehensive comparison.
Overall, the MD simulations reveal that several, if not all, of the Kelch domain binding peptides analyzed contain β-turn like LMs at their binding sites. These structures likely display resemblance to their bound state conformations to certain extents. It is anticipated that preformed structures are important features in regulating the binding affinities and other thermodynamic parameters of the different interactions.
Binding affinity correlations
Because all experiments and simulations were conducted with the same conditions for each peptide, it was possible to assess correlations between the affinities of binding and their physical properties (Fig. 5). A good correlation (r2 = 0.77) was found between Kyte Doolittle hydropathy index (higher values indicates more hydrophobic content in the sequence) and the free energy of binding (ΔG) (Fig. 5a). This analysis helps to gauge how hydrophobic residues flanking the common binding motif may stabilize free state structure and affect binding affinity. The two main outliers in this correlation are the p62 and WTX pS286 peptides. For p62, this deviation is probably due to the lack of an E in position i + 2. The overestimated affinity of the WTX pS286 peptide is understandable considering the Kyte Doolittle scale does not include values for phosphorylated amino acids35. Although phosphoserine was assigned the maximum negative value on the scale (−4.5), a more negative value is probably appropriate.
Figure 5. Correlations between ΔG of binding and hydropathy, disorderness, and circular variances.

ΔG values were plotted against Kyte Doolittle hydropathy indexes35 (a), average circular variances (b), combined circular variances and Kyte Doolittle indexes (c), and PONDR-FIT disorder predictions (d) of the peptides shown in Fig. 1. For the WTX pS286 peptide, phosphoserine was assigned the maximum hydrophilic value on the scale (−4.5). The circular variances were calculated over the last 0.5 μs of the MD trajectories. Disorder predictions were performed on the full-length sequences and the average values for the segments in Fig. 1a were plotted. For the WTX pS286 peptide, phosphoserine was changed to glutamic acid.
Circular variance values, a measure of the amplitude of backbone dihedral angle fluctuations, extracted from the MD simulations, also correlate well (r2 = 0.75) with ΔG (Fig. 5b). The trend clearly illustrates that the binding affinity is heavily determined by the free state dynamics. By combining hydropathy indices with circular variances (Fig. 5c), the correlation was moderately improved (r2 = 0.81). Interestingly, the binding affinities correlate to a lesser extent (r2 = 0.50) to the PONDR-FIT disorder tendencies (Fig. 5d). The major outlier here is PGAM5, with an average disorder probability of 0.36 for the 20-mer region of its sequence (Fig. 5d).
A higher affinity Kelch domain interacting peptide
Based on the findings from our analysis of the site 1-type proteins, a peptide with a higher affinity for the Kelch domain than any of the natural peptides (identified to date) was generated. This peptide aided our interpretation and understanding of the thermodynamics of interaction with the Kelch domain and, even more importantly, it may be a potential therapeutic agent36. Based on the results obtained here and in our previous work11, we hypothesize that by restricting a peptide to sample preferentially the bound state like conformation, the entropic cost of binding may be reduced, potentially increasing binding affinity. Therefore, our attempt at designing a higher affinity peptide focused on reducing free state entropy.
MD data indicated that β-turn formation at the Kelch domain binding sites is a common feature of the various peptides, therefore, we aimed to increase the turn propensity of this region. The likely sites of turn formation in the site 1-type binders are the ‘DEET'-like regions11 (Fig. 1a). The residue-specific and overall turn potentials of the ‘DEET'-like regions of the various proteins show that the high turn potentials of p62, FAC1 and WTX result primarily from their sequences containing proline at position i + 1 (Supplementary Table 5). Based upon this analysis, a single point mutation, E78P, was made to the natural, 20-mer NRF2 site 1 peptide (Fig. 1a). This single mutation increases the turn potential to 6.13, possibly enriching the population of molecules with defined structure in solution. ITC measurement showed that the E78P mutation indeed increases the binding affinity of the peptide (7 ± 1 nM) compared to the natural sequence by 3–4 fold (Table 1). Notably, this increase in binding affinity arises primarily due to a decreased entropic cost of binding. MD simulations were then used to examine the free state structures of the E78P peptide. The data confirms that the E78P peptide is able to adopt a hairpin structure with similar cross-strand contacts, region of compactness and bound state resemblance as the natural peptide (Fig. 2,3,4 and Supplementary Fig. 4–6).
Discussion
Using a combined experimental and computational approach, we have revealed factors that govern the binding affinity and specificity of different disordered partners to the Kelch hub. Our findings provide insights into the biological roles of the various protein-protein interactions and importantly, led to the design of a high affinity Kelch-binding peptide, which may have therapeutic purposes.
We have grouped the proteins based on their binding affinities to Kelch as this may be helpful in deciphering the relationships between binding affinity and the biological functions of the various protein-protein interactions. Tier 1 consists of NRF2 only, which is the master regulator of the cellular oxidative stress response pathway14,37. Tier 2 consists of PALB2, PGAM5 and WTX, which have Kd's in the ~100–200 nM range. These proteins have been shown to promote NRF2-mediated cytoprotective gene expression, by presumably, disrupting the low affinity (Kd of ~1 μM) site 2-Kelch domain interaction16. The last group of proteins, consisting of FAC1, p62 and PTMA, have dissociation constants > 1000 nM. PTMA contains a nuclear localization signal, and is thought to function as a vehicle for shuttling Keap1 into the nucleus38. The transient nature of its shuttling role may explain its lower affinity. It should be noted that while the proteins discussed here interact with the Kelch domain of Keap1, the purpose of many of the interactions are not well established. The binding parameters reported here and hypothesis presented in several recent review articles may give insights into their possible roles39,40.
The development of higher affinity Kelch domain ligands, which can compete with NRF2, is an area of active research. Several NRF2 inducers are currently in development or clinical trials for the treatment of chronic kidney disease, diabetes, cancer prevention, multiple sclerosis and oxidative tissue damage41,42,43,44,45. However, many of these compounds do not disrupt the NRF2-Kelch domain interaction directly. Compounds, such as our E78P peptide, which specifically bind to the NRF2 binding site on the Kelch domain can be alternative therapeutic agents. Head-to-tail cyclization or attachment of our E78P peptide to a cell-penetration peptide may further improve this peptide as a drug candidate63.
In conclusion, our findings suggest that intrinsic disorder coupled with a preformed β-turn resembling LM located at the binding site is a common feature among many of the Kelch domain interacting proteins. The LMs are differentially stabilized by intramolecular contacts flanking and in close proximity to the Kelch binding region. The extent of motif stabilization is likely an important factor in modulating binding affinity. We found that the hydropathic indices and free state dynamics of the peptides were well correlated with the measured free energy of binding. These parameters will be useful for predicting the affinities of other possible Kelch domain interactions. Based on this knowledge, we have selectively mutated the turn region of the NRF2 site 1 peptide to increase its binding affinity by reducing the conformational freedom of the free state. Importantly, this modified higher affinity peptide may have potential therapeutic applications. Our results provide insight into the biological roles of the various Kelch domain interactions and development of specific NRF2 inducers.
Methods
Protein purification and peptide synthesis
The Kelch domain of human Keap1 (residues 321–609), subcloned into the pET15b expression vector (from Dr. Mark Hannink, University of Missouri-Columbia), was expressed as an N-terminally His-tagged protein in Escherichia coli BL21 (DE3) and grown in minimal M9 medium. Protein expression was induced by addition of 0.5 mM IPTG at 18°C for 24 h. The protein was purified from the crude cell lysate by affinity chromatography using Ni Sepharose™ 6 Fast Flow beads (Amersham Biosciences). The His-tag was then cleaved by incubation with human α-thrombin (Haematologic Technologies Inc.) overnight at 4°C. The Kelch domain was purified from the cleavage mixture using a HiLoad Superdex-75 size-exclusion column (GE Healthcare) equilibrated with 50 mM sodium phosphate buffer, 100 mM NaCl, 1 mM DTT at pH 7.
Isothermal titration calorimetry (ITC) experiments
ITC experiments were carried out on a VP-ITC instrument (MicroCal) at 25°C. The protein and peptide samples were dialyzed into a buffer containing 50 mM sodium phosphate, 100 mM NaCl, 1 mM DTT at pH 7 and degassed before the experiments. ~40 μM Kelch was added to the 1.4 mL sample cell and subjected to stepwise titration with 5 μL aliquots of ~500 μM peptide. Concentrations of Kelch and peptides were determined by amino acid analysis (Advanced Protein Technology Centre, The Hospital for Sick Children, Toronto, ON). The dissociation constant (Kd), molar binding stoichiometry (n) and the binding enthalpy (ΔH), entropy (ΔS) and Gibbs free energy (ΔG) were determined by fitting the binding isotherm to a single-binding-site model with Origin7 software (MicroCal). All ITC experiments were performed in duplicate.
MD simulations
The amino acid sequences of all simulated peptides were the same as those used in the ITC experiments (Fig. 1a & Supplementary Table 4). We used the Crystallography & NMR System (CNS)46 to generate an extended structure from each sequence. Simulated annealing was performed on each structure and resulting conformations that did not resemble the Neh2 domain site 1 region bound to the Kelch domain (PDB id: 2FLU) were used as starting structures30. The N- and C-terminus of each structure was capped with acetyl (ACE) and NH2 groups, respectively, using Chimera47. For the WTX peptide with S286 phosphorylated (WTX pS286), a dianionic phosphate group (PO42−) was modeled onto S286 of the non-phosphorylated WTX peptide structure.
The MD simulations were performed using GROMACS (GROningen MAchine for Chemical Simulations) version 4.548. GROMOS96 53a6 force field49,50 was used (Except in the WTX pS286 peptide simulation, where the GROMOS96 43a1p51 force field was used). This force field has been extensively tested and shown to perform well in simulations of IDPs10,11. The starting structures were solvated in cubic boxes of linear size of 6 nm with periodic boundary conditions applied in all directions. The SPC (simple point charge) water model was used52. Protonation states of all ionizable residues were chosen based on their most probable state at pH 7. Histidine residues were protonated on ND1 only (a set of trajectories using the NE2 only histidine protonation state were highly similar; data for these simulations is shown in Supplementary Fig 7). Each system was overall charge-neutral and brought to an ionic strength of 0.1 M with sodium (Na+) and chloride (Cl−) ions. The simulations followed the protocols established in10,11,53,54 to avoid physical artifacts. Protein and non-protein atoms were coupled to their own temperature baths at 310 K using the Parrinello-Donadio-Bussi v-rescale algorithm55. Pressure was maintained isotropically at 1 bar using the Parrinello-Rahman barostat56. A 2 fs timestep was used. Prior to the simulations, the energy of each system was minimized using the steepest descents algorithm. This was followed by 2 ps of position-restrained dynamics with all non-hydrogen atoms restrained with a 1000 kJ mol−1 force constant. Initial atom velocities were taken from a Maxwellian distribution at 310 K. All bond lengths were constrained using the LINCS algorithm57. A 1.0 nm cut-off was used for Lennard-Jones interactions. Dispersion corrections for energy and pressure were applied. Electrostatic interactions were calculated using the Particle-Mesh Ewald (PME) method58 with 0.12 nm grid-spacing and a 1.0 nm real-space cut-off. Charge groups were not used (single atom charge groups)54. Data was collected at 4 ps intervals and each simulation was run for at least 1 μs. The total simulation time was 10 μs.
Peptide dynamics were quantified by analyzing the circular variance (C.V.) of the φ and ψ dihedral angles over time. C.V. is defined as59:
where m is the number of structures included in the analysis, and R is calculated using:
The value of C.V. ranges between 0 and 1. Lower values represent tighter clustering about the mean and higher values are indicative of greater φ and ψ variability.
Author Contributions
Conceived and designed the experiments: E.A.C., M.K., W.Y.C. Performed the experiments: E.A.C., R.C.K. Analyzed the data: E.A.C., R.C.K. Wrote the paper: E.A.C., M.K., W.Y.C.
Supplementary Material
Supplementary Information
Acknowledgments
We thank SharcNet (www.sharcnet.ca), the SciNet HPC Consortium and Compute Canada for providing the computational resources. Financial support was provided by the Ontario Early Researcher Award Program, Canadian Institutes of Health Research (CIHR; MOP 74679), University of Waterloo and Natural Sciences and Engineering Research Council of Canada (NSERC).
References
- Rual J. F. et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 (2005). [DOI] [PubMed] [Google Scholar]
- Stelzl U. et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 (2005). [DOI] [PubMed] [Google Scholar]
- Ekman D., Light S., Bjorklund A. K. & Elofsson A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol 7, R45 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. D. et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93 (2004). [DOI] [PubMed] [Google Scholar]
- Vidal M., Cusick M. E. & Barabasi A. L. Interactome networks and human disease. Cell 144, 986–998 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K., Cortese M. S., Romero P., Iakoucheva L. M. & Uversky V. N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 272, 5129–5148 (2005). [DOI] [PubMed] [Google Scholar]
- Das R. K., Mao A. H. & Pappu R. V. Unmasking functional motifs within disordered regions of proteins. Sci Signal 5, pe17 (2012). [DOI] [PubMed] [Google Scholar]
- Davey N. E. et al. Attributes of short linear motifs. Mol Biosyst 8, 268–281 (2012). [DOI] [PubMed] [Google Scholar]
- Mészáros B., Tompa P., Simon I. & Dosztányi Z. Molecular principles of the interactions of disordered proteins. J Mol Biol 372, 549–561 (2007). [DOI] [PubMed] [Google Scholar]
- Cino E. A., Choy W.-Y. & Karttunen M. Comparison of Secondary Structure Formation Using 10 Different Force Fields in Microsecond Molecular Dynamics Simulations. J Chem Theory Comput 8, 2725–2740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cino E. A., Wong-Ekkabut J., Karttunen M. & Choy W. Y. Microsecond molecular dynamics simulations of intrinsically disordered proteins involved in the oxidative stress response. PLoS One 6, e27371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M., Simon I., Friedrich P. & Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol 338, 1015–1026 (2004). [DOI] [PubMed] [Google Scholar]
- Mohan A. et al. Analysis of molecular recognition features (MoRFs). J Mol Biol 362, 1043–1059 (2006). [DOI] [PubMed] [Google Scholar]
- Itoh K. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13, 76–86 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K. I. et al. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 26, 2887–2900 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K. I., Kobayashi A., Katsuoka F. & Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem 387, 1311–1320 (2006). [DOI] [PubMed] [Google Scholar]
- Camp N. D. et al. Wilms Tumor Gene on X Chromosome (WTX) Inhibits Degradation of NRF2 Protein through Competitive Binding to KEAP1 Protein. J Biol Chem 287, 6539–6550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12, 213–223 (2010). [DOI] [PubMed] [Google Scholar]
- Lo S.-C. & Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem 281, 37893–903 (2006). [DOI] [PubMed] [Google Scholar]
- Ma J. et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol 32, 1506–1517 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan G. D. et al. Fetal Alz-50 clone 1 interacts with the human orthologue of the Kelch-like Ech-associated protein. Biochemistry 43, 12113–12122 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan B., Nakamura Y. & Yokoyama S. Structural analysis of the complex of Keap1 with a prothymosin alpha peptide. Acta Crystallogr Sect F Struct Biol Cryst Commun 64, 233–238 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-E. et al. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal 22, 1645–1654 (2010). [DOI] [PubMed] [Google Scholar]
- Niture S. K. & Jaiswal A. K. INrf2 (Keap1) targets Bcl-2 degradation and controls cellular apoptosis. Cell Death Differ 18, 439–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Velichkova M. et al. A human homologue of Drosophila kelch associates with myosin-VIIa in specialized adhesion junctions. Cell Motil Cytoskeleton 51, 147–164 (2002). [DOI] [PubMed] [Google Scholar]
- Yi S., Boys B. L., Brickenden A., Konermann L. & Choy W. Y. Effects of zinc binding on the structure and dynamics of the intrinsically disordered protein prothymosin alpha: evidence for metalation as an entropic switch. Biochemistry 46, 13120–13130 (2007). [DOI] [PubMed] [Google Scholar]
- Xue B., Dunbrack R. L., Williams R. W., Dunker A. K. & Uversky V. N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804, 996–1010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia M. & Kukol A. Structure and dynamics of the kinase IKK-β--A key regulator of the NF-kappa B transcription factor. J Struct Biol 176, 133–142 (2011). [DOI] [PubMed] [Google Scholar]
- Petros A. M. et al. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci U S A 98, 3012–3017 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S.-C., Li X., Henzl M. T., Beamer L. J. & Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J 25, 3605–3617 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan B. et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 21, 689–700 (2006). [DOI] [PubMed] [Google Scholar]
- Tong K. I. et al. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol 27, 7511–7521 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M., Ulmschneider J. P., Ulmschneider M. B. & White S. H. Conformational states of melittin at a bilayer interface. Biophys J 104, L12–14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal J., Yoo T. H., Georgiou G. & Truskett T. M. Structural ensemble of an intrinsically disordered polypeptide. J Phys Chem B 117, 118–124 (2013). [DOI] [PubMed] [Google Scholar]
- Kyte J. & Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J Mol Biol 157, 105–132 (1982). [DOI] [PubMed] [Google Scholar]
- Hancock R. et al. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic Biol Med 52, 444–451 (2012). [DOI] [PubMed] [Google Scholar]
- Itoh K. et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236, 313–322 (1997). [DOI] [PubMed] [Google Scholar]
- Niture S. K. & Jaiswal A. K. Prothymosin-alpha mediates nuclear import of the INrf2/Cul3 Rbx1 complex to degrade nuclear Nrf2. J Biol Chem 284, 13856–13868 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Stępkowski T. M. & Kruszewski M. K. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med 50, 1186–1195 (2011). [DOI] [PubMed] [Google Scholar]
- Tian H. et al. Keap1: One stone kills three birds Nrf2, IKKβ and Bcl-2/Bcl-xL. Cancer Lett 325, 26–34 (2012). [DOI] [PubMed] [Google Scholar]
- Li Y., Paonessa J. D. & Zhang Y. Mechanism of chemical activation of Nrf2. PLoS One 7, e35122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Rivera J., Ortiz A. & Egido J. Antioxidants in kidney diseases: the impact of bardoxolone methyl. Int J Nephrol 2012, 321714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn M. B. & Liby K. T. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 12, 564–571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn M. B. et al. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod 74, 537–545 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. & Gordon G. B. A strategy for cancer prevention: stimulation of the Nrf2-ARE signaling pathway. Mol Cancer Ther 3, 885–893 (2004). [PubMed] [Google Scholar]
- Brunger A. T. Version 1.2 of the Crystallography and NMR system. Nat Protoc 2, 2728–2733 (2007). [DOI] [PubMed] [Google Scholar]
- Pettersen E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- Hess B., Kutzner C., van der Spoel D. & Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 4, 435–447 (2008). [DOI] [PubMed] [Google Scholar]
- Oostenbrink C., Soares T. A., van der Vegt N. F. A. & van Gunsteren W. F. Validation of the 53A6 GROMOS force field. Eur Biophys J 34, 273–284 (2005). [DOI] [PubMed] [Google Scholar]
- Oostenbrink C., Villa A., Mark A. E. & van Gunsteren W. F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J Chem Theory Comput 25, 1656–1676 (2004). [DOI] [PubMed] [Google Scholar]
- Hansson T., Nordlund P. & Aqvist J. Energetics of nucleophile activation in a protein tyrosine phosphatase. J Mol Biol 265, 118–127 (1997). [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C., Postma J. P. M. & Gusteren W. F. Interaction models for water in relation to protein hydration. in Intermolecular forces 331–342 (1981). [Google Scholar]
- Wong-Ekkabut J. & Karttunen M. Assessment of common simulation protocols for simulations of nanopores, membrane proteins and channels. J Chem Theory Comput 8, 2905–2911 (2012). [DOI] [PubMed] [Google Scholar]
- Wong-Ekkabut J., Miettinen M. S., Dias C. & Karttunen M. Static charges cannot drive a continuous flow of water molecules through a carbon nanotube. Nat Nanotechnol 5, 555–557 (2010). [DOI] [PubMed] [Google Scholar]
- Bussi G., Donadio D. & Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- Parrinello M. & Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys 52, 7182–7190 (1981). [Google Scholar]
- Hess B., Bekker H., Berendsen H. J. C. & Johannes J. G. E. M. A linear constraint solver for molecular simulations. J Comput Chem 18, 1463–1472 (1997). [Google Scholar]
- Darden T., York D. & Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys 98, 10089–10093 (1993). [Google Scholar]
- MacArthur M. W. & Thornton J. M. Conformational analysis of protein structures derived from NMR data. Proteins 17, 232–251 (1993). [DOI] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J.-M. & Brenner S. E. WebLogo: a sequence logo generator. Genome Res 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A. & Schulten K. VMD: visual molecular dynamics. J Mol Graph 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- Zhao J., Redell J. B., Moore A. N. & Dash P. K. A novel strategy to activate cytoprotective genes in the injured brain. Biochem Biophys Res Commun 407, 501–506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information