

Abstract
Catalytic activity is primarily a surface phenomenon, however, little is known about Co3O4 nanocrystals in terms of the relationship between the oxygen reduction reaction (ORR) catalytic activity and surface structure, especially when dispersed on a highly conducting support to improve the electrical conductivity and so to enhance the catalytic activity. Herein, we report a controllable synthesis of Co3O4 nanorods (NR), nanocubes (NC) and nano-octahedrons (OC) with the different exposed nanocrystalline surfaces ({110}, {100}, and {111}), uniformly anchored on graphene sheets, which has allowed us to investigate the effects of the surface structure on the ORR activity. Results show that the catalytically active sites for ORR should be the surface Co2+ ions, whereas the surface Co3+ ions catalyze CO oxidation, and the catalytic ability is closely related to the density of the catalytically active sites. These results underscore the importance of morphological control in the design of highly efficient ORR catalysts.
The advent of nanotechnology and the ability to synthesize a marvelous panoply of nanocrystals have breathed a new life to the catalysis science1,2,3,4,5. The notion that catalysts are necessarily nanomaterials is rooted in the importance of surface in activating chemical bonds. Although there have been numerous reports on the catalytic activities of nanomaterials, detailed understanding of how surface structure affects catalytic performance is still lacking. There is therefore a need to systematically study the catalytic activity as a function of nanocrystalline morphology other than the size since the surface structure is tunable by varying the morphology. The prerequisite is the selective synthesis of differently shaped nanocrystal catalysts with uniform crystal surfaces, preferably dispersed on a supporting substrate.
The spinel type Co3O4, in which the Co2+ and Co3+ ions occupy the tetrahedral and octahedral sites, respectively6, is known to be a promising catalytic material7,8,9,10,11,12. It has been reported that different morphologies of Co3O4 nanocrystals have a direct bearing on their catalytic activities for CO oxidation. For example, the {110} faces of Co3O4 nanocrystals have a higher catalytic activity for CO oxidation than {100} and {111}, because of the more abundant catalytically active Co3+ sites on the former12. For the CH4 combustion, however, the catalytic activity of the nanocrystalline surfaces was found to be in the order of {112} > {011} ≫ {001}, depending instead on the surface energy13. In the main, the catalytic activity of a given catalyst is therefore determined by the nature of adsorption/activation/desorption of the reactants and products on the catalytically active sites12,13,14,15.
The spinel-type Co3O4 nanocrystals are also a potential alternate for the high cost Pt and its alloys to catalyze the oxygen reduction reaction (ORR), a critical reaction which underlies a battery of renewable-energy technologies such as fuel cell. To our knowledge, however, no study has been reported on the correlation between the shape and the ORR catalytic activities of Co3O4 nanocrystals. Such a study requires anchoring the Co3O4 nanocrystals onto a substrate, which is preferably conductive and thus can enhance the ORR activity and stabilize the catalyst system. As a relatively new class of carbon-based nanomaterials, graphene and carbon nanotube (CNT) have high electrical conductivity, large surface area, high mechanical strength, and structural flexibility, making them ideal substrates for supporting such nanocrystal catalysts. Indeed, graphene and CNT supported Co-based electro-catalysts have already been used for ORR with improved catalytic activity and stability16,17,18,19. However, shape-controllable synthesis of Co3O4 nanocrystals on graphene and CNT as composites is still an unmet challenge.
In this paper, we report the controllable synthesis of Co3O4 nanorods, nanocubes and nano-octahedrons with difference exposed surfaces uniformly immobilized in situ on graphene sheets. This series of nanocrystals showed much enhanced ORR catalytic activity when dispersed on graphene. More significantly, the quantitative catalytic activity depends on the detailed nanocrystalline morphology and thus the surface structure of the nanocrystals, namely, {111} > {100} > {110}, pointing to the Co2+ ions as the ORR active sites.
Results
Shape-selective synthesis of Co3O4 nanocrystals
Detailed procedures for the synthesis of Co3O4 nanoparticles (NP), nanorods (NR), nanocubes (NC) and nano-octahedrons (OC) on the surface of reduced graphene oxides (RGO) have been given in the experimental section, and are here illustrated in Scheme 1. The crystalline phases of these nanocomposites were ascertained by XRD patterns (Figure SI-1), with the help of the standard crystal structure of Co3O4 (JCPDS 65-3103). Co3O4 NP around 10 nm across were formed by thermally decomposing the precursors nucleated from the supersaturated metal bicarbonate solution accompanied by the slow release of CO2 (Figure SI-2), as we reported previously20,21,22.
Presumably, through the Ostwald ripening process, the initially nucleated precursors were transformed into cobalt carbonate hydroxide (Co(CO3)0.5OH) nanorods with a length of several hundred nanometers and a diameter of ~10 nm (Figure SI-3)21. The subsequent calcination process caused a spontaneous transformation into the Co3O4 NR with the overall morphology conserved. Figure 1A shows a typical low-magnification transmission electron microscopy (TEM) image of the synthesized Co3O4 NR, densely distributed on the surface of RGO sheets. We further examined the crystallographic nature of the individual Co3O4 NR through high resolution TEM (HRTEM) observations. Shown in Figure 1B is a section of the Co3O4 NR observed in the [001] orientation, which extends along the [110] direction. The side walls are parallel to the (2–20) plane (Figure 1B, C). When the nanorod was titled to the [1–10] zone axis, the (004) side planes and the (222) crystal planes could also be clearly observed (Figure 1D, E). On the basis of these results, the nanorod morphology can be approximately laid out as shown in Figure 1F: the nanorod assumes its axial direction along [110] and is bounded by the side planes of {001} and {1–10}. Similar Co3O4 NR were prepared previously by the calcination of cobalt carbonate hydroxide nanorod precursors obtained by the precipitation of cobalt acetate and sodium carbonate in ethylene glycol, and used for low temperature catalytic CO oxidation12.
Figure 1. Co3O4 nanorods anchored on the RGO sheets.

Low magnification TEM image (A); HRTEM images viewed along the (B, C) [001] and (D, E) [1–10] axes; (F) Schematic illustration of the nanorod morphology highlighting the exposed surfaces. Note: [uvw] is a crystal axis index, (hkl) is a crystal plane index.
When H2O2 was added, during the Ostwald ripening process, the cobalt carbonate hydroxide (Co(CO3)0.5OH•0.11H2O) nanorods were gradually transformed into Co3O4 nanocrystals, as seen from SEM images of the intermediates (Figure SI-4). After complete transformation, well-defined Co3O4 OC were uniformly dispersed on the surface of graphene sheets (Figure 2A). To reveal the exposed surfaces, we refer to the TEM images in Figure 2. According to the HRTEM image of a single Co3O4 OC particle taken along the [001] direction (Figure 2B), the two-dimensional (2D) projection appears to be square (see the model octahedron in the bottom left inset in Figure 2B). By tilting the Co3O4 OC to the [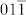 ] zone axis, the 2D projection of the corresponding TEM image (Figure 2C) becomes diamond-like. These results are consistent with the octahedron morphology of the Co3O4 nanocrystals anchored on the surface of RGO sheets with eight exposed {111} surfaces (see the model structure in Figure 2D).
] zone axis, the 2D projection of the corresponding TEM image (Figure 2C) becomes diamond-like. These results are consistent with the octahedron morphology of the Co3O4 nanocrystals anchored on the surface of RGO sheets with eight exposed {111} surfaces (see the model structure in Figure 2D).
Figure 2. Co3O4 nano-octahedrons anchored on the RGO sheets.

(A) Low magnification TEM image; (B) HRTEM image along the [001] direction; (C) HRTEM image along the [01-1] direction; and (D) Schematic illustration of a nano-octahedron bounded by the eight {111} surfaces.
Finally, Co3O4 NC were formed on the surface of RGO sheets by co-precipitation of Co2+ and ammonia followed by hydrothermal treatment in the presence of H2O2. TEM observations revealed that the as-formed Co3O4 nanocrystals have a perfect cubic morphology and a uniform crystallite size of about 10 nm (Figure 3A). Excellent crystallinity of the Co3O4 NC was confirmed by the HRTEM image in Figure 3B. The lattice fringes of d200 (0.418 nm) and d220 (0.285 nm) of Co3O4 are clearly observed from the [001] direction, indicating the nanocubes bounded by the (001) facets (see the constructed nanocube model in Figure 3C).
Figure 3. Co3O4 nanocubes grown on the RGO sheets.

(A) Low magnification TEM image; (B) HRTEM image; and (C) Schematic illustration of the nanocube morphology with the six exposed {100} surfaces.
The composition of the prepared cobalt oxides was investigated by XPS spectra, which are shown in Figure SI-5A. The Co 2p spectra all show a doublet consisting of a low energy band (Co 2p3/2 at 780. 6 eV) and a high energy band (Co 2p1/2 at 796.0 eV) for the Co3O4 NP, NR, NC and OC, in agreement with the standard spectra of Co3O423,24. The energy difference between the peak of Co 2p3/2 and 2p1/2 splitting is approximately 15 eV, indicating the presence of both Co2+/Co3+ species in the cobalt oxides samples23,24,25. The RGO contents in the composites were obtained from the TGA curves (Figure SI-5B), and calculated based on the weight loss below 400°C. According to that analysis, the mass percentages of RGO are around 16.1 wt% for Co3O4 NP/RGO, 16.0 wt% for Co3O4 NR/RGO, 17.4 wt% for Co3O4 NC/RGO, and 14.0 wt% for Co3O4 OC/RGO.
Shape-dependent ORR catalytic activity of the Co3O4 nanocrystals
To assess their ORR catalytic activity, the nanocrystal materials were loaded (with equal mass loading) onto glassy carbon electrodes. The electrodes were interrogated by cyclic voltammetry (CV) in O2- and for reference, N2-, saturated 0.1 M KOH solutions, and the data are shown in Fig. 4A. The Co3O4 NP (size ~10 nm) on the surface of RGO sheets exhibited very poor ORR activity: with an onset potential of ~−0.25 V vs. Hg/HgO. The Co3O4 NR/RGO hybrids with exposed surfaces dominated by {110} showed a much more positive ORR onset potential (~−0.1 V), suggesting higher ORR catalytic activity than Co3O4 NP/RGO. Remarkably, the Co3O4 NC/RGO with the six exposed {100} surfaces and the Co3O4 OC/RGO nanocomposites with the eight exposed {111} surfaces achieved even more positive onset potentials, e.g., ~−0.06 V for Co3O4 NC/RGO, and ~−0.04 V for Co3O4 OC/RGO, closely approaching that of Pt/C, the gold standard for ORR catalysts.
Figure 4.

(A) CV curves of Co3O4 nanocrystals/RGO composites on glassy carbon electrodes in N2-saturated (solid line) or O2-saturated 0.1 M KOH (dash line); (B) Rotating-disk voltammograms, (C) The electron transfer number (n) profiles obtained from (B), and (D) Tafel plots for the Co3O4 /RGO composite electrodes and the commercial Pt/C electrode; (E) J-T chronoamperometric responses at −0.40 V versus Hg/HgO reference electrode at a rotating rate of 2400 rpm. The 0.1 M KOH solution electrolyte is firstly bubbled by N2 for 30 min, and then is introduced by O2 gas for around 3000 s, and is finally added by 20 vol% of methanol; (F) Chronoamperometric responses (percentage of current retained versus operation time) of the kept at −0.40 V versus Hg/HgO reference electrode in O2-saturated 0.1 M KOH electrolyte at a rotating rate of 2400 rpm.
The ORR kinetics of the Co3O4/RGO composites was investigated using the rotating-disk electrode (RDE) technique in O2-saturated 0.1 M KOH electrolyte. As can be seen from the LSV curves in Figure 4B, the ORR process is diffusion controlled when the potential is negative to −0.20 V, mixed diffusion kinetic controlled in the potential region from −0.20 to −0.10 V, and kinetic controlled in the potential range from −0.01 to 0 V. Unsupported Co3O4 NP prepared by thermal decomposition of Co(CO3)1/2OH precursors, as we reported previously26, exhibited much lower onset potential and diffusion-limited current density than those of the present four Co3O4/RGO composite electrodes, suggesting the positive effect of RGO on the ORR catalytic activity of Co3O417. Among the four Co3O4/RGO composite electrodes, the Co3O4 NP/RGO composite catalyst shows the lowest onset potential, whereas the Co3O4 OC/RGO composite shows the highest one. The half-wave potentials of the composite catalysts are in the sequence of Co3O4 OC/RGO (−0.14 V) > Co3O4 NC/RGO (−0.16 V) > Co3O4 NR/RGO (−0.20 V) > Co3O4 NP/RGO (−0.33 V). In the diffusion controlled region, the diffusion-limited current densities follow the trend of the half-wave potentials for the composite catalyst series. Together, these results suggest that the Co3O4 OC/RGO composite catalyst exhibits the highest ORR catalytic activity among the four samples, and the different catalytic activities can be attributed to the different morphologies of the Co3O4 nanocrystals in the composites.
A series of rotating disk voltammograms of oxygen reduction are shown in Figure SI-(6–9)A with the commercial Pt/C and the Co3O4/RGO composite catalysts at different rotation rates in O2-saturated 0.1 M KOH electrolyte. The RDE data were analyzed using the Koutecky-Levich equation (Eq. 1 in the experimental section), according to which a plot of the inverse current density J−1 versus Ω−1/2, shown in Figure SI-(6–9)B, should yield a straight line with the intercept corresponding to Jk and the slope reflecting the so-called B factor. The electron transfer number for the O2 reduction process can be calculated from the B factor according to Eq. 2 (see the experimental section). The linearity of the Koutecky-Levich plots and the near parallelism of the fitting lines are consistent with the first-order reaction kinetics with respect to the concentration of the dissolved oxygen and implicate similar electron transfer numbers for the ORR at different potentials in the region of −0.30 V to −0.50 V. The calculated electron transfer numbers (n) for the commercial Pt/C and the Co3O4/RGO composite catalysts in the potential region of −0.30 to −0.50 V are shown in Figure 4C. We can see that the Co3O4 OC/RGO composite electrode can catalyze the ORR via a 4 e process, in much the same way as a high-quality commercial Pt/C catalyst does, which is impressive for a non-Pt catalyst. However, the ORR electron transfer number for the Co3O4 NR/RGO and Co3O4 NC/RGO composite catalysts were calculated to be ~3.5, suggesting incomplete reduction of oxygen, but still domination by the 4e process.
The ORR catalytic activity of the Co3O4/RGO hybrid catalysts can also be gleaned from the Tafel slopes at low and high overpotentials in O2-saturated 0.1 M KOH aqueous solution. The Tafel data are shown in Figure 4D. The E versus log (-J) curves of the samples similarly show two Tafel slopes at low and high overpotentials, respectively, indicating a similar change in reaction mechanisms with the potential. The two slopes can be explained in term of the isotherms at two different O2 coverages; i) the Temkin isotherm (high O2 coverage) associated with an intermediate oxide coverage arising from ORR at low overpotential, whereby the first electron transfer step involving an adsorbed product such as OH− is the rate-determining step; and ii) the Langmuir isotherm (low O2 coverage) at high overpotential wherein significant oxide coverage ceases to exist, which is commonly the case when a two-electron transfer reaction is the rate-determining step. This is a characteristic feature of ORR on mixed valence spinel oxide, e.g., Co-based ORR catalysts25,27. As can be observed in Figure 4D, the Co3O4 NC (83.7 mV/decade) and Co3O4 OC (101.4 mV/decade) on RGO sheets exhibit smaller Tafel slopes at the over-potentials from −0.05 V to −0.10 V than the Co3O4 NR/RGO hybrid (115.6 mV/decade) in 0.1 M KOH electrolyte, demonstrating high ORR catalytic activities close to that of the commercial Pt/C catalyst (93.9 mV/decade).
Other important performance metric for an ORR catalysts include the tolerance of the commonly used fuel molecules and cycle stability, which are especially relevant for fuel cells such as direct methanol fuel cell. To examine the possible crossover effect for the catalytic performance, we measured the electrocatalytic selectivity of the Co3O4/RGO composite electrode against electro-oxidation of methanol molecules. The current density-time (J-T) chronoamperometric response profiles are shown in Figure 4E. For the commercial Pt/C electrode, a sharp decrease (~85%) in current density was observed upon methanol addition (20 vol%) into the O2-saturated 0.1 M KOH electrolyte. However, the amperometric responses of the Co3O4/RGO composite electrodes are strong and stable, showing a retention ratio of at least 80% after the addition of methanol. Such high selectivity of the Co3O4/RGO composite electrodes toward the ORR and the remarkably good tolerance to crossover effect can be attributed to the much lower ORR potential than required for oxidation of the fuel molecules28. Moreover, the Co3O4/RGO hybrid electrodes also exhibited excellent stability as measured by chronoamperometric measurements (Figure 4F). At a constant voltage of −0.40 V vs Hg/HgO, the ORR current density produced in the hybrid catalysts almost had no decay over 10000 s of continuous operation, whereas the commercial Pt/C catalyst exhibited ~22% decrease in current density. Thus, in comparison with the commercial Pt/C catalyst, our Co3O4/RGO composite electrodes are more insensitive to methanol molecules, thus more resistive to poisoning by the possible methanol crossover from the anode of a fuel cell, and are more stable under operating conditions.
Discussion
Co3O4 has the normal-spinel structure Co2+Co23+O4, in which the Co2+ ion in the formula unit occupies the tetrahedral site, while the two Co3+ ions occupy the octahedral sites6, as shown in Figure 5A. Figure 5B–D depict the close-packed planes of {001}, {111} and {110}, and their surface atomic configurations of the spinel-type Co3O4 crystals. Experimental and theoretical measurements have demonstrated that the three low Miller index planes ({100}, {110} and {111}) of such metallic oxide particles with a fcc structure differ not only in the surface atomic density but also in the electronic structure, geometric bonding and chemical reactivity29. As a result, those planes have different surface energies, following the order of γ{111} < γ{100} < γ{110}, which is closely parallel to the catalytic activities for CO and CH4 oxidation12,13,30,31.
Figure 5. Structure models of the spinel Co3O4 nanocrystals.

(A) Three-dimensional atomic arrangement and (B–D) Surface atomic configurations in the {100}, {111} and {110} planes.
For catalyzing CO oxidation, the CO molecule interacts preferably with the surface Co3+ cations, which is the only favorable site for CO adsorption, as confirmed both theoretically32 and experimentally33,34. The oxidation of the adsorbed CO then occurs by abstracting the surface oxygen that had been coordinated with the Co3+ cations. The partially reduced cobalt site, i.e., Co2+ cation with a neighboring oxygen vacancy, is re-oxidized by a gas-phase oxygen molecule back to the active Co3+ form. Consequently, the surface Co3+ cations are regarded as the active sites for CO oxidation, whereas the surface Co2+ cations are almost inactive12,31,35,36,37. It is known that in the Co3O4 crystal structure, the {001} and {111} planes contain only Co2+ cations, while the {110} plane is composed mainly of Co3+ cations (Figure 5B–D). This scenario has been proved by surface differential diffraction studies concluding that the Co3+ cations are present solely on the {110} plane38,39. Similarly, in our own experiment with the Co3O4 NR/RGO composite catalyst, the initial transformation temperatures for CO oxidation is 60°C, considerably lower than that with Co3O4 NC/RGO (100°C) and Co3O4 OC/RGO (120°C) (Figure SI-10). Although the catalytic activities of the Co3O4/RGO composites for CO oxidation are by no means optimized, our study suffices to conclude that the Co3O4 NRs with the predominantly {110} exposed surfaces have higher catalytic activity for CO oxidation than the Co3O4 NCs with the sole six {100} exposed surfaces and the OCs with the only eight {111} exposed surfaces, in excellent agreement with the literature reports12,30,31.
In sharp contrast, for ORR catalysis, the Co3O4 OC enclosed by the eight {111} facets on the RGO sheets was found to exhibit the highest catalytic activity among the four Co3O4/RGO nanocomposite catalysts we have studied in the present work, followed by Co3O4 NC/RGO, and then Co3O4 NR/RGO, with Co3O4 NP/RGO being the least active (Figure 4B). Surprisingly, this ORR catalytic activity order correlates very well with the surface Co2+ density order of the corresponding nanocrystals on RGO excepting the unsupported nanoparticles, namely, {111} > {100} > {110}. This strongly suggests that the surface Co2+ ions are the catalytically active sites for ORR. Note that the measured specific surface areas of the composites are 139.4 m2 g−1 for Co3O4 NP/RGO, 110.0 m2 g−1 for Co3O4 NR/RGO, 116.6 m2 g−1 for Co3O4 NC/RGO, and 98.9 m2 g−1 for Co3O4 OC/RGO composites (see the N2 adsorption isotherms in Figure SI-11). And the contents of Co3O4 in the composites are around 83.9 wt% for Co3O4 NP/RGO, 84.0 wt% for Co3O4 NR/RGO, 82.6 wt% for Co3O4 NC/RGO, and 86.0 wt% for Co3O4 OC/RGO (Figure SI-5B). The similar specific surface areas together with the similar amounts of catalysts used for the ORR testing exclude the possibility of the specific surface area being an important factor that determines the ORR catalytic activity. Thus, it is the exposed crystal planes of the Co3O4 nanocrystals that play a vital role in determining the ORR catalytic activity.
Assuming that the adsorption/desorption process of O2 on the catalytic active sites is involved in the rate-determined step of the ORR, the surface bonding of O2 to the composite catalysts should be critical. In general, O2 molecules with the bond length of 0.12 nm adsorbs on the catalytic active sites mainly via three modes (Griffths, Bridge, and Pauling)40. For the mode of Griffths, the angle of the adsorbed Oads…Co…Oads is around 36°. In comparison with the angle of O-Co-O in the bulk of Co3O4 (95.5° for O-Co3+-O, and 109.5° for O-Co2+-O), such a huge angle mismatch would induce a large intra-molecular stress, resulting in the weak adsorption of O2 on the catalytic active sites. In the Bridge mode (Co…(Oads = Oads)…Co), the bond length of O2 molecules (~0.12 nm) fails to match the distance between the adjacent catalytically active sites (0.1956 nm for Co3+-O, and 0.1902 for Co2+-O). Thus we are left with the possibility that O2 molecules be preferably adsorbed on the catalytically active sites via the Pauling mode (Co…(Oads = O)). Conceivably, when O2 molecules are absorbed on the catalytically active sites via the Pauling mode, it is the surface Co2+ (3d54s2) cations rather than the surface Co3+ (3d54s1) cations which prefer to transfer electrons to the absorbed O2 molecules to weaken and to assist breaking the O-O bond, meanwhile leaving themselves oxidized to Co3+. This suggests that the surface Co2+ sites should be the catalytically active sites instead of Co3+ sites for ORR, and can naturally explain why the Co3O4 nanocrystals with the predominant {111} and {110} exposed surfaces exhibited higher catalytic activity for ORR than that with the {110} exposed surfaces. Such explanation also applies to the observation that CoO exhibits better catalytic ORR performance than Co3O416,41. In addition, the density of Co2+ cations in {111} planes (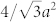 ) is higher than that in {100} planes (
) is higher than that in {100} planes (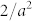 ), resulting in catalytic activity of the Co3O4 OC/RGO composite catalysts enclosed by the eight {111} facets than that of the Co3O4 NC/RGO composite catalyst surrounded by the six {100} exposed surfaces.
), resulting in catalytic activity of the Co3O4 OC/RGO composite catalysts enclosed by the eight {111} facets than that of the Co3O4 NC/RGO composite catalyst surrounded by the six {100} exposed surfaces.
It is widely known that the RGO can serve as both a supporter for the catalyst dispersion and a conduction path for shuttling electrons involved in redox reactions. Here it is clearly the case as well, and this would indiscriminately enhance the catalytic activity of the different nanocomposite catalysts in our study. It is also possible, however, that due to the specific interaction of the different crystal faces with the RGO, the catalytic activity enhancement may be different for different nanocomposites. In particular, the interaction of the Co2+-rich surface with the RGO may be more beneficial to the ORR catalysis. Indeed, some studies along thins line have appeared in recent years16,17. Nevertheless, this aspect still underlines the role of the nanocrystal surfaces in ORR catalysis and adds to source of inspiration for tuning nanocrystal morphologies for optimizing catalytic efficiency.
In sum, we have demonstrated the morphological control of Co3O4 nanocrystals uniformly immobilized in situ on RGO sheets by judiciously choosing the oxidant and tuning the reaction conditions such as pH value. The resulting nanorods predominantly exposing the {110} surfaces, nanocubes surrounded by the six {100} facets, and nano-octahedrons enclosed by the eight {111} facets have allowed us to further investigate the crystal face effects on the ORR catalytic activity. We found that, while the surface Co3+ ions are the catalytically active sites for CO oxidation, and the surface Co2+ ions act as the catalytically active sites for ORR. Additionally, the density of the catalytically active sites on the surface is closely related to the catalytic activity. Accordingly, we have established that the catalytic activity for ORR of these crystalline facets decreases in the sequence of {111} > {100} ≫ {110}. This fundamental understanding shows that morphological control of metal oxide catalysts is a promising surface engineering strategy for the development of nanostructured catalysts in general and non-precious metal free nano-catalysts in particular for ORR in alkaline media.
Methods
Materials and reagents
Graphite flake (natural, ~325 mesh, Alfa Aesar), potassium permanganate (KMnO4, Riedel-de Haën), hydrogen peroxide solution (30 wt%, H2O2, BDH), nickel chloride hexahydrate (NiCl2·6H2O, Fisher), cobalt chloride hexahydrate (CoCl2·6H2O), sodium hydrogen carbonate (NaHCO3, BDH), ammonia water (28 ~ 29 wt%) and hydrazine monohydrate (min 98.0 wt%, N2H4·H2O, Wako) were used without further purification.
Synthesis of graphene oxide (GO) sheets
Graphene oxide sheets were synthesized from natural graphite by a modified Hummers method42. Briefly, 0.5 g of graphite (~325 mesh, Alfa Aesar) and NaNO3 (0.5 g; Aldrich, >99%) were dispersed into concentrated H2SO4 (20 mL; Fisher Scientific, 98%) with an ice bath. Under vigorous stirring, KMnO4 (2.0 g; Riedel-de Haën, >99%) was then added gradually. After removing the ice bath, the mixture was stirred at room temperature for 24 h. As the reaction progressed, the mixture became pasty with a brownish color. Successively, 20 mL of H2O was slowly added to the pasty mixture while keeping the mixture in an ice bath, since the addition of water into the concentrated H2SO4 medium will release a large amount of heat. After dilution with 40 mL of H2O, 5 mL of 30% H2O2 (VMR) was added to the mixture, accompanied by bubbling and changing to brilliant yellow color. After continuously stirring for 2 h, the mixture was filtered and washed with DI water. Then, the products were dispersed in 10 wt% HCl aqueous solution, and washed with DI water again three times to remove impurity ions. Finally, the products were dispersed in DI water via ultrasonication, and then centrifuged at 7000 rpm for 1 h. The supernatant was collected as a GO aqueous solution with a concentration of ~1 mg mL−1.
Controllable synthesis of differently shaped Co3O4 nanocrystals on reduced graphene oxide (RGO) sheets
To start with, 20 mM of CoCl2·6H2O and 40 mM of NaHCO3 were dissolved into 90 mL of DI water and mixed with 10 mL of GO solution, and the mixture was flushed with gaseous CO2 for 2 h forming Solution A. Solution A was stirred at room temperature for 12 h, and subsequently refluxed at 100°C for 10.0 h after adding in 0.1 mL N2H4, forming Solution B. The precipitates in Solution B were centrifuged and washed with DI water three times, and then freeze dried. Finally, the products were thermally treated at 400°C for 1 h in a N2 atmosphere with a heating rate of 5°C/min, to form the Co3O4 nanoparticles/RGO (Co3O4 NP/RGO) composites. In parallel experiments, when Solution B was poured into a 70 mL capacity autoclave with Teflon liner and then hydrothermally treated at 100°C for 12 h before the thermal treatment process, the as-formed products were labeled as Co3O4 nanorods/RGO (Co3O4 NR/RGO) composites. When Solution B and 5.0 mL of 30 wt% hydrogen dioxide (H2O2) were poured into a 70 mL capacity autoclave with Teflon liner and then hydrothermally treated at 100°C for 12 h before the thermal treatment process, the final products were labeled as Co3O4 octahedrons/RGO (Co3O4 OC/RGO) composites.
As for Co3O4 NC on RGO sheets, a typical synthesis is as follows. First, 1 mmol CoCl2·6H2O was dissolved in a mixture of 1 mL of 30 wt% hydrogen peroxide (H2O2) and 40 mL of distilled water. When the solution was clarified, the solution was maintained at pH 9.0 by adding ammonia solution (25 ~ 28 wt%). Then 5 mL of GO solution was added into the above solution, followed by stirring for 1 h. The reaction mixture was then charged into a 70 mL capacity autoclave with Teflon liner, which was then kept at 180°C for 12 h. After the reaction was completed, the autoclave was allowed to cool down to room temperature naturally and opened for product collection. The precipitates were washed with DI water three times and freeze dried. Finally, the products were thermally treated at 400°C for 1 h in a N2 atmosphere with a heating rate of 5°C min−1, to form the Co3O4 nanocubes/RGO (Co3O4 NC/RGO) composites.
General Materials Characterization
The product morphologies were directly examined by scanning electron microscopy (SEM) using JEOL JSM-6700F at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) observations were carried out on a JEOL 2010 microscope operating both at 200 kV. X-ray diffraction (XRD) was performed on a Philips PW-1830 X-ray diffractometer with Cu kα irradiation (λ = 1.5406 Å). The step size and scan rate are set as 0.05° and 0.025°/s, respectively. X-ray photoelectron spectroscopy (XPS) was measured on a Perkin-Elmer model PHI 5600 XPS system with a resolution of 0.3–0.5 eV from a monochromated aluminum anode X-ray source with Kα radiation (1486.6 eV). The thermogravimetric analysis (TGA) was performed from 30 to 700°C on a TGA Q5000 (TA Instruments Ltd) at a heating rate of 5°C min−1 under an air flow of 25 mL min−1. Brunauer-Emmett-Teller (BET) surface areas were measured on a Coulter SA 3100 surface area analyzer.
Catalytic measurements for oxygen reduction reaction (ORR)
Electrochemical measurements were carried out by cyclic voltammetry (CV) on a CHI 660D electrochemical workstation. A conventional, three-electrode cell consisting of glassy carbon electrode (GCE) with an area of 0.125 cm2 was used as the working electrode, Pt foil was employed as the counter electrode and Hg/HgO (1.0 M KOH) (MMO, 0.098 V vs. SHE) was used as the reference electrode. The working electrode was modified with a catalyst layer by dropping a suitable amount of catalyst ink on the GCE. The catalyst ink was prepared by ultrasonically dispersing 10 mg of the carbon supported catalysts in a 2.0 mL solution (1.9 mL of ethanol and 0.1 mL of 5 wt% Nafion solution) for 30 min to obtain a homogeneous solution. 10 μL of the dispersion was pipetted out and dropped onto a glassy carbon rotating disk electrode of 3 mm in diameter, which was then dried in air. CV experiments were conducted at room temperature in 0.1 M KOH solution saturated with nitrogen. For all of the experiments, stable voltammogram curves were recorded after scanning for 20 cycles in the potential region from 0 to 0.6 V in 0.1 M KOH solution. Polarization curves for the oxygen reduction reaction (ORR) were obtained in 0.1 M KOH solution using the rotating ring disk electrode (RRDE-3A). Before the RRDE study, the electrodes were cycled at 50 mV s−1 between 0 and 0.6 V until reproducible cyclic voltammograms were obtained. Normalized currents are given in terms of geometric weight (mA cm−2). The working electrode was scanned cathodically at a rate of 5 mV s−1 with varying rotating speed from 400 rpm to 2400 rpm. Koutecky-Levich plots (J−1 vs. ω−1/2) were analyzed at various electrode potentials. The slopes of their best linear fit lines were used to calculate the number of electrons transferred (n) on the basis of the Koutecky-Levich equation:
Where J is the measured current density, Jk and JL are the kinetic- and diffusion-limiting current densities, ω is the angular velocity, n is transferred electron number, F is the Faraday constant (96485 C mol−1), Co is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), ν is the kinematic viscosity of the electrolyte (0.01 cm2 s−1), Do is the O2 diffusion coefficient (1.9 × 10−5 cm2 s−1), and k is the electron-transfer rate constant.
Catalytic measurements for CO oxidation
The catalytic activity toward CO oxidation was evaluated in a continuous flow reactor. In brief, the reaction gas, 5% CO in nitrogen (99.999%) (10 mL min−1) and air (99.999%) (40 mL min−1) was fed to a catalyst (22.5 mg) containing fixed-bed flow reactor made of glass with an inner diameter of 2.4 mm. Steady-state catalytic activity was measured at each chosen temperature, from room temperature to 200°C in a step of 20°C. The effluent gas was analyzed on-line by an on-stream gas chromatograph (Ramiin GC 2060) equipped with a TDX-01 column.
Author Contributions
S.Y. designed the experiments. J.X. carried out the experiments. Q.K. carried out the CO oxidation experiment. J.X. and S.Y. analyzed the data and wrote the manuscript. Q.K., F.X., S.W. and L.G. contributed to the data analysis. All the authors discussed the research.
Supplementary Material
Supplementary information
Acknowledgments
This work was supported by the HK-RGC General Research Funds (GRF no. HKUST 605710 and 604809), and the Fundamental Research Funds for the Central Universities (Project No. 2013QN158).
References
- Tian N., Zhou Z.-Y., Sun S.-G., Ding Y. & Wang Z. L. Synthesis of tetrahexahedral platinum nanocrystals with high-index facets and high electro-oxidation activity. Science 316, 732–735 (2007). [DOI] [PubMed] [Google Scholar]
- Burda C., Chen X. B., Narayanan R. & El-Sayed M. A. Chemistry and properties of nanocrystals of different shapes. Chem. Rev. 105, 1025–1102 (2005). [DOI] [PubMed] [Google Scholar]
- Zhou K. & Li Y. Catalysis Based on Nanocrystals with Well-Defined Facets. Angew. Chem. In. Ed. 51, 602–613 (2012). [DOI] [PubMed] [Google Scholar]
- Zhou Z.-Y., Tian N., Li J.-T., Broadwell I. & Sun S.-G. Nanomaterials of high surface energy with exceptional properties in catalysis and energy storage. Chem. Soc. Rev. 40, 4167–4185 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang H., Jin M. & Xia Y. Enhancing the catalytic and electrocatalytic properties of Pt-based catalysts by forming bimetallic nanocrystals with Pd. Chem. Soc. Rev. 41, 8035–8049 (2012). [DOI] [PubMed] [Google Scholar]
- Spencer C. D. & Schroeer D. Mössbauer study of several cobalt spinels using Co57 and Fe57. Phys. Rev. B. 9, 3658–3665 (1974). [Google Scholar]
- Zavyalova U., Scholz P. & Ondruschka B. Influence of cobalt precursor and fuels on the performance of combustion synthesized Co3O4/gamma-Al2O3 catalysts for total oxidation of methane. Appl. Catal. A-Gen. 323, 226–233 (2007). [Google Scholar]
- Liotta L. F., Di Carlo G., Pantaleo G. & Deganello G. Catalytic performance of Co3O4/CeO2 and Co3O4/CeO2-ZrO2 composite oxides for methane combustion: Influence of catalyst pretreatment temperature and oxygen concentration in the reaction mixture. Appl. Catal. B-Environ. 70, 314–322 (2007). [Google Scholar]
- Xiao T. C., Ji S. F., Wang H. T., Coleman K. S. & Green M. L. H. Methane combustion over supported cobalt catalysts. J. Mol. Catal. A-Chem. 175, 111–123 (2001). [Google Scholar]
- Zwinkels M. F. M., Jaras S. G., Menon P. G. & Griffin T. A. Catalytic Materials for High Temperature Combustion. Catal. Rev. Sci. Eng. 35, 319–358 (1993). [Google Scholar]
- Alvarez A., Ivanova S., Centeno M. A. & Odriozola J. A. Sub-ambient CO oxidation over mesoporous Co3O4: Effect of morphology on its reduction behavior and catalytic performance. Appl. Catal. A-Gen. 431, 9–17 (2012). [Google Scholar]
- Xie X. W., Li Y., Liu Z. Q., Haruta M. & Shen W. J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 458, 746–749 (2009). [DOI] [PubMed] [Google Scholar]
- Hu L. H., Peng Q. & Li Y. D. Selective Synthesis of Co3O4 Nanocrystal with Different Shape and Crystal Plane Effect on Catalytic Property for Methane Combustion. J. Am. Chem. Soc. 130, 16136–16367 (2008). [DOI] [PubMed] [Google Scholar]
- Suntivich J. et al. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal-air batteries. Nat. Chem. 3, 546–550 (2011). [DOI] [PubMed] [Google Scholar]
- Suntivich J., May K. J., Gasteiger H. A., Goodenough J. B. & Shao-Horn Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 334, 1383–1385 (2011). [DOI] [PubMed] [Google Scholar]
- Liang Y. Y. et al. Oxygen Reduction Electrocatalyst Based on Strongly Coupled Cobalt Oxide Nanocrystals and Carbon Nanotubes. J. Am. Chem. Soc. 134, 15849–15857 (2012). [DOI] [PubMed] [Google Scholar]
- Liang Y. et al. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780–786 (2011). [DOI] [PubMed] [Google Scholar]
- Liang Y. Y. et al. Covalent Hybrid of Spinel Manganese-Cobalt Oxide and Graphene as Advanced Oxygen Reduction Electrocatalysts. J. Am. Chem. Soc. 134, 3517–3523 (2012). [DOI] [PubMed] [Google Scholar]
- Xiao J. W., Xu G. L., Sun S. G. & Yang S. H. MFe2O4 and MFe@oxide Core-shell Nanoparticles Anchored on N-doped Graphene Sheets for Synergistically Enhancing Lithium Storage Performance and Electrocatalytic Activity for Oxygen Reduction Reactions. Part. Part. Syst. Charact. 10.1002/ppsc.201300105 (2013). [Google Scholar]
- Xiao J. & Yang S. Nanocomposites of Ni(OH)2/Reduced Graphene Oxides with Controllable Composition, Size, and Morphology: Performance Variations as Pseudocapacitor Electrodes. ChemPlusChem 77, 807–816 (2012). [Google Scholar]
- Xiao J. W. & Yang S. H. Bio-inspired synthesis of NaCl-type CoxNi1-xO (0 < = x < 1) nanorods on reduced graphene oxide sheets and screening for asymmetric electrochemical capacitors. J. Mater. Chem. 22, 12253–12262 (2012). [Google Scholar]
- Xiao J. W. & Yang S. H. Bio-inspired synthesis: understanding and exploitation of the crystallization process from amorphous precursors. Nanoscale 4, 54–65 (2012). [DOI] [PubMed] [Google Scholar]
- Fu L. et al. Beaded cobalt oxide nanoparticles along carbon nanotubes: Towards more highly integrated electronic devices. Adv. Mater. 17, 217–221 (2005). [Google Scholar]
- Ernst B., Libs S., Chaumette P. & Kiennemann A. Preparation and characterization of Fischer-Tropsch active Co/SiO2 catalysts. Appl. Catal. A-Gen. 186, 145–168 (1999). [Google Scholar]
- Xu J., Gao P. & Zhao T. S. Non-precious Co3O4 nano-rod electrocatalyst for oxygen reduction reaction in anion-exchange membrane fuel cells. Energy Environ. Sci. 5, 5333–5339 (2012). [Google Scholar]
- Xiao J. & Yang S. Sequential crystallization of sea urchin-like bimetallic (Ni, Co) carbonate hydroxide and its morphology conserved conversion to porous NiCo2O4 spinel for pseudocapacitors. RSC Adv. 1, 588–595 (2011). [Google Scholar]
- De Koninck M. & Marsan B. MnxCu1-xCo2O4 used as bifunctional electrocatalyst in alkaline medium. Electrochim. Acta 53, 7012–7021 (2008). [Google Scholar]
- Cui H. F., Ye J. S., Liu X., Zhang W. D. & Sheu F. S. Pt-Pb alloy nanoparticle/carbon nanotube nanocomposite: a strong electrocatalyst for glucose oxidation. Nanotechnology 17, 2334–2339 (2006). [Google Scholar]
- Tang X. F., Li J. H. & Hao J. M. Synthesis and characterization of spinel Co3O4 octahedra enclosed by the {111} facets. Mater. Res. Bull. 43, 2912–2918 (2008). [Google Scholar]
- Hu L., Sun K., Peng Q., Xu B. & Li Y. Surface Active Sites on Co3O4 Nanobelt and Nanocube Model Catalysts for CO Oxidation. Nano Res. 3, 363–368 (2010). [Google Scholar]
- Sun Y. et al. Ultrathin Co3O4 nanowires with high catalytic oxidation of CO. Chem. Commun. 47, 11279–11281 (2011). [DOI] [PubMed] [Google Scholar]
- Broqvist P., Panas I. & Persson H. A DFT study on CO oxidation over Co3O4. J. Catal. 210, 198–206 (2002). [Google Scholar]
- Grillo F., Natile M. M. & Glisenti A. Low temperature oxidation of carbon monoxide: the influence of water and oxygen on the reactivity of a Co3O4 powder surface. Appl. Catal. B-Environ. 48, 267–274 (2004). [Google Scholar]
- Jansson J. Low-temperature CO oxidation over Co3O4/Al2O3. J. Catal. 194, 55–60 (2000). [Google Scholar]
- Petitto S. C., Marsh E. M., Carson G. A. & Langell M. A. Cobalt oxide surface chemistry: The interaction of CoO(100), Co3O4(110) and Co3O4(111) with oxygen and water. J. Mol. Catal. A-Chem. 281, 49–58 (2008). [Google Scholar]
- Jansson J. et al. On the catalytic activity of Co3O4 in low-temperature CO oxidation. J. Catal. 211, 387–397 (2002). [Google Scholar]
- Omata K., Takada T., Kasahara S. & Yamada M. Active site of substituted cobalt spinel oxide for selective oxidation of CO/H-2.2. Appl. Catal. A-Gen. 146, 255–267 (1996). [Google Scholar]
- Beaufils J. P. & Barbaux Y. Study of adsorption on powders by surface differential diffraction measurements. Argon on Co3O4. J. Appl. Cryst. 15, 301–307 (1982). [Google Scholar]
- Ziolkowski J. & Barbaux Y. Identification of Sites Active in Oxidation of Butene-1 to Butadiene and CO2 on Co3O4 in Terms of the Crystallochemical Model of Solid-Surfaces. J. Mol. Catal. 67, 199–215 (1991). [Google Scholar]
- Yeager E. Dioxygen electrocatalysis: mechanisms in relation to catalyst structure. J. Mol. Catal. 38, 5–25 (1986). [Google Scholar]
- Guo S., Zhang S., Wu L. & Sun S. Co/CoO Nanoparticles Assembled on Graphene for Electrochemical Reduction of Oxygen. Angew. Chem. In. Ed. 51, 11770–11773 (2012). [DOI] [PubMed] [Google Scholar]
- Hummers W. S. & Offeman R. E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 80, 1339 (1958). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information