

Abstract
Focal adhesion kinase (FAK) is an attachment complex protein associated with the regulation of muscle mass through as-of-yet unclear mechanisms. We tested whether FAK is functionally important for muscle hypertrophy, with the hypothesis that FAK knockdown (FAK-KD) would impede cell growth associated with a trophic stimulus. C2C12 skeletal muscle cells harboring FAK-targeted (FAK-KD) or scrambled (SCR) shRNA were created using lentiviral transfection techniques. Both FAK-KD and SCR myotubes were incubated for 24 h with IGF-I (10 ng/ml), and additional SCR cells (±IGF-1) were incubated with a FAK kinase inhibitor before assay of cell growth. Muscle protein synthesis (MPS) and putative FAK signaling mechanisms (immunoblotting and coimmunoprecipitation) were assessed. IGF-I-induced increases in myotube width (+41 ± 7% vs. non-IGF-I-treated) and total protein (+44 ± 6%) were, after 24 h, attenuated in FAK-KD cells, whereas MPS was suppressed in FAK-KD vs. SCR after 4 h. These blunted responses were associated with attenuated IGF-I-induced FAK Tyr397 phosphorylation and markedly suppressed phosphorylation of tuberous sclerosis complex 2 (TSC2) and critical downstream mTOR signaling (ribosomal S6 kinase, eIF4F assembly) in FAK shRNA cells (all P < 0.05 vs. IGF-I-treated SCR cells). However, binding of FAK to TSC2 or its phosphatase Shp-2 was not affected by IGF-I or cell phenotype. Finally, FAK-KD-mediated suppression of cell growth was recapitulated by direct inhibition of FAK kinase activity in SCR cells. We conclude that FAK is required for IGF-I-induced muscle hypertrophy, signaling through a TSC2/mTOR/S6K1-dependent pathway via means requiring the kinase activity of FAK but not altered FAK-TSC2 or FAK-Shp-2 binding.
Keywords: focal adhesion kinase, hypertrophy, insulin-like growth factor-I, tuberous sclerosis complex 2, mammalian target of rapamycin, S6 kinase 1, skeletal muscle
attachment complexes, or focal adhesion complexes, are macromolecular structures situated in the sarcolemma of muscle fibers that link the extracellular matrix (ECM) to the cytoplasmic cytoskeleton and consist of a variety of ECM receptors/integrins and intracellular cytoskeletal and signaling molecules (7, 30). Interactions of ECM proteins with integrin receptors stimulate intracellular signaling pathways that are important in cell growth and migration (51), and in adult skeletal muscle, focal adhesion complexes play a crucial part in the transmission of lateral forces during contraction (41). Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase that localizes to focal adhesion complexes and represents a key component of integrin-mediated signaling (9). Engagement of integrin receptors induces phosphorylation of FAK at Tyr397, which correlates with its activation (8), and a growing body of evidence has associated FAK activation with the hypertrophic response to mechanical stress in skeletal muscle. Indeed, expression patterns of FAK have been reported to be load dependent; i.e., phosphorylation of FAK was lowered following hindlimb suspension in rodents (28) and immobilization in humans (13), and increased in models of chronic overload (18, 28), and following hypertrophic resistance exercise training in humans (58). Finally, local overexpression of FAK (pCMV-FAK plasmid electroporation) in vivo in rodents was shown to stimulate muscle hypertrophy (16) and induce S6K1 phosphorylation (37), suggesting that FAK is a key component of muscle hypertrophy.
FAK is activated not only by integrin engagement but also through stimulation by hormones and growth factors, including insulin and insulin-like growth factor-I (IGF-I) (3, 10, 25). In fibroblasts, it was demonstrated that FAK physically interacted with the IGF-I receptor, which was proposed to be important for the stabilization and phosphorylation of the IGF-I receptor (2). This stabilization impacted on downstream Akt/extracellular signal-related kinase (ERK) signaling, indicating a direct association between FAK and IGF-I receptor signaling. The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway is a critical regulator of IGF-I-mediated skeletal muscle hypertrophy (5, 48), and cardiac hypertrophy due to FAK overexpression in mouse hearts was associated with stimulation of PI3K/Akt signaling pathways (11). FAK was also reported to modulate the activity of this pathway through increased phosphorylation of S6 kinase 1 (S6K1) (37), which is a potential regulator of in vivo muscle protein synthesis (MPS) downstream of mTOR (47, 56). In a recent study (40), activation of mTOR signaling and subsequent hypertrophy in response to mechanical stress (stretch) in rat neonatal cardiomyocytes was reported to occur via activation of FAK, which was itself subject to negative regulation by the protein tyrosine phosphatase SH2 domain-containing protein tyrosine phosphatase (Shp-2). Thus, these data implicate a central regulatory role for FAK in both mechanical activity- and growth factor-induced hypertrophy in skeletal muscle; however, the degree of reliance on intact FAK signaling for hypertrophy-related signaling in skeletal muscle remains to be defined.
In C2C12 myotubes, IGF-I incubation is a classical model for studying muscle hypertrophy (i.e., increases in myotube diameter) through activation of the canonical PI3K/Akt/mTOR signaling pathway (39, 48). Herein, we used this approach to assess whether FAK is required for myocyte growth and to define some of the mechanisms involved; we hypothesized that reducing FAK expression by short hairpin (sh)RNA would restrict growth in response to IGF-I. More specifically, we sought to explore whether the role of FAK in IGF-I-mediated growth of C2C12 myotubes was due to a structural property of FAK in the adhesion complex or through its catalytic role as a kinase, using a reportedly specific inhibitor of FAK that targets its phosphorylation at Tyr397 (26).
MATERIALS AND METHODS
Cell culture.
Murine C2C12 myoblasts (passage nos. 6-9; ECACC, Salisbury, UK) were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Paisley, UK) containing 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; Lonza BioWhittaker, Verviers, Belgium), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (250 mg/ml; all from Sigma-Aldrich, Poole, UK) at 37°C and in a 5% CO2 atmosphere. Proliferating myoblasts were seeded onto six-well plates (Nunclon Delta; Thermo Scientific), and after 2–4 days (at ∼95% confluency), the medium was switched to differentiation medium consisting of DMEM containing 2% (vol/vol) horse serum (Sigma-Aldrich) as well as the above mixture of antimycotics and antibiotics to induce differentiation of the myoblasts into multinucleated myotubes. Differentiation medium was changed every 24 h, and experiments were performed on days 4 and 5 postinduction of differentiation. Experiments where signaling was measured were carried out >24 h following a medium change.
shRNA interference.
The lentiviral plasmid used (pLKO.1-mFAK) was obtained from OpenBiosystems (Huntsville, AL; Clone ID: RMM4534-NM_001130409) and targeted the mouse sequence 5′-CAA CCT TAA TAG AGA AGA AA-3′; the scrambled shRNA (SCR) was used as a negative control, as reported previously (35), with a hairpin sequence: CCT AAG GTT AAG TCG CCC TCG CTC TAG CGA GGG CGA CTT AAC CTT AGG (Addgene plasmid 1864; Addgene, Cambridge, MA). The plasmids were transformed in DH5α cells and isolated. The actual DNA sequence was confirmed at the Pennsylvania State University College of Medicine DNA sequencing core facility. Packaging plasmids psPAX2 and envelope protein plasmid pMD2.G were a gift from Trono Lab (Addgene plasmids 12260 and 12259 respectively). Human embryonic kidney-293FT cells (Invitrogen, Carlsbad, CA) were grown in DMEM; 80–85% confluent plates were rinsed once with Opti-MEM (Invitrogen) and then incubated with Opti-MEM for 4 h before transfections. psPAX2 and pMD2.G along with either scramble or pLKO.1-mFAK were added after mixing with Lipofectamine 2000 as per the manufacturer's instructions (Invitrogen). Opti-MEM media was changed after overnight incubation with DMEM containing 10% FBS, without antibiotics to allow cells to take up the plasmids and recover. Culture media were collected at 36 and 72 h posttransfection for viral particles. Viral particles present in the supernatant were harvested after a 15-min spin at 1,500 g to remove cellular debris. The supernatant was further filtered using a 0.45-μm syringe filter. A supernatant-containing virus was stored at −80°C for long-term storage. C2C12 cells at 60% confluence were infected twice overnight with 3 ml of viral supernatant containing 8 μg/ml polybrene in serum-free, antibiotic-free DMEM. Fresh DMEM containing 10% FBS, antibiotics, and 2 μg/ml puromycin (Sigma, St. Louis, MO) was added the next day. Cells were selected for two to five generations in puromycin, and no selection was used in the generation where cells were to be used experimentally. Cells that survived under puromycin selection were either harvested (as stable cells) and stored or used as myotubes following differentiation. For FAK-KD, three independent shRNAs targeting FAK mRNA were designed, and the one that gave the highest degree of knockdown was selected to proceed with experimentation.
IGF-I and FAK14 inhibitor incubations.
Experiments using IGF-I were carried out on days 4 and 5 postinduction of differentiation. For acute experiments, the medium was changed 24 h prior to treatments, after which cells were incubated in the presence of 10 ng/ml long R3 IGF-I (Sigma-Aldrich) for 2, 4, and 8 h. For chronic IGF-I treatment, the medium was changed immediately before cells were incubated in 10 ng/ml of IGF-I for 24 h. Long R3 IGF-I was chosen due to its low affinity for IGF-I-binding proteins and used at a dose shown previously to induce hypertrophy in C2C12 cells (39). For inhibitor experiments, cells were incubated for 30 min prior to IGF-I administration with 1 μmol/l of FAK14 inhibitor (F14; Tocris Bioscience, Bristol, UK). The dose of F14 was chosen based upon initial experiments demonstrating that 1 μmol/l of F14 effectively suppressed basal FAK Tyr397 phosphorylation after 2 h of treatment (data not shown). At the end of the experiments the medium was retained, and cells were washed twice in ice-cold phosphate-buffered saline before being harvested in sodium hydroxide for protein/DNA measurements or extraction buffer for immunoblotting (see below).
Myotube diameter.
Following 24 h of IGF-I treatment, light microscope images were taken, and myotube diameter was assessed using Image J software (National Institutes of Health, Frederick, MD), measuring 10 myotubes per field and using five random fields. Data were expressed relative to the 0-h time point in the SCR control group.
Protein/DNA measurements.
Total alkaline-soluble protein and DNA were measured in cells following 24 h of IGF-I treatment. Cells were harvested in 0.3 mol/l NaOH and incubated at 37°C for 30 min before an aliquot was removed for measurement of total protein using the Bradford assay. The remaining sample was incubated at 4°C for 10 min in the presence of 1 mol/l perchloric acid (PCA), before samples were centrifuged at 3,000 g for 10 min. The resultant pellet was washed with 0.2 mol/l PCA before being incubated at 70°C for 1 h in 2 mol/l PCA. After further centrifugation of samples at 5,000 g for 10 min, the DNA-containing supernatant was collected, and DNA was quantified by spectrophotometric measurement of the absorbance at 260 and 280 nm (NanoVue; GE Healthcare, Little Chalfont, UK).
MPS measurements.
For acute measurements of MPS, the surface sensing of translation technique was used (27, 52). This method involves incubation of cells with the antibiotic puromycin (a tyrosyl-tRNA analog) and subsequent immunoblotting using anti-puromycin antibodies to assess levels of incorporation of puromycin into newly synthesized polypeptide chains. Puromycin (1 μmol/l) was added to cells in the last 30 min of IGF-I treatment, and cells were harvested in extraction buffer for measurement of puromycin-labeled peptides by immunoblotting (see below), using mouse monoclonal puromycin antibody (12D10).
Chronic (24 h) measurements of MPS were performed using the stable isotope tracer deuterium oxide (D2O), a method adapted specifically for assessing longer-term rates of MPS (22). Medium was changed immediately prior to cells being incubated either with or without 10 ng/ml IGF-I for 24 h and replaced with fresh medium enriched with 5% D2O. Following incubations, 1 ml of medium was sampled from each well, and cells were harvested in 1 ml of PCA (1 mol/l). Following homogenization, samples were centrifuged (10,000 rpm, 10 min). The mixed muscle pellet was washed twice in 70% ethanol and hydrolyzed overnight at 110°C in 1 ml of HCl (0.1 mol/l) and 1 ml of +H dowex resin. Hydrolyzed amino acids were eluted into 2 mol/l NH4OH and evaporated to dryness. Deuterium labeling of protein-bound alanine was determined using gas chromatography-mass spectrometry following conversion to its tert-butyldimethylsilyl derivative and single ion monitoring (SIM) of m/z 260 and 261. Medium sampled from each well was analyzed for D2O enrichment using a modification of the acetone exchange method (59). Briefly, 2 μl of 10 N NaOH was added to 100 μl of medium, and following a 15-s vortex mix, 1 μl of acetone was added. This was incubated for 24 h to allow full hydrogen/deuterium exchange. The acetone was extracted into 200 μl of n-heptane, the n-heptane layer was transferred to an autosampler vial, and 0.5 μl was injected into the gas chromatograph-mass spectrophotometer. D2O enrichment was determined via SIM of m/z 58 and 59 with reference to a standard curve of known D2O enrichments. Fractional synthesis rate (FSR) was calculated using the following equation:
where MPEAla represents protein bound alanine enrichment, MPEMW represents medium water enrichment, and t represents time in hours. From this, the absolute rate of protein synthesis (ASR) was calculated by
Immunoblotting.
Cells were homogenized in extraction buffer (50 mM Tris·HCl, pH 7.5, 1 mM EDTA, 1 mM EGTA, 10 mM β-glycerophosphate, 50 mM NaF and complete protease inhibitor tablet; Roche, West Sussex, UK) by repeated pipetting using gel-loading tips and were centrifuged at 10,000 g for 10 min at 4°C. The supernatant was used for immunoblotting. Protein was quantified by Bradford assays, and samples were diluted and boiled in 1 × Laemmli loading buffer to the same concentration. Samples (5 μg of total protein) were loaded onto Criterion XT Bis-Tris 4–12% SDS-PAGE gels (Bio-Rad, Hemel Hempstead, UK) for electrophoresis at 200 V for 1 h. Samples were transferred to polyvinylidene difluoride membranes for 45 min at 100 V. Membranes were subsequently blocked in 2.5% low-fat milk [diluted in Tris-buffered saline and 0.1% Tween-20 (TBS-T)] for 1 h at room temperature and then incubated overnight at 4°C in the presence of the following primary antibodies: diluted 1:2,000 FAK Tyr397 (no. 8556), total FAK (no. 3285), Akt Ser473 (no. 4058) and Thr308 (no. 4056), ERK 1/2 Thr202/Tyr204 (no. 4377), proline-rich Akt substrate of 40 kDa (PRAS40) Thr246 (no. 2640), tuberous sclerosis complex 2 (TSC2) Thr1462 (no. 3611), S6K1 Thr389 (no. 9205) and Thr421/Ser424 (no. 9204), eukaryotic initiation factor (eIF)4E-binding protein 1 (4E-BP1) Thr37/46 (no. 2855), eukaryotic elongation factor 2 (eEF2) Thr56 (no. 2331), and eIF4G Ser1108 (no. 2441) (all from Cell Signaling Technology, Beverly, MA). The following day, membranes were washed for 3 × 5 min with TBS-T and incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-rabbit secondary antibody (New England Biolabs, Hitchin, UK). Membranes were washed for 3 × 5 min in TBS-T and incubated for 5 min with enhanced chemiluminescence reagent (Millipore, Watford, UK) before being visualized using a Chemidoc XRS system (Bio-Rad). Bands were quantified by measurement of peak density and normalization against Coomassie brilliant blue staining of the membrane (57). Since our principal objective was to compare the effects of FAK-KD on hypertrophic responses to IGF-I incubation, the full time course of immunoblotting data for each cell phenotype was run on separate gels and quantified relative to the 0-h time point in corresponding SCR or FAK-KD groups. Nonetheless, additional experiments indicated that there were no significant differences in basal phosphorylation of selected targets (S6K1 Thr389, Akt Ser473, and 4E-BP1 Thr37/46) between FAK-KD and scrambled shRNA cells when samples from each cell phenotype were run on the same gel (data not shown).
m7GTP-sepharose eIF4E affinity purification.
m7GTP-Sepharose affinity purification was used to assess interactions between components of the eIF4F complex, namely associations between eIF4G and eIF4E and between 4E-BP1 and eIF4E. Protein lysate (300 μg) was incubated with 30 μl of m7GTP-Sepharose 4B bead slurry (GE Healthcare, Little Chalfont, UK) at 4°C with continuous rotation overnight. Beads were subsequently collected by centrifugation (1,000 g, 2 min) and washed twice with buffer (50 mM Tris·HCl, pH 7.4, 140 mM NaCl, 1 mM EGTA, 0.5 mM NaVO4, 50 mM NaF, and complete protease inhibitor tablet; Roche). Bound proteins were eluted by boiling samples at 100°C for 7 min in the presence of Laemmli buffer. Samples were centrifuged to recover protein and were loaded onto 4–12% polyacrylamide gels for immunoblotting, as described above, with measurement of total levels of eIF4E (sc-13963), 4E-BP1 (sc-6936), and eIF4G (sc-11373) (all from Santa Cruz Biotechnology).
Coimmunoprecipitation.
Protein lysate (100 μg) from scrambled shRNA cells treated with IGF-I for 2 and 4 h were used for immunoprecipitation with FAK antibody and protein G-agarose according to the manufacturer's protocol (Sigma-Aldrich). Immunoprecipitated samples were used for immunoblotting (see above) with antibodies against Shp-2 (no. 3752) and TSC2 (no. 3612) (Cell Signaling Technology).
Statistical analysis.
Results were analyzed by unpaired Student's t-test for two group comparisons, one-way ANOVA with Tukey's post hoc testing for multiple group comparisons and two-way repeated-measures ANOVA with Bonferroni posttests for multiple groups with multiple time points, with statistical significance set at P < 0.05 (GraphPad Prism version 5.0; GraphPad Software, La Jolla, CA). Data are presented as means ± SE and represent six replicates (wells) per experiment.
RESULTS
Efficacy of lentiviral-mediated knockdown of FAK protein.
As shown in Figure 1A, efficacy of lentiviral-mediated knockdown of FAK was confirmed at the protein level in myotubes on day 4 postinduction of differentiation (−87 ± 1% vs. scrambled shRNA cells, P < 0.0001). The average myotube diameter of FAK-KD cells was consistently lower than in scrambled shRNA cells (−35 ± 9%, P < 0.05; Fig. 1, B and C).
Fig. 1.
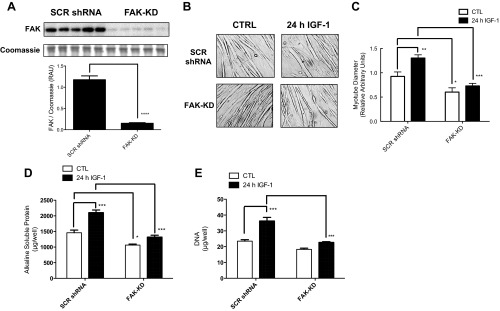
Protein expression of total focal adhesion kinase (FAK) in nontreated scrambled (SCR) short hairpin (sh)RNA vs. FAK-knockdown (FAK-KD) C2C12 cells (A), light microscope images (B), mean myotube diameter (C), total protein per well (D), and total DNA per well (E) following chronic insulin-like growth factor-I (IGF-I) treatment in FAK vs. scrambled shRNA cells. Protein expression and myotube width data are presented as relative arbitrary units ± SE (n = 6–12 replicates/group). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 vs. corresponding non-IGF-I-treated or scrambled shRNA groups. CTL, control.
Effect of 24-h IGF-I treatment on myotube diameter and protein/DNA content in FAK vs. scrambled shRNA C2C12 cells.
IGF-I treatment for 24 h increased average myotube width (+41 ± 7%, P < 0.01, vs. non-IGF-I-treated controls; Fig. 1, B and C) as well as total protein (+44 ± 6%, P < 0.001; Fig. 1C) and DNA content (+54 ± 9%, P < 0.001; Fig. 1E) per well in scrambled shRNA cells. IGF-I-mediated increases in myotube diameter, total protein, and DNA in FAK-KD cells (all nonsignificant vs. non-IGF-I-treated controls) were significantly attenuated compared with scrambled shRNA cells (Fig. 1, C–E).
FAK and Akt/mTOR signaling after IGF-I treatment in FAK vs. scrambled shRNA C2C12 cells.
Initial experiments indicated that baseline phosphorylation of measured targets was similar between FAK-KD and scrambled shRNA cells (data not shown). Phosphorylation of FAK at Tyr397 was increased significantly after 2 (+30 ± 5%) and 4 h (+28 ± 6%) of IGF-I treatment in scrambled shRNA cells, returning to basal levels by 8 h (Fig. 2A). Predictably, FAK Tyr397 phosphorylation was not increased in FAK-KD cells following IGF-I administration (Fig. 2A). Total FAK protein expression was unaltered with IGF-I, except for at the 8-h time point, where total FAK was similarly reduced in FAK-KD and scrambled shRNA cells (−22 ± 3%; Fig. 2B). At all time points, IGF-I increased Akt Ser473 phosphorylation significantly, with a peak increase at 2 h (+306 ± 6%; Fig. 2C). Increases in Akt Ser473 were similar in FAK knockdown cells, with the exception of the 2-h time point, where the increase was somewhat attenuated (+212 ± 7; Fig. 2C). Similarly, IGF-I increased Akt Thr308 phosphorylation at each time point in scrambled shRNA cells, with the largest increases observed at 2 (+460 ± 22%) and 8 h (+438 ± 17%) (Fig. 2D). Increases in Akt Thr308 phosphorylation with IGF-I at 8 h in FAK-KD cells were significantly blunted compared with control cells (+271 ± 27%), albeit still significantly elevated above non-IGF-I-treated controls.
Fig. 2.
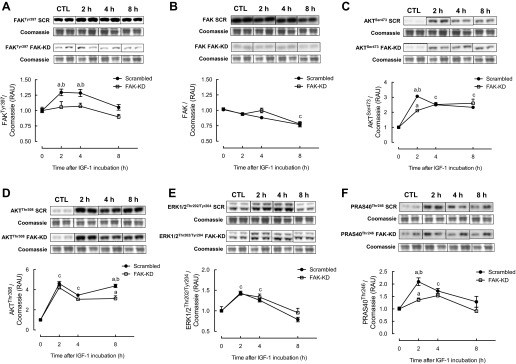
Changes in phosphorylation of FAK (A), total FAK protein expression (B), phosphorylation of Akt (C and D), extracellular signal-related kinase 1/2 (ERK1/2; E), and proline-rich Akt substrate of 40 kDa (PRAS40; F) in response to IGF-I treatment in FAK vs. scrambled shRNA C2C12 cells. Data are presented as relative arbitrary units (RAU) ± SE normalized to Coomassie staining of the membrane (n = 12 replicates/group). aP < 0.05 vs. respective basal group; bP < 0.05 between groups at that time point; cP < 0.05 from basal at that time point for both groups.
Phosphorylation of ERK1/2 at Thr202/Tyr204 was induced following IGF-I treatment similarly in both scrambled and FAK-KD cells (+40 ± 5% at 2 h; Fig. 2E). In scrambled shRNA cells, PRAS40 Thr246 phosphorylation was increased after 2 (+209 ± 15%) and 4 h (+172 ± 10%) of IGF-I treatment. PRAS40 phosphorylation was increased in FAK-KD cells in response to IGF-I; however, the increase at 2 h was significantly attenuated compared with the scrambled shRNA group (+135 ± 7%; Fig. 2F). At all time points, IGF-I increased phosphorylation of TSC2 significantly at Thr1462 in scrambled shRNA cells (+384 ± 17% at 2 h; Fig. 3A), and this was markedly attenuated in FAK knockdown cells, with only a significant increase observed at 2 h (+189 ± 12%). As shown in Fig. 3B, in scrambled shRNA myotubes, S6K1 Thr389 phosphorylation was elevated at all time points with IGF-I (+187 ± 2% at 2 h; Fig. 3B), but there was no change in S6K1 Thr389 in FAK knockdown cells. Similarly, IGF-I increased S6K1 Thr421/Ser424 in scrambled shRNA cells (+265 ± 16% at 2 h; Fig. 3C), whereas this increase was significantly lower in FAK-KD cells. Phosphorylation of 4E-BP1 at Thr37/46 was increased at all time points with IGF-I (+60 ± 3% at 2 h), whereas in FAK-KD cells there was a reduction in 4E-BP1 phosphorylation with IGF-I (Fig. 3D). In both scrambled and FAK-KD cells, IGF-I resulted in a significant reduction in eEF2 Thr56 phosphorylation (−23 ± 2% at 2 h; Fig. 3E), returning to control levels by 8 h. Finally, eIF4G Ser1108 phosphorylation was increased after 2 h of IGF-I treatment in both scrambled (+76 ± 12%) and FAK (+60 ± 3%) shRNA cells (Fig. 3F), and levels remained elevated in both groups at subsequent time points.
Fig. 3.
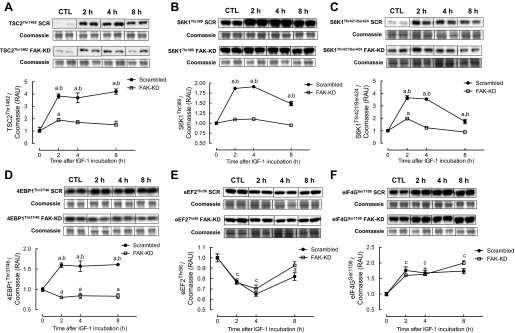
Changes in phosphorylation of tuberous sclerosis 2 (TSC2; A), ribosomal protein S6 kinase (S6K1; B and C), eukaryotic initiation factor (eIF)4E-binding protein 1 (4E-BP1; D), eukaryotic elongation factor 2 (eEF2; E), and eIF4G (F) in response to IGF-I in FAK vs. SCR shRNA C2C12 cells. Data are presented as RAU ± SE normalized to Coomassie staining of the membrane (n = 12 replicates/group). aP < 0.05 vs. respective basal group; bP < 0.05 between groups at that time point; cP < 0.05 from basal at that time point for both groups.
Changes in translational regulators, MPS, and binding of FAK to Shp-2 and TSC2 following IGF-I treatment.
Reduced binding of 4E-BP1 to eIF4E was seen following IGF-I treatment in scrambled shRNA cells (−474 ± 40% at 4 h; Fig. 4A), and in FAK-KD cells a reduction was observed, but this was significantly less than in scrambled shRNA cells (−163 ± 49% at 4 h). IGF-I increased binding of eIF4G to eIF4E in scrambled shRNA cells, with the largest increase detected at 4 h (+547 ± 28%; Fig. 4B). Increased binding of eIF4G to eIF4E was observed in FAK-KD cells, but was significantly lower than in control cells (+194 ± 35% at 4 h). IGF-I-induced increases in protein synthesis over 24 h (+73 ± 20% in scrambled shRNA cells), as measured by changes in D2O-bound alanine, were suppressed in FAK shRNA cells (Fig. 4C). Additionally, IGF-I increased MPS significantly after 4 h in scrambled shRNA cells (+135 ± 6%), as measured by incorporation of puromycin into newly synthesized peptide chains, but this was attenuated in FAK-KD cells (Fig. 4, D and E). Although Shp-2 and TSC2 both coimmunoprecipitated with FAK, there were no significant changes in binding of Shp-2 or TSC2 to FAK following 2 or 4 h of IGF-I treatment in scrambled shRNA cells (Fig. 4F).
Fig. 4.
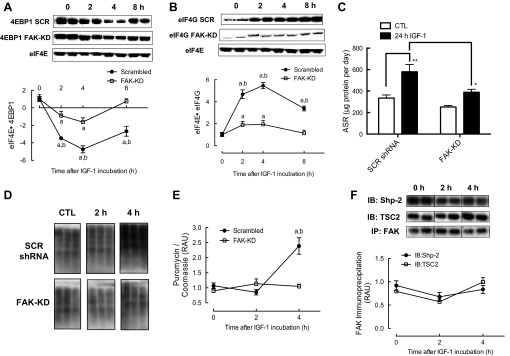
Changes in binding of 4E-BP1 to eIF4E (A) and eIF4G to eIF4E (B), changes in protein synthesis as measured by deuterium oxide (C) and puromycin (D and E) in response to IGF-I in FAK vs. SCR shRNA C2C12 cells, and changes in binding of FAK to SH2 domain-containing protein tyrosine phosphatase-2 (Shp-2) and TSC2 (F) in response to IGF-I in scrambled shRNA cells. Data are presented as RAU ± SE (n = 6 replicates/group). aDifferent from respective basal group; bdifferent between groups at that time point. *P < 0.05 and **P < 0.01 vs. respective control group. ASR, absolute synthesis rate; IB, immunoblot; IP, immunoprecipitation.
Changes in protein, DNA, and Akt/mTOR signaling with inhibition of FAK kinase activity following IGF-I treatment.
IGF-I-induced increases in total protein (+34 ± 3%) and DNA (+23 ± 4%) after 24 h were significantly attenuated when scrambled shRNA cells were incubated in the presence of F14, an inhibitor of FAK Tyr397 phosphorylation (Fig. 5, A and B). IGF-I treatment for 2 h (a time point where signaling responses to IGF-I were predominantly at their largest) induced an increase in FAK Tyr397 (+28 ± 9%), and this was significantly suppressed with inhibition of FAK phosphorylation (Fig. 5C). Total FAK protein expression was unchanged with IGF-I or F14 (Fig. 5D). IGF-I induced increases in Akt Ser473 (+528 ± 49%), S6K1 Thr389 (+110 ± 9%), and 4E-BP1 Thr37/46 (+88 ± 7%), and these were significantly suppressed with inhibition of FAK phosphorylation (Fig. 5, E–G).
Fig. 5.
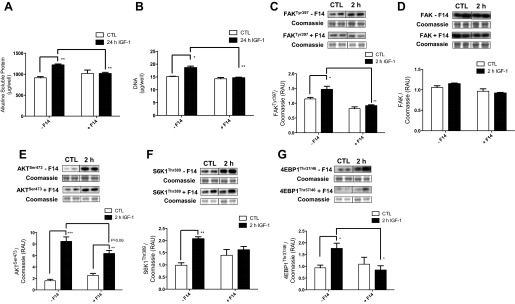
Protein content (A), DNA content (B), and changes in phosphorylation of focal FAK (C), total FAK protein expression (D) and phosphorylation of Akt (E), S6K1 (F), and 4E-BP1 (G) in response to IGF-I and FAK inhibitor 14 (F14) in SCR shRNA C2C12 cells. Data are presented as RAU ± SE (n = 6 replicates/group). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. respective control group.
DISCUSSION
Integrins are cell surface receptors that interact with ECM molecules and mediate intracellular signal transduction through the formation of multiprotein complexes known as focal adhesion complexes, and FAK is thought to be a key player in this process (34). Integrin-mediated signaling is a central mediator of mechanical load-induced hypertrophy, and there are several lines of evidence implicating activation of FAK in load-mediated induction of protein synthesis and hypertrophy in rodent skeletal muscle in vivo (16, 37). Moreover, FAK can also be modulated by nonintegrin stimuli such as insulin and IGF-I (3, 25) and may be involved in PI3K/Akt activation (2). However, no studies to date have elucidated the requirement or molecular mechanisms by which FAK mediates growth in skeletal muscle. In the present study, we demonstrated that shRNA-mediated stable knockdown of FAK in C2C12 cells was associated with a marked attenuation of the growth response associated with IGF-I, and this was associated with FAK-mediated stimulation of the TSC2/mTOR pathway (Fig. 6). Critically, this appears to be related to the catalytic action of FAK rather than to its structural properties.
Fig. 6.
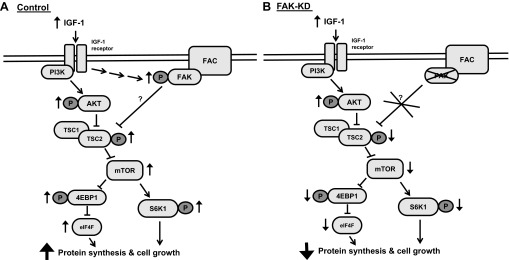
Proposed mechanism of FAK-mediated growth of C2C12 cells in response to IGF-I. A: IGF-I induces phosphorylation/activation of FAK. FAK facilitates in activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway by inhibiting TSC2, ultimately resulting in activation of mammalian target of rapamycin (mTOR), and subsequent activation of S6K1 and 4E-BP1, 2 key regulators of muscle protein synthesis. In FAK-KD cells (B), cell growth is inhibited through reduced activation of mTOR, 4E-BP1, and S6K1, secondary to reduced inhibition of TSC2. FAC, focal adhesion complex; P, phosphorylation.
Skeletal muscle unloading has been associated with reduced levels of FAK phosphorylation both in rodent soleus muscle after 7-day tail suspension (28) and following leg immobilization in humans (13, 24). Furthermore, increased FAK phosphorylation and protein content have been observed after training in animal models (18, 28) and in humans (58) and has been associated with the activation of anabolic signaling pathways (37) and hypertrophy in muscle (16). In this study, we demonstrated that IGF-I-induced cell growth (as estimated by changes in myotube diameter and total protein; Fig. 1) was markedly suppressed in C2C12 myotubes harboring FAK-targeted shRNA. Furthermore, untreated FAK-KD myotubes were generally thinner than control cells (Fig. 1), suggesting that FAK is also required for normal cell growth. IGF-I also increased DNA content significantly in scrambled shRNA cells, and this increase was observed consistently whether cells were treated with IGF-I at the onset of differentiation or when myotubes were fully formed (data not shown). The observed increases in DNA content with IGF-I treatment could have potentially been due to IGF-I forcing remaining quiescent undifferentiated myoblasts into the cell cycle, highlighting the fact that IGF-I represents a potent growth factor on a population of quiescent myoblasts among differentiated myotubes. However, it has been shown previously (43) that treatment with DNA synthesis inhibitor arabinofuranosyl cytidine did not affect the ability of IGF-I to induce increases in myotube diameter in C2C12 cells, indicating that the hypertrophic effects of IGF-I may have been independent of its mitogenic effects. Nevertheless, increases in total protein and DNA with IGF-I appeared to be dependent upon FAK, suggesting governance over both postmitotic and mitotic cell growth.
IGF-I-mediated myotube growth in vitro occurs via activation of anabolic signaling pathways, particularly the PI3K/Akt/mTOR pathway (5, 48). Recent studies have suggested that the integrin-associated protein FAK could be an important component of the IGF-I/PI3K/Akt pathway in muscle (37, 40). In the current study, IGF-I induced an acute, transient increase in FAK Tyr397 phosphorylation that has been linked with its activation (8). Increases in Akt Ser473 and Thr308 phosphorylation with IGF-I were similar in both scrambled and FAK shRNA cells, apart from at the 2-h time point for Ser473 [residue under regulation by mTORc2 (50)] and at 8 h for Thr308 [residue downstream of PI3K (1)], where there was a suppression in FAK knockdown cells. Whether the suppression of Akt in FAK-KD cells at the time points observed in the present study was important in the blunted anabolic response to IGF-I remains to be established. However, there is evidence that, although Akt Thr308 may be important for mTOR regulation, Ser473 phosphorylation might be more important in the regulation of forkhead box O transcription factors (29, 33). Thus the suppression in Akt Ser473 in FAK-KD cells after 2 h of IGF-I treatment is unlikely to have impacted on downstream mTOR signaling. Nevertheless, the suppressed phosphorylation of Akt Ser473 at 2 h paralleled changes in Akt substrate PRAS40 Thr246 phosphorylation in FAK-KD cells. Phosphorylation at this residue is thought to relieve PRAS40 inhibition of mTOR, which in nonstimulated conditions occurs through binding to raptor, preventing Ras homolog enriched in brain (Rheb)-mediated activation of mTOR (49). It has been demonstrated previously that activation of mTORc1 can cause feedback inhibition of insulin receptor substrate-1, resulting in reduced Akt phosphorylation at Thr308 (54). This was not observed in the present study, although Akt phosphorylation was not measured beyond 8 h of IGF-I treatment. Nonetheless, it is feasible that feedback inhibition from mTORC1 activation to proximal IGF-I signaling is not active, at least during IGF-I-induced muscle growth. In contrast to the changes in Akt phosphorylation, increased ERK1/2 Thr202/Tyr204 phosphorylation in response to IGF-I did not differ in FAK-KD and control shRNA cells. These data contrast with previous studies showing that ERK1/2 activation in cardiac myocytes paralleled activation of FAK in response to mechanical stress (15, 19), suggesting a role for ERK1/2 signaling in the hypertrophic effects of FAK in response to mechanical signals as opposed to growth factor stimulation and/or in cardiac but not skeletal myocytes.
Following stimulation by growth factors, Akt inhibits the GTPase activating protein activity of TSC2 through phosphorylation at Thr1462, leading to activation of the GTPase Rheb, a positive regulator of mTOR (21, 31, 60). In this study, IGF-I stimulation of TSC2 Thr1462 was ablated in FAK knockdown cells, suggesting that the mechanism by which FAK mediated IGF-I-induced growth occurred in a TSC2-dependent manner. It is not clear whether FAK was directly responsible for the increased phosphorylation at this residue on TSC2, as it is thought to be downstream of Akt (as described above). There is evidence to suggest that FAK may positively regulate anabolic signaling pathways through direct interaction with TSC2, albeit in nonmuscle cells (20), but whereas in the present study coimmunoprecipitation experiments revealed binding of FAK to TSC2 in scrambled shRNA cells, there were no differences in interactions between IGF-I-treated and control cells (Fig. 6). Furthermore, there was no evidence of tyrosine phosphorylation of TSC2 under any of the present conditions (data not shown), suggesting that the blunting of IGF-I-induced TSC2 Thr1462 phosphorylation in FAK-KD cells was independent of TSC2 tyrosine phosphorylation.
Concurrent with an inhibition of TSC2 phosphorylation in FAK-KD cells, IGF-I-induced increases in S6K1 phosphorylation at both Thr389 and Thr421/Ser424 [both required for the full activation of p70 kinase activity (46) and an indirect marker of mTOR activity (32)] were completely suppressed in FAK-KD cells. Moreover, increases in Thr37/46 phosphorylation of 4E-BP1 [another substrate of mTOR (6)] seen with IGF-I treatment in control cells were completely attenuated in FAK-KD cells. S6K1 regulates multiple targets involved in protein translation, including eEF2 (56), and its activation has been correlated with increases in MPS in human subjects (38). 4E-BP1 is also important for cap-dependent protein translation, since phosphorylation of 4E-BP1 results in the release of translation factor eIF4E (36). Thus, FAK appeared to be important for full activation of anabolic signaling targets downstream of mTOR, namely S6K1 and 4E-BP1. This was associated with inhibition of TSC2 and occurred potentially via an Akt-independent mechanism (see Fig. 6).
In contrast to the differences in S6K1 and 4E-BP1 between FAK knockdown and control cells, decreases in eEF2 Thr56 and increases in eIF4G Ser1108 after IGF-I were similar in both FAK and scramble shRNA cells. It is thought that eEF2 catalyzes translocation of peptidyl-tRNA during elongation and is regulated by S6K1, whereas phosphorylation at Thr56 inactivates eEF2 via eEF2 kinase (56). However, in light of other results these changes may not represent important indicators of translation under the current conditions. Indeed, the observed changes in TSC2 and S6K1 strongly agree with our findings that increased association of eIF4E and eIF4G with IGF-I and reduced association of eIF4E with 4E-BP1 were attenuated in FAK knockdown cells. A crucial component of protein translation initiation is the binding of mRNA to the 43S preinitiation complex, and this is mediated by formation of the eIF4F complex (42). Binding of eIF4E to eIF4G enables the 40S ribosomal subunit to associate with the m7GTP cap structure on mRNA, and the ability of eIF4E to interact with eIF4G is dependent on the extent of its binding to 4E-BP1. Phosphorylation of 4E-BP1 reduces interactions between eIF4E and 4E-BP1, enabling formation of eIF4F and activating protein translation initiation. Thus, attenuation of increased eIF4E·eIF4G and reduced eIF4E·4E-BP1 interactions is consistent with impairment in activation of protein translation following IGF-I stimulation in FAK knockdown cells. The lack of differences in eIF4G phosphorylation between FAK-KD and control groups suggests that impairment of FAK activation was not important for activation of eIF4G and that the impairment of IGF-I-mediated activation of translation initiation through FAK occurred through impaired activation of eIF4E and upstream factors. We also measured changes in MPS after 2 and 4 h of IGF-I treatment (time points where signaling responses were generally at their greatest). Increases in MPS after 4 h of IGF-I treatment were significantly suppressed in FAK knockdown cells. The lack of detectable changes in MPS during the first 2 h of IGF-I treatment, despite activation of multiple anabolic signaling targets in control cells, indicates that there may have been some delay in the onset of IGF-I-induced increases in MPS. Nevertheless, increases in MPS over 24 h, as assessed by D2O incorporation into protein-bound alanine, were attenuated in FAK-KD cells.
Increased phosphorylation of FAK at Tyr397 correlates with increased catalytic activity in response to mechanical stretch of muscle (40). Thus, we questioned whether the kinase activity of FAK was required for IGF-I-mediated growth in muscle cells or whether the effects were a result of its structural role in the costamere. Scrambled shRNA myotubes were incubated with F14, an ATP-dependent competitive inhibitor of FAK phosphorylation at Tyr397, such that FAK was present but unable to be phosphorylated at its key tyrosine residue and thus become activated. Inhibition of FAK Tyr397 phosphorylation using F14 suppressed IGF-I-mediated growth as well as acute (2 h) IGF-I-induced increases in Akt and S6K1 phosphorylation, providing strong evidence that FAK kinase activity is required for IGF-I-induced cell growth in skeletal muscle cells.
It is not clear how IGF-I activates FAK; however, it has emerged that FAK may be subject to regulation by the protein phosphatase Shp-2. Indeed, during muscle differentiation, Shp-2-mediated dephosphorylation of FAK is required for myoblasts to withdraw from the cell cycle and undergo differentiation into myotubes (14). Also, in cardiac myocytes, activation of mTOR signaling and hypertrophic growth in response to cyclic stretch was associated with activation of FAK, secondary to reduced inhibition by Shp-2. Thus, mTOR and S6K1 appear to be critical downstream effectors of anabolic signaling mediated by FAK following IGF-I stimulation, most likely via direct binding to TSC2, and potentially secondary to reduced inhibition by Shp-2. However, although coimmunoprecipitation analysis in the present study revealed direct binding of Shp-2 to FAK in scramble shRNA myotubes, there were no changes in the amount of binding in response to IGF-I. Thus, the mechanism by which IGF-I induced activation of FAK appeared not to have occurred via reduced binding/dephosphorylation by Shp-2.
We have identified that FAK represents a key component of IGF-I-mediated growth in C2C12 cells. Whether FAK plays an important part in human skeletal muscle mass regulation in vivo remains to be determined, but increasing evidence suggests its central role (13, 58). Furthermore, we hypothesize that dysregulation of the attachment complex could represent a crucial feature of pathological muscle-wasting conditions beyond genetic dystrophy disorders, e.g., in sarcopenia. Indeed, dystrophic phenotypes have been observed in rodents deficient in various attachment proteins in muscle, including talin (12) and various integrin subunits (23, 53). Furthermore, acute knockdown, by RNAi, of various attachment proteins in fully developed worm (17) and fly (44) muscle yields dystrophic phenotypes. However, it has also been reported that the costamere-associated protein integrin-linked kinase is an important component of activation of IGF-I receptor and Akt signaling in response to mechanical stress in skeletal muscle (55), demonstrating that molecular redundancy will almost certainly exist in vivo. Finally, it should also be highlighted that recent studies have demonstrated a complex relationship between mTORC1 signaling and skeletal muscle growth in vivo, where more mTORC1 activity does not necessarily translate into more growth (4, 45) such that although FAK represents a key component of IGF-I-mediated hypertrophy via mTORC1, this is just one component of a complex network of genes regulating hypertrophy in vivo. In conclusion, the findings of the present study provide support for the importance of attachment complex proteins such as FAK in muscle growth, although further investigation into their potential roles in aging and in wasting diseases in human skeletal muscle in vivo is warranted.
GRANTS
This work was supported by the National Institute for Arthritis and Musculoskeletal and Skin Diseases (Grant R01-AR-054342) and General Medical Sciences (Grant R01-GM-38032) and the Medical Research Council (G0801271).
DISCLOSURES
The authors declare that they have no conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
H.C., A.A.K., P.P., N.J.S., and P.J.A. contributed to the conception and design of the research; H.C., A.A.K., D.J.W., and P.J.A. performed the experiments; H.C., D.J.W., and P.J.A. analyzed the data; H.C., A.A.K., C.H.L., J.A.T., D.J.W., K.S., N.J.S., and P.J.A. interpreted the results of the experiments; H.C. prepared the figures; H.C. drafted the manuscript; H.C., A.A.K., C.H.L., J.A.T., K.S., N.J.S., and P.J.A. edited and revised the manuscript; H.C., A.A.K., C.H.L., J.A.T., P.P., D.J.W., K.S., N.J.S., and P.J.A. approved the final version of the manuscript.
REFERENCES
- 1. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551, 1996 [PMC free article] [PubMed] [Google Scholar]
- 2. Andersson S, D'Arcy P, Larsson O, Sehat B. Focal adhesion kinase (FAK) activates and stabilizes IGF-1 receptor. Biochem Biophys Res Commun 387: 36–41, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Baron V, Calleja V, Ferrari P, Alengrin F, Van Obberghen E. p125Fak focal adhesion kinase is a substrate for the insulin and insulin-like growth factor-I tyrosine kinase receptors. J Biol Chem 273: 7162–7168, 1998 [DOI] [PubMed] [Google Scholar]
- 4. Bentzinger CF, Lin S, Romanino K, Castets P, Guridi M, Summermatter S, Handschin C, Tintignac LA, Hall MN, Rüegg MA. Differential response of skeletal muscles to mTORC1 signaling during atrophy and hypertrophy. Skelet Muscle 3: 6, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Brunn GJ, Hudson CC, Sekulić A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277: 99–101, 1997 [DOI] [PubMed] [Google Scholar]
- 7. Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol 12: 463–518, 1996 [DOI] [PubMed] [Google Scholar]
- 8. Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 15: 954–963, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci 4: D102–D113, 1999 [DOI] [PubMed] [Google Scholar]
- 10. Casamassima A, Rozengurt E. Insulin-like growth factor I stimulates tyrosine phosphorylation of p130(Cas), focal adhesion kinase, and paxillin. Role of phosphatidylinositol 3′-kinase and formation of a p130(Cas) Crk complex. J Biol Chem 273: 26149–26156, 1998 [DOI] [PubMed] [Google Scholar]
- 11. Clemente CF, Xavier-Neto J, Dalla Costa AP, Consonni SR, Antunes JE, Rocco SA, Pereira MB, Judice CC, Strauss B, Joazeiro PP, Matos-Souza JR, Franchini KG. Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J Mol Cell Cardiol 52: 493–501, 2012 [DOI] [PubMed] [Google Scholar]
- 12. Conti FJ, Felder A, Monkley S, Schwander M, Wood MR, Lieber R, Critchley D, Muller U. Progressive myopathy and defects in the maintenance of myotendinous junctions in mice that lack talin 1 in skeletal muscle. Development 135: 2043–2053, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol 585: 241–251, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Oliveira MV, Marin TM, Clemente CF, Costa AP, Judice CC, Franchini KG. SHP-2 regulates myogenesis by coupling to FAK signaling pathway. FEBS Lett 583: 2975–2981, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Domingos PP, Fonseca PM, Nadruz W, Jr, Franchini KG. Load-induced focal adhesion kinase activation in the myocardium: role of stretch and contractile activity. Am J Physiol Heart Circ Physiol 282: H556–H564, 2002 [DOI] [PubMed] [Google Scholar]
- 16. Durieux AC, D'Antona G, Desplanches D, Freyssenet D, Klossner S, Bottinelli R, Fluck M. Focal adhesion kinase is a load-dependent governor of the slow contractile and oxidative muscle phenotype. J Physiol 587: 3703–3717, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Etheridge T, Oczypok EA, Lehmann S, Fields BD, Shephard F, Jacobson LA, Szewczyk NJ. Calpains mediate integrin attachment complex maintenance of adult muscle in Caenorhabditis elegans. PLoS Genet 8: e1002471, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fluck M, Carson JA, Gordon SE, Ziemiecki A, Booth FW. Focal adhesion proteins FAK and paxillin increase in hypertrophied skeletal muscle. Am J Physiol Cell Physiol 277: C152–C162, 1999 [DOI] [PubMed] [Google Scholar]
- 19. Franchini KG, Torsoni AS, Soares PH, Saad MJ. Early activation of the multicomponent signaling complex associated with focal adhesion kinase induced by pressure overload in the rat heart. Circ Res 87: 558–565, 2000 [DOI] [PubMed] [Google Scholar]
- 20. Gan B, Yoo Y, Guan JL. Association of focal adhesion kinase with tuberous sclerosis complex 2 in the regulation of s6 kinase activation and cell growth. J Biol Chem 281: 37321–37329, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 4: 699–704, 2002 [DOI] [PubMed] [Google Scholar]
- 22. Gasier HG, Fluckey JD, Previs SF. The application of 2H2O to measure skeletal muscle protein synthesis. Nutr Metab (Lond) 7: 31, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gheyara AL, Vallejo-Illarramendi A, Zang K, Mei L, St-Arnaud R, Dedhar S, Reichardt LF. Deletion of integrin-linked kinase from skeletal muscles of mice resembles muscular dystrophy due to alpha 7 beta 1-integrin deficiency. Am J Pathol 171: 1966–1977, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, Smith K, Rennie MJ. Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol 586: 6049–6061, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goel HL, Dey CS. Focal adhesion kinase tyrosine phosphorylation is associated with myogenesis and modulated by insulin. Cell Prolif 35: 131–142, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance WG. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem 51: 7405–7416, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, Hornberger TA. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 25: 1028–1039, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gordon SE, Flück M, Booth FW. Selected Contribution: Skeletal muscle focal adhesion kinase, paxillin, and serum response factor are loading dependent. J Appl Physiol 90: 1174–1183; discussion 1165, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11: 859–871, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Hynes RO. Cell adhesion: old and new questions. Trends Cell Biol 9: M33–M37, 1999 [PubMed] [Google Scholar]
- 31. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Isotani S, Hara K, Tokunaga C, Inoue H, Avruch J, Yonezawa K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J Biol Chem 274: 34493–34498, 1999 [DOI] [PubMed] [Google Scholar]
- 33. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125–137, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Juliano RL. Signal transduction by cell adhesion receptors and the cytoskeleton: functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu Rev Pharmacol Toxicol 42: 283–323, 2002 [DOI] [PubMed] [Google Scholar]
- 35. Kazi AA, Lang CH. PRAS40 regulates protein synthesis and cell cycle in C2C12 myoblasts. Mol Med 16: 359–371, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kimball SR, Shantz LM, Horetsky RL, Jefferson LS. Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J Biol Chem 274: 11647–11652, 1999 [DOI] [PubMed] [Google Scholar]
- 37. Klossner S, Durieux AC, Freyssenet D, Flueck M. Mechano-transduction to muscle protein synthesis is modulated by FAK. Eur J Appl Physiol 106: 389–398, 2009 [DOI] [PubMed] [Google Scholar]
- 38. Kumar V, Selby A, Rankin D, Patel R, Atherton P, Hildebrandt W, Williams J, Smith K, Seynnes O, Hiscock N, Rennie MJ. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. J Physiol 587: 211–217, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 280: 2737–2744, 2005 [DOI] [PubMed] [Google Scholar]
- 40. Marin TM, Clemente CF, Santos AM, Picardi PK, Pascoal VD, Lopes-Cendes I, Saad MJ, Franchini KG. Shp2 negatively regulates growth in cardiomyocytes by controlling focal adhesion kinase/Src and mTOR pathways. Circ Res 103: 813–824, 2008 [DOI] [PubMed] [Google Scholar]
- 41. Monti RJ, Roy RR, Hodgson JA, Edgerton VR. Transmission of forces within mammalian skeletal muscles. J Biomech 32: 371–380, 1999 [DOI] [PubMed] [Google Scholar]
- 42. Pain VM. Initiation of protein synthesis in eukaryotic cells. Eur J Biochem 236: 747–771, 1996 [DOI] [PubMed] [Google Scholar]
- 43. Park IH, Erbay E, Nuzzi P, Chen J. Skeletal myocyte hypertrophy requires mTOR kinase activity and S6K1. Exp Cell Res 309: 211–219, 2005 [DOI] [PubMed] [Google Scholar]
- 44. Perkins AD, Ellis SJ, Asghari P, Shamsian A, Moore ED, Tanentzapf G. Integrin-mediated adhesion maintains sarcomeric integrity. Dev Biol 338: 15–27, 2010 [DOI] [PubMed] [Google Scholar]
- 45. Phillips BE, Williams JP, Gustafsson T, Bouchard C, Rankinen T, Knudsen S, Smith K, Timmons JA, Atherton PJ. Molecular networks of human muscle adaptation to exercise and age. PLoS Genet 9: e1003389, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pullen N, Thomas G. The modular phosphorylation and activation of p70s6k. FEBS Lett 410: 78–82, 1997 [DOI] [PubMed] [Google Scholar]
- 47. Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 23: 1761–1769, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 3: 1009–1013, 2001 [DOI] [PubMed] [Google Scholar]
- 49. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903–915, 2007 [DOI] [PubMed] [Google Scholar]
- 50. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101, 2005 [DOI] [PubMed] [Google Scholar]
- 51. Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol 71: 435–478, 1999 [DOI] [PubMed] [Google Scholar]
- 52. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6: 275–277, 2009 [DOI] [PubMed] [Google Scholar]
- 53. Taverna D, Disatnik MH, Rayburn H, Bronson RT, Yang J, Rando TA, Hynes RO. Dystrophic muscle in mice chimeric for expression of alpha5 integrin. J Cell Biol 143: 849–859, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26: 1932–1940, 2007 [DOI] [PubMed] [Google Scholar]
- 55. Wang HV, Chang LW, Brixius K, Wickstrom SA, Montanez E, Thievessen I, Schwander M, Muller U, Bloch W, Mayer U, Fassler R. Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J Cell Biol 180: 1037–1049, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20: 4370–4379, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Welinder C, Ekblad L. Coomassie staining as loading control in Western blot analysis. J Proteome Res 10: 1416–1419, 2011 [DOI] [PubMed] [Google Scholar]
- 58. Wilkinson SB, Phillips SM, Atherton PJ, Patel R, Yarasheski KE, Tarnopolsky MA, Rennie MJ. Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J Physiol 586: 3701–3717, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yang D, Diraison F, Beylot M, Brunengraber DZ, Samols MA, Anderson VE, Brunengraber H. Assay of low deuterium enrichment of water by isotopic exchange with [U-13C3]acetone and gas chromatography-mass spectrometry. Anal Biochem 258: 315–321, 1998 [DOI] [PubMed] [Google Scholar]
- 60. Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5: 578–581, 2003 [DOI] [PubMed] [Google Scholar]