

Abstract
Liver-specific thyroid hormone receptor-β (TRβ)-specific agonists are potent lipid-lowering drugs that also hold promise for treating nonalcoholic fatty liver disease and hepatic insulin resistance. We investigated the effect of two TRβ agonists (GC-1 and KB-2115) in high-fat-fed male Sprague-Dawley rats treated for 10 days. GC-1 treatment reduced hepatic triglyceride content by 75%, but the rats developed fasting hyperglycemia and hyperinsulinemia, attributable to increased endogenous glucose production (EGP) and diminished hepatic insulin sensitivity. GC-1 also increased white adipose tissue lipolysis; the resulting increase in glycerol flux may have contributed to the increase in EGP. KB-2115, a more TRβ- and liver-specific thyromimetic, also prevented hepatic steatosis but did not induce fasting hyperglycemia, increase basal EGP rate, or diminish hepatic insulin sensitivity. Surprisingly, insulin-stimulated peripheral glucose disposal was diminished because of a decrease in insulin-stimulated skeletal muscle glucose uptake. Skeletal muscle insulin signaling was unaffected. Instead, KB-2115 treatment was associated with a decrease in GLUT4 protein content. Thus, although both GC-1 and KB-2115 potently treat hepatic steatosis in fat-fed rats, they each worsen insulin action via specific and discrete mechanisms. The development of future TRβ agonists must consider the potential adverse effects on insulin sensitivity.
Keywords: thyroid hormone receptor-β agonists, insulin resistance, hepatic steatosis
the burgeoning epidemics of obesity and diabetes necessitate novel therapies. Thyroid hormone agonism remains a tantalizing modality (1, 54). It has the potential to mediate favorable effects on lipid metabolism and energy expenditure, decreasing plasma low-density lipoprotein (LDL) and increasing energy expenditure through futile substrate cycling (34, 35, 46) and increasing thermogenesis (21). However, generalized thyroid hormone excess has a number of untoward effects, including tachyarrhythmias, pulmonary hypertension, osteoporosis, agitation, and even psychosis.
Thyroid hormones act primarily via three nuclear receptors, TRα1, TRβ1, and TRβ2; most tissues express both TRα and TRβ, although one isoform often predominates (1, 10, 26, 31, 55). For example, TRα1 expression predominates in the cardiac and skeletal muscle, osteoclast, and many regions of the brain. In comparison, TRβ1 predominates in liver, and TRβ2 plays a critical role in the regulation of the hypothalamic-pituitary-thyroid axis (8, 10, 19, 27). Activating TRβ in the liver could be therapeutic for metabolic diseases by lowering plasma and intrahepatic lipid content. However, natural thyroid hormones have nearly equivalent affinities for TRβ and TRα [KD(TRα1)/KD(TRβ1) = 0.7 (11)]. Thus, any attempts to reap the benefits from activating TRβ with thyroid hormones will be limited by the toxicities associated with TRα activation. The tissue-specific expression of TR isoforms does provide the opportunity for selective thyroid hormone action in specific tissues. Specifically, liver-selective TRβ-specific agonists can potentially uncouple the beneficial effect of thyroid hormone on hepatic and plasma lipids without the deleterious effects on heart and bone.
Several TRβ agonists have demonstrated efficacy in decreasing plasma and hepatic lipids in both preclinical and clinical trials (2, 3, 24, 33, 51). These compounds weakly bind to and activate TRα, thus enhancing the therapeutic dose range. They can be administered at high enough doses to enhance TRβ activation without the untoward effects from TRα activation that would be seen if thyroid hormone was given at similar doses. There are multiple mechanisms that may account for the reduction in hepatic lipids, including induction of hepatic carnitine palmitoyltransferase (CPT)-1α leading to an increase in hepatic lipid oxidation (28). We hypothesized that by amelioration of fatty liver, TRβ agonists would also improve lipid-induced hepatic insulin resistance. We studied two thyroid hormone analogs: GC-1 (sobetirome), a first-generation TRβ agonist with fivefold selectivity for TRβ over TRα (51, 53), and KB-2115 (eprotirome), a more β-selective TRβ agonist that is reported to be more hepatic specific than GC-1 (3, 24).
MATERIALS AND METHODS
Animals.
Male Sprague-Dawley rats (275–300 g) were received from Harlan Laboratories and acclimated for at least 3 days. Rats were housed on a 12:12-h light-dark cycle and received food and water ad libitum. Rats underwent the placement of jugular venous (for blood sampling) and carotid artery (for infusion) catheters 3–5 days before the initiation of TRβ agonist treatment. Rats were placed on a high-fat diet (Dyets 112245: 26% carbohydrate, 59% fat, 15% protein calories; Dyets, Bethlehem, PA) during the treatment period. TRβ agonists or vehicle (5% DMSO in phosphate-buffered saline) were injected intraperitoneally at a dose of 0.164 mg·kg-1·day-1 GC-1 or 0.1 mg·kg-1·day-1 KB-2115 for 10 days. Body weight was monitored three to four times weekly. Experiments were performed 16–24 h after the final injection of TRβ agonist. All procedures were approved by the Institutional Animal Care and Use Committee of the Yale University School of Medicine and the Veterans' Affairs Connecticut Healthcare System.
Quantitative PCR.
Total RNA was extracted from ∼15 mg liver, ∼100 mg white adipose tissue, or ∼100 mg skeletal muscle using an RNeasy mini kit (Qiagen, Valencia, CA). RNA was reverse-transcribed into cDNA with the use of the QuantiTect Reverse Transcription Kit (Qiagen). The abundance of transcripts was assessed by real-time PCR on an Applied Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA) with a SYBR Green detection system (Stratagene, La Jolla, CA). The expression data for each gene of interest were against β-actin as the invariant control and relative expression determined using amplification efficiencies (38). Primer sequences are shown in Table 1.
Table 1.
qPCR primer sequences
| Forward | Reverse | |
|---|---|---|
| β-Actin | CCAGATCATGTTTGAGACCTTC | CATGAGGTAGTCTGTCAGGTCC |
| Adiponectin | GCAACCGAAGGGCCAGGAGC | TCTGCCATCACGGCCCGGTA |
| Deiodinase 1 | TGGTTCGTCCTGAAGGTCCGC | TCAACACCAGGGGTCTGCTGC |
| Deiodinase 2 | ATGGGACTCCTCAGCGTAGA | GCACAGGCAAAGTCAAGAAG |
| G-6-Pase | TGCAAGGGAGAACTCAGCAA | GGACCAAGGAAGCCACAATG |
| GLUT4 | GCAGCGAGTGACTGGGCA | CCAGCCACGTTGCATTGTAG |
| Hairless | GCCCAGCGTATCCGTCGCTTT | AACCTGCGTCGCAACCCTGA |
| Malic enzyme 1 | ATGGAGAAGGAAGGTTTATCAAAG | GGCTTCTAGGTTCTTCATTTCTTC |
| PEPCK | ATGACACCCTCCTCCTGCAT | CAGGAAGTGAGGAAGTTTGTGG |
| PC | AGATGCACTTCCATCCCAAG | CCTTGGTCACGTGAACCTTT |
| SERCA 1 | GGTTTGGCAGGAACGGAAT | GGTGGATTTGATGGAGAGGAT |
| Sortilin 1 | CGATGACATGGTGTTCATGCA | CCTCGGTCATCAGAGGTAAAGATT |
| Tbp | GGACTCCTGTCTCCCCTACC | CTCAGTGCAGAGGAGGGAAC |
| Twist1 | CAGGGCGTGGGGCACACTTT | GGTGATGCCGTTGCCTCTGGG |
G-6-Pase, glucose-6-phosphatase; PEPCK, phosphenolpyruvate carboxykinase; PC, pyruvate carboxylase; Tbp, TATA box-binding protein.
Western blotting.
Tissue (∼100 mg) was homogenized in 1 ml ice-cold homogenization buffer [20 mM Tris·HCl, pH 7.4, 5 mM EDTA, 0.25 mM EGTA, 10 mM Na4P2O7, 1% Tergitol-type NP-40, protease and phosphatase inhibitor cocktails (Roche Diagnostics)] and centrifuged at 6,000 relative centrifugal force at 4°C for 15 min. The supernatant was taken, and protein concentration was determined by the Bradford method (Bio-Rad, Hercules, CA). Protein (100 μg) was loaded and resolved by SDS-PAGE using a 4–12% gradient gel and electroblotted onto a polyvinylidene difluoride membrane (DuPont, Boston, MA) using a wet-transfer cell. The membrane was then blocked for 60 min at room temperature in wash buffer containing 5% (wt/vol) nonfat dried milk and incubated overnight with primary antibody. After being washed, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Bio-Rad) for 60 min. Detection was performed with enhanced chemiluminescence.
Protein kinase B (Akt), phosphorylated Akt (Ser473), and phosphorylated Akt substrate of 160 kDa (AS160) (Thr642) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Glucose 6-phosphate catalytic subunit, pyruvate carboxylase, phosphoenolpyruvate carboxykinase (PEPCK), and GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). AS160 antibody was purchased from Upstate/Millipore (Billerica, MA). Sortilin antibody was purchased from Abcam (Cambridge, MA). β-Actin antibody was purchased from Sigma (St. Louis, MO). GLUT4 and COOH-terminal tether containing a UBX domain, for GLUT4 (TUG) antisera were previously described (5, 57).
Gluconeogenic enzyme activity assays.
Liver tissue (∼100 mg) was homogenized in 1 ml ice-cold homogenization buffer [10 mM HEPES, pH 7.4, 300 mM sucrose, 1 mM EDTA, and protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN)], and mitochondrial, microsomal, and cytoplasmic fractions were separated by ultracentrifugation at 4°C. Before activity assays, protein concentration of each fraction was determined by the Bradford method (Bio-Rad).
PEPCK activities were measured in the cytoplasmic fraction by a spectrophotometric assay, coupling production of oxaloacetate from phosphoenolpyruvate (PEP) by PEPCK to oxidation of NADH by malate dehydrogenase, as previously described (48). Dithiothreitol (2 mM) was added to the cytoplasmic fraction after ultracentrifugation. Reactions were performed in 96-well plates with a 200-μl final volume containing 110 mM imidazole chloride, pH 6.8, 3 mM MgSO4, 3 mM MnCl2, 13 mM NaF, 30 mM NaHCO3 freshly gassed with 5% CO2, 0.3 mM NADH, 6 U/ml malate dehydrogenase, 2 mM PEP, 0.5 mM dGDP, and 5 μg cytoplasmic protein. Control samples were run simultaneously in the absence of dGDP. The reaction was initiated with dGDP, and the drop in NADH absorption at 340 nm was followed at 25°C for 20 min at 10-s intervals.
Glucose-6-phosphatase catalytic subunit activity was measured in the microsomal fraction by a spectrophotometric assay measuring the production of inorganic phosphate from the hydrolysis of glucose 6-phosphate or mannose 6-phosphate, as previously described (47). The latency of microsomal preparations was determined by assaying mannose 6-phosphate hydrolysis in intact vs. detergent-disrupted (0.2% sodium deoxycholate treatment for 20 min) microsomes. The glucose-6-phosphatase activity of intact microsomes was corrected for the portion of activity resulting from the disrupted vesicles within untreated preparations. Reactions were carried out for 15 min at 30°C in 50 mM sodium cacodylate (pH 6.5), 2 mM EDTA, and 5 mM glucose 6-phosphate or mannose 6-phosphate, with intact microsomes at 600 μg/ml or disrupted microsomes at 60 μg/ml. Reactions were terminated by addition of phosphate detection reagent (6 parts 0.42% ammonium molybdate tetrahydrate in 1 M H2SO4, 2 parts 2% SDS, and 1 part 1% ascorbic acid). Samples were incubated at 45°C for 25 min to take the colorization of the phosphate detection reagent to completion, and absorbance was measured at 820 nm.
Pyruvate carboxylase activity was assayed in the mitochondrial fraction after disruption by sonication. Pyruvate carboxylase activity was measured by a coupled spectrophotometric assay, coupling oxaloacetate production from pyruvate by pyruvate carboxylase to NADH oxidation by malate dehydrogenase (32). The reactions were performed at 30°C in 96-well plates with 50 mM Tris, pH 8.0, 10 mM NaHCO3, 10 mM MgCl2, 1 mM NADH, 4 mU/ml malate dehydrogenase, 10 mM ATP, 20 μM acetyl-CoA, and 25 μg/ml disrupted mitochondrial protein. Reactions were initiated with 2 mM pyruvate, and negative controls were performed without the addition of pyruvate. Absorption at 340 nm was measured every 10 s for 10 min.
Tissue triglyceride isolation and measurement.
Triglycerides were extracted using ∼200 mg tissue. Tissues were homogenized in ice-cold 2:1 chloroform-methanol, and lipids were extracted with shaking at room temperature for 3–4 h. H2SO4 was added to ∼100 mM, and samples were vortexed and then centrifuged to achieve phase separation. The organic phase was collected, dried down, and resuspended in chloroform. Triglyceride content was measured using the Genzyme Triglyceride-SL kit (Genzyme Diagnostics).
Hyperinsulinemic-euglycemic clamp studies.
Clamp studies were performed after an overnight fast as previously described (42). The basal period began with a prime (2.0 mg/kg over 3 min) of 99% labeled [6,6-2H]glucose, followed by a continuous infusion at a rate of 0.2 mg·kg-1·min-1 for 2 h to assess basal glucose turnover. After the basal period, the hyperinsulinemic-euglycemic clamping was conducted for 120 min with a primed continuous infusion of human insulin (40 mU·kg-1·min-1 for 3 min/4 mU·kg-1·min-1) (Novo Nordisk) and a variable infusion of ∼20% dextrose to maintain euglycemia (∼110 mg/dl). The 20% dextrose was enriched with [6,6-2H]glucose to ∼2.5% to match the enrichment in the plasma achieved after the basal period (i.e., “hot-GINF”). A 20-μCi bolus of 2-deoxy-d-[1-14C]glucose (Perkin-Elmer) was injected at 120 min in the clamp to evaluate the rate of insulin-stimulated tissue glucose uptake. At the end of the clamp, rats were anesthetized with pentobarbital sodium injection (75 mg/kg), and all tissues were taken within 3 min, frozen immediately using cooled aluminum tongs in liquid N2, and stored at -80°C for the subsequent analysis.
2-Deoxyglucose uptake.
Tissue 2-deoxyglucose (2-DG) uptake studies were performed as previously described (17). After time = 120 min during the hyperinsulinemic-euglycemic clamp, 20 mCi of 2-deoxy-d-[1-14C]glucose were given as a single intravenous bolus. Plasma was collected at 122, 125, 130, and 140 min to determine 14C activity and plasma glucose concentration. Epididymal white adipose tissue and gastrocnemius samples were homogenized, and 2-deoxy-[14C]glucose 6-phosphate was separated from 2-deoxy-[14C]glucose by ion exchange chromatography. 2-DG uptake was calculated based on the intracellular 2-DG content and the plasma 2-DG area under the curve.
Biochemical analysis and calculations.
Plasma glucose concentrations were measured using a Beckman Glucose Analyzer II (Beckman Coulter). Plasma insulin and glucagon concentrations were determined by radioimmunoassay using the LINCOplex Assay system (Millipore). Nonesterified fatty acids were measured using the NEFA-HR Color A and B reagent test kit (Wako Chemicals, Richmond, VA). Serum total thyroxine (T4) and triiodothyronine (T3) concentrations were measured by coated tube RIAs (Siemens Medical Solution Diagnostics, Los Angeles, CA) as previously described (14, 39). Procedures were adapted for measurements in rat serum. Thyroid-stimulating hormone (TSH) was measured using a sensitive, heterologous, disequilibrium, double-antibody precipitation RIA developed for mice (39). A series of experiments demonstrated that TRβ agonists interfere with the measurement of triiodothyronine (data not shown); thus, we were unable to assess the effect of KB-2115 and GC-1 on total T3 levels.
Statistical analysis.
Statistical analysis of the data, other than thyroid function test data, was performed using Graph-Pad Prism 5.0.3. Data were compared using Student's unpaired t-test. All data are expressed as means ± SE unless otherwise indicated. P values <0.05 were considered significant. The thyroid function tests were compared with the Mann-Whitney U-test, testing the null hypothesis that the median values of the thyroid function tests were equal. P values <0.05 were considered significant.
RESULTS
The effects of TRβ agonists on hepatic steatosis and on whole body glucose metabolism were assessed in high-fat-fed male Sprague Dawley rats treated with either vehicle or thyromimetic for 10 days. At the end of 10 days, we performed hyperinsulinemic-euglycemic clamp studies and molecular analyses.
GC-1 treatment reduces hepatic steatosis but causes hyperglycemia and insulin resistance.
Ten days of GC-1 treatment did not alter body weight in high-fat-fed animals (Table 2). However, GC-1 treatment prevented the development of hepatic steatosis, as reflected by a 75% reduction in hepatic triglyceride content in GC-1-treated animals compared with vehicle-treated animals (P < 0.0001; Fig. 1A). GC-1 treatment also led to a 26% increase in fasting plasma glucose concentration (P < 0.0001; Table 2). This was associated with a threefold increase in fasting plasma insulin concentration (P = 0.0003), suggesting insulin resistance. Additionally, we observed a 19% increase in fasting plasma glucagon concentration (P = 0.04; Table 2).
Table 2.
Fasting parameters, GC-1 vs. vehicle treatment
| Vehicle | GC-1 | P | |
|---|---|---|---|
| Body wt, g | 394 ± 9 | 387 ± 11 | 0.62 |
| Basal glucose, mg/dl | 108 ± 2 | 135 ± 4 | <0.0001 |
| Basal insulin, μU/ml | 23 ± 5 | 64 ± 7 | 0.0003 |
| Basal glucagon, pg/ml | 41 ± 2 | 48 ± 3 | 0.04 |
| NEFA, mM | 0.35 ± 0.04 | 0.47 ± 0.03 | <0.04 |
Values are means ± SE. NEFA, nonesterified fatty acid.
Fig. 1.

Physiological comparison between 10 days GC-1 treatment (filled bars) vs. vehicle (open bars). A: hepatic tissue triglyceride (TG) content. B: glucose infusion rate (GINF) during hyperinsulinemic period of hyperinsulinemic-euglycemic clamp. C: whole body insulin-stimulated glucose disposal. D: endogenous glucose production under basal (open bars, vehicle; filled bars, GC-1) and clamped (light hatched bars, vehicle; dark hatched bars, GC-1) conditions. E: basal glycerol turnover (*P < 0.04 vs. vehicle; ***P < 0.0004 vs. vehicle).
Hyperinsulinemic-euglycemic clamp studies were performed to quantify changes in endogenous glucose production (EGP), insulin stimulated-glucose disposal, and hepatic insulin sensitivity. GC-1 treatment increased EGP by 56% (P = 0.01; Fig. 1D). The glucose infusion rate required to maintain euglycemia in GC-1-treated animals was decreased by 31% vs. vehicle-treated animals (P < 0.03; Fig. 1B), reflecting a decrease in insulin sensitivity. Whole body insulin-stimulated glucose disposal was similar between the two groups (Fig. 1C). Instead, GC-1 treatment resulted in hepatic insulin resistance, as reflected by impairment in insulin-mediated suppression of hepatic glucose production (Fig. 1D).
Thyroid hormone can enhance adipose tissue lipolysis, and glycerol turnover (an index of whole body lipolysis) was quantified during the basal period of the clamp. Glycerol turnover was increased by 22% in GC-1-treated rats (P = 0.03; Fig. 1E). Consistent with this increase in glycerol turnover, fasting plasma nonesterified fatty acids were increased by 33% in the GC-1-treated group (P = 0.04; Table 2).
GC-1 treatment results in reduction in hepatic insulin signaling but has only small effect on gluconeogenic gene expression.
Thyroid hormone can increase transcription of key gluconeogenic enzymes, notably PEPCK and glucose-6-phosphatase. Thus, we evaluated gluconeogenic gene expression in the livers of GC-1-treated animals (Fig. 2, A and B). We observed no change in PEPCK mRNA expression (P = 0.21) and no change in PEPCK protein abundance. In contrast, PEPCK activity in a cytoplasmic fraction was increased 70% by GC-1 therapy (6.8 ± 0.2 mmol·mg-1·min-1 vs. 11.6 ± 0.9 mmol·mg-1·min-1, P < 0.0005). There was a trend toward decreased glucose-6-phosphatase mRNA expression (P = 0.058), protein abundance was unchanged, and catalytic subunit specific activity was unchanged (vehicle treated: 6.4 ± 1.2 pmol·μg-1·min-1; GC-1 treated: 6.7 ± 0.8 pmol·μg-1·min-1). Pyruvate carboxylase mRNA increased by 44% (P = 0.047) with GC-1 treatment, but without a significant difference in protein by Western, and difference in pyruvate carboxylase activity was not significant (vehicle treated: 6.5 ± 0.2 nmol·mg-1·s-1; GC-1 treated: 7.2 ± 0.2 nmol·mg-1·s-1, P = 0.27).
Fig. 2.

Molecular comparison between 10 days GC-1 (filled bars) vs. vehicle (open bars). A: hepatic expression of genes associated with gluconeogenesis [phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase (G6Pase), pyruvate carboxylase (PC)]. mRNA levels were measured by qPCR, and gluconeogenic genes were normalized to β-actin and expressed as a fraction of the level seen in vehicle-treated animals. B: protein abundance of gluconeogenic enzymes in liver, assessed by Western blot. Values were normalized against β-actin levels. Samples from vehicle-treated and GC-1-treated animals are from noncontiguous lanes of the same blot. C: specific activities of gluconeogenic enzymes in liver. D: hepatic protein kinase B (Akt) phosphorylation assessed by Western blot, phosphor (p)-Akt normalized to total Akt abundance (*P < 0.04 vs. vehicle; ***P < 0.0004 vs. vehicle).
We assessed hepatic Akt phosphorylation in basal and clamped animals to further explore the mechanism for hepatic insulin resistance following GC-1 treatment. Compared with the increase in insulin-stimulated Akt phosphorylation in vehicle-treated animals, Akt phosphorylation was completely suppressed by GC-1 treatment (Fig. 2C). Thus, although GC-1 treatment prevents the development of fatty liver in fat-fed rats, it impaired insulin-stimulated Akt activation.
KB-2115 reduces hepatic steatosis but attenuates insulin-stimulated peripheral glucose uptake.
KB-2115 is a more TRβ-specific compound that has been shown to effectively lower plasma lipids in humans. We tested KB-2115 in high-fat-fed rats to determine if this would be a more effective therapy for nonalcoholic fatty liver disease (NAFLD) and hepatic insulin resistance. Ten days of KB-2115 (0.1 mg·kg-1·day-1) had no effect on body weight (Table 3). KB-2115 prevented the development of hepatic steatosis in high-fat-fed rats as effectively as GC-1 (Fig. 3A). Unlike GC-1 treatment, KB-2115 treatment did not lead to fasting hyperglycemia, although it still resulted in fasting hyperinsulinemia; insulin concentrations were almost fourfold greater with KB-2115 treatment than with vehicle treatment (Table 3). Fasting plasma glucagon concentration was unchanged.
Table 3.
Fasting parameters, KB-2115 vs. vehicle treatment
| Vehicle | KB-2115 | P | |
|---|---|---|---|
| Body wt, g | 397 ± 14 | 421 ± 8 | 0.15 |
| Basal glucose, mg/dl | 116 ± 6 | 112 ± 4 | 0.55 |
| Basal insulin, μU/ml | 11 ± 3 | 39 ± 10 | 0.02 |
| Basal glucagon, pg/ml | 56 ± 7 | 58 ± 6 | 0.77 |
| NEFA, mM | 0.39 ± 0.03 | 0.40 ± 0.04 | 0.78 |
Values are means ± SE.
Fig. 3.

Physiological comparison between 10 days KB-2115 (filled bars) treatment vs. vehicle (open bars). A: hepatic tissue triglyceride content. B: glucose infusion rate during hyperinsulinemic period of hyperinsulinemic-euglycemic clamp. C: whole body insulin-stimulated glucose disposal. D: endogenous glucose production under basal (open bars, vehicle; filled bars, KB-2115) and clamped (light hatched bars, vehicle; dark hatched bars, KB-2115) conditions. E: basal glycerol turnover. F and G: tissue-specific glucose uptake as assessed by 2-deoxyglucose (2-DG) uptake in skeletal muscle (F) and white adipose tissue (G). (*P < 0.04 vs. vehicle; **P < 0.04 vs. vehicle; ***P < 0.0004 vs. vehicle).
We quantified the rate of EGP and insulin-stimulated glucose disposal with hyperinsulinemic-euglycemic clamp studies. In contrast to GC-1, KB-2115 treatment did not increase the basal rate of EGP (P = 0.24; Fig. 3D). However, KB-2115-treated high-fat-fed rats were markedly more insulin resistant than vehicle-treated high-fat-fed rats, as reflected by a 58% decrease in glucose infusion rate required to maintain euglycemia under hyperinsulinemic conditions (P = 0.0002; Fig. 3B). This difference was not attributable to a difference in hepatic insulin sensitivity, since clamped EGF was not different between KB-2115 and vehicle-treated animals (P = 0.66; Fig. 3D).
Instead, there was a 46% decrease in whole body insulin-stimulated glucose disposal compared with vehicle-treated rats (P = 0.001; Fig. 3C). This decrease in insulin-stimulated peripheral glucose disposal was accounted for by a decrease in skeletal muscle glucose uptake, as quantified by 2-DG uptake (Fig. 3, F and G). Insulin-stimulated skeletal muscle 2-DG uptake was reduced by KB-2115 treatment (68% decrease; P = 0.03), whereas epididymal white adipose tissue 2-DG uptake was unaffected. KB-2115 treatment did not cause a significant change in glycerol turnover (P = 0.39; Fig. 3E), or in plasma nonesterified fatty acid concentration (4% increase; P = 0.78; Table 3), in contrast to GC-1 treatment.
KB-2115 treatment induces small changes in gluconeogenic gene expression and hepatic insulin signaling.
Hepatic gluconeogenic gene mRNA expression was evaluated in fasted rats (Fig. 4A). Glucose-6-phosphatase gene expression was increased by 50% (P = 0.027), but there were no significant changes in PEPCK and pyruvate carboxylase expression. Similar to changes seen in specific activity after GC-1 treatment, PEPCK activity was increased by 80% (P < 0.0005), glucose-6-phosphatase activity was unchanged (vehicle treated: 5.4 ± 0.4 pmol·μg-1·min-1; KB-2115 treated: 4.6 ± 0.5 pmol·μg-1·min-1), and pyruvate carboxylase activity was not significantly changed (vehicle treated: 4.2 ± 0.2 nmol·mg-1·s-1; KB-2115 treated: 4.5 ± 0.2 nmol·mg-1·s-1, P = 0.3). Hepatic insulin signaling was preserved in KB-2115-treated animals. Insulin-stimulated Akt phosphorylation increased approximately twofold in KB-2115-treated animals compared with the greater than threefold increase in vehicle-treated animals (P = 0.25 comparing the fold increase in Akt phosphorylation; Fig. 4B).
Fig. 4.
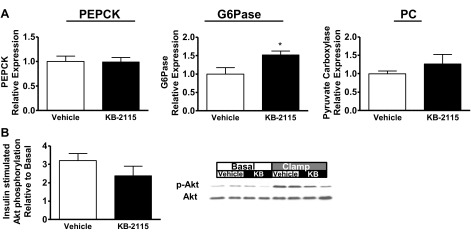
Molecular comparison between livers from animals after 10 days KB-2115 (filled bars) vs. vehicle (open bars). A: hepatic expression of genes associated with gluconeogenesis (PEPCK, glucose-6-phosphatase, pyruvate carboxylase). mRNA levels were measured by qPCR, and gluconeogenic genes were normalized to β-actin and expressed as a fraction of the level seen in vehicle-treated animals. B: Akt phosphorylation assessed by Western blot, phospho-Akt normalized to total Akt abundance (*P < 0.04 vs. vehicle).
KB-2115 treatment decreases skeletal muscle GLUT4 content.
We investigated potential explanations for the decrease in insulin-stimulated skeletal muscle glucose uptake following KB-2115 treatment. First, we considered whether KB-2115 led to ectopic lipid accumulation. However, skeletal muscle triglyceride content had a tendency to be lower in the KB-2115 treatment group (Fig. 5A). Insulin signaling was evaluated by assessing insulin-stimulated Akt phosphorylation and AS160 phosphorylation (Fig. 5, B and C). Skeletal muscle insulin-stimulated Akt phosphorylation was unaffected, and there was a trend toward increased AS160 phosphorylation (P = 0.1). These data suggest that KB-2115 did not alter muscle insulin signaling. We assessed the abundance of the insulin-stimulated glucose transporter GLUT4 at the level of mRNA and protein (Fig. 5, D and E). KB-2115 treatment did not cause any difference in GLUT4 mRNA expression (P = 0.52) but resulted in an ∼36% reduction in GLUT4 protein abundance (P = 0.018). The stability of GLUT4 protein is influenced by its targeting among intracellular membranes, and GLUT4 normally accumulates in a stable pool of intracellular vesicles termed “GLUT4 storage vesicles” (GSVs) (4). The TUG protein traps GSVs as an intracellular, insulin-responsive pool, and TUG depletion can accelerate degradation of GLUT4 protein (7, 57). We observed that TUG abundance was decreased by 45% in skeletal muscle of KB-2115-treated rats (Fig. 5F). This result suggested that accumulation of GLUT4 in GSVs is markedly decreased by KB-2115 treatment. In principle, a reduction in the number of GSVs could be because of impaired formation or accelerated loss of these vesicles. We therefore examined sortilin, a protein critical for GSV formation (6). Sortilin abundance was unchanged at the level of mRNA or protein (Fig. 5, G and H), suggesting that the decrease in GSVs is not the result of reduced formation. The data support the idea that GSVs are not efficiently trapped in an insulin-responsive pool in skeletal muscle of KB-2115-treated animals and that GLUT4 degradation is consequently accelerated. In contrast, insulin-stimulated glucose disposal was not impaired by GC-1 treatment. Although there was a trend toward increased GLUT4 mRNA in the skeletal muscle of these animals (relative GLUT4 mRNA expression: vehicle treatment, 1.00 ± 0.09; GC-1 treatment, 1.67 ± 0.33; P = 0.06), GLUT4 protein abundance was unchanged (relative protein abundance by Western: vehicle treatment, 1.0 ± 0.1; GC-1 treatment, 0.92 ± 0.09; P = 0.6).
Fig. 5.

Connecting the thyroid hormone receptor-β agonism of KB-2115 with decreased skeletal muscle glucose uptake (open bars, vehicle; filled bars, KB-2115). A: skeletal muscle tissue triglyceride content. B and C: assessment of skeletal muscle insulin signaling at the level of Akt phosphorylation (B) and AS-160 phosphorylation (C). D: skeletal muscle GLUT4 mRNA expression. E: skeletal muscle GLUT4 protein abundance measured by Western blot. F: skeletal muscle tether containing a UBX domain, for GLUT4 (TUG) protein abundance measured by Western blot (*P < 0.04 vs. vehicle). G: skeletal muscle sortilin 1 mRNA expression. H: skeletal muscle sortilin protein abundance measured by Western blot.
Assessment of tissue-specific thyroid hormone action.
The difference in physiological response to GC-1 vs. KB-2115 may be because of differences in the affinities of these compounds for TRβ and/or tissue specificity. We assessed this by measuring the expression of key TR-responsive genes, deiodinase 1 (30) in liver and hairless (49) in white adipose and skeletal muscle (Fig. 6). GC-1 and KB-2115 treatment led to similar increases in these markers in TRβ predominant liver and in white adipose (a tissue with both TRβ and TRα receptors). Hepatic deiodinase 1 expression was increased fourfold by GC-1 treatment (P < 0.0001) and threefold by KB-2115 treatment (P = 0.0002). White adipose tissue hairless expression was increased by approximately twofold by GC-1 treatment (P = 0.009) and approximately threefold by KB-2115 treatment (P < 0.04). In contrast, there was a striking difference between the effect of GC-1 and KB-2115 on TRα-predominant skeletal muscle. Skeletal muscle hairless expression was increased by >25-fold by GC-1 treatment (P < 0.0001) but was not significantly increased by KB-2115 treatment (26% increase; P = 0.37). Two additional thyroid hormone-regulated genes, sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) 1 and malic enzyme 1, were evaluated in KB-2115-treated skeletal muscle and found to be unaffected by treatment.
Fig. 6.

Assessment of thyroid hormone action in multiple tissues. Expression of thyroid hormone-responsive genes after GC-1 treatment (A) or KB-2115 treatment (B). Genes examined: deiodinase 1 in liver, hairless in white adipose tissue, hairless in skeletal muscle, deiodinase 2 in skeletal muscle, sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) 1 in skeletal muscle, and malic enzyme 1 (ME1) in skeletal muscle (open bars, vehicle; filled bars, thyromimetic; *P < 0.04 vs. vehicle; **P < 0.04 vs. vehicle; ***P < 0.0004 vs. vehicle).
Local tissue T3 levels are significantly influenced by local deiodinase 2 levels, which can further complicate the interpretation of the effects of thyroid hormone mimetics. Deiodinase 2 mRNA expression was not significantly different in either GC-1- or KB-2115-treated gastrocnemius.
Finally, to evaluate the effect of TRβ agonist treatment on the hypothalamic-pituitary-thyroid axis, thyroid function tests were measured. In contrast to the minimal effect seen when used at lower doses in humans (3, 24), 0.164 mg/kg GC-1 and 0.1 mg/kg KB-2115 in Sprague-Dawley rats suppressed endogenous hypothalamic-pituitary-thyroid axis function by both suppressing TSH and total T4 (Fig. 7). GC-1 treatment led to significant differences between groups for TSH (z = 2.76; UA = 0; P = 0.0058) and total T4 (z = 2.76; UA = 0; P = 0.0058). Similarly, KB-2115 treatment also led to significant differences in plasma TSH (z = 3.73; UA = 3; P = 0.0002) and total T4 (z = 3.92; UA = 0; P = 0.0001).
Fig. 7.

Serum thyroid-stimulating hormone (TSH) and total thyroxine (T4) levels after vehicle, GC-1, and KB-2115 treatment. The horizontal dashed line represents the lower limit of detection.
DISCUSSION
Liver-specific TRβ agonists are promising therapeutic agents for treating hyperlipidemia (1, 2). While the underlying mechanisms for lowering plasma lipids are not fully established, possible mechanisms include increased LDL receptor expression (15, 29), decreased sterol response element-binding protein (SREBP)-1c expression (18, 20), increased SREBP-2 expression (45), and increased reverse cholesterol transport, with increased cytochrome p450 7A1 activity in rodent models (15, 20). Additionally, as a result of their ability to increase hepatic lipid oxidation, hepatic and TRβ-specific thyroid hormone analogs have the potential to decrease NAFLD. Treatments targeting NAFLD may decrease the incidence of nonalcoholic steatohepatitis, which is projected to eventually overtake hepatitis C as the leading cause of liver transplantation (9). Treatment of NAFLD may also reduce the burden of diseases associated with insulin resistance, since decreased hepatic ectopic lipid accumulation should lead to decreased hepatic insulin resistance (23, 36, 37, 43). In the present set of studies, both TRβ agonists effectively prevented the development of hepatic steatosis in fat-fed rats. However, both TRβ agonists tested caused insulin resistance through discrete mechanisms.
GC-1 had notable effects on hepatic glucose metabolism. Specifically, GC-1 increased hepatic glucose output and reduced hepatic insulin sensitivity. Although thyroid hormone can induce expression of key gluconeogenic genes (16, 28), and PEPCK expression was significantly increased in a prior model of rodent thyrotoxicosis (22), changes in gluconeogenic enzymes can only partially explain the observed phenotype. There was a slight increase in the expression of pyruvate carboxylase mRNA, although without significant increases in pyruvate carboxylase protein or activity. There was no change in glucose-6-phosphatase expression or activity. In contrast, while GC-1 did not alter cytosolic PEPCK mRNA or protein expression, there was a slight increase in enzyme activity. A similar increase was seen with KB-2115 treatment, suggesting nongenomic effects on gluconeogenesis. In addition to this increase in PEPCK activity, GC-1 also increased basal glycerol turnover. Glycerol enters gluconeogenesis at the level of dihydroxyacetone phosphate, bypassing regulatory checkpoints for other three- and four-carbon precursors at the pyruvate carboxylase and PEPCK steps. Increasing glycerol flux can increase the rate of EGP (40) and may represent an additional mechanism accounting for the increase in basal EGP. In addition to the changes in basal PEPCK activity and glycerol flux, GC-1 also impaired hepatic insulin sensitivity, as reflected by the diminished suppression of EGP during the hyperinsulinemic-euglycemic clamp. This was associated with impaired insulin-stimulated phosphorylation of Akt, suggesting a defect in insulin signaling. Thus, the combined contributions of increased PEPCK activity, increased glycerol influx, and impaired insulin signaling may account for the fasting hyperglycemia and hyperinsulinemia seen in the GC-1-treated animals. In contrast, while KB-2115 also increased PEPCK activity to a similar degree, it did not alter glycerol flux or hepatic insulin sensitivity and thus did not cause fasting hyperglycemia.
KB-2115 is considered a more liver-specific agonist, and this may account for some of the differences from GC-1 (3, 24). As with GC-1, KB-2115 treatment effectively prevented the development of fatty liver in high-fat-fed rats but did so without causing fasting hyperglycemia. Still, the development of fasting hyperinsulinemia suggested that KB-2115 caused insulin resistance. Also, while KB-2115 led to a similar decrease in glucose infusion rates during a hyperinsulinemic-euglycemic clamp to those seen in the GC-1-treated rats, the underlying mechanism for insulin resistance was different. KB-2115 primarily decreased insulin-stimulated peripheral glucose disposal, which was largely attributable to decreased skeletal muscle glucose uptake.
We further explored the mechanisms that may account for the skeletal muscle insulin resistance in KB-2115-treated rats. There was no difference in skeletal muscle lipid content, and skeletal muscle triglyceride content actually tended to be lower in KB-2115-treated animals. Furthermore, insulin signaling was not perturbed, with appropriate increases in Akt and AS160 phosphorylation in response to insulin. Instead, we found a surprising decrease in GLUT4 transporter protein, which was also associated with a similar decrease in TUG abundance. These data are consistent with decreased GSV content. Thus, while skeletal muscle insulin signaling remained intact, the reduction in total GLUT4 available for insulin-stimulated translocation to the membrane reduced the maximum capacity for insulin-stimulated glucose transport into skeletal muscle, thereby accounting for the decline in peripheral glucose disposal. The decrease in GLUT4 protein is not a direct effect on GLUT4 transcription, since GLUT4 mRNA was not reduced in the KB-2115-treated animals. Rather, GSV abundance may be decreased at a posttranslational step. Abundance of sortilin, a key protein involved in GSV assembly, was unchanged. This suggests that GSV assembly was not affected by KB-2115 treatment. Instead, the data suggest that GSVs may not be appropriately sequestered in an intracellular pool, and therefore subject to increased turnover. Taken together, our data suggest that KB-2115 treatment decreases skeletal muscle GSV abundance and, as a result, decreases insulin-stimulated skeletal muscle glucose uptake in KB-2115-treated animals.
We evaluated the possibility that KB-2115 treatment may directly act in skeletal muscle. Such an effect could be via off-target binding of KB-2115 to the TRα receptor, from decreased endogenous thyroid hormone production by action of KB-2115 on the hypothalamic-pituitary-thyroid axis thereby decreasing TRH or TSH levels, or from differential interactions of this drug with coactivators and/or corepressors that interact with the thyroid hormone receptor (TR). Skeletal muscle thyroid hormone action was evaluated by assessing the expression of key downstream genes. Expression of key thyroid hormone-responsive genes, hairless, SERCA 1, and malic enzyme 1 (13, 49, 52), was not changed, arguing against alterations in skeletal muscle thyroid hormone action with KB-2115. Thus, the effects in skeletal muscle are likely an indirect, off-target effect of KB-2115. Interestingly, GC-1 markedly increased expression of hairless and induced a strong tendency toward increased expression of GLUT4, which also contains a thyroid hormone response element in its promoter (44, 50). These findings suggest that this compound acts directly in skeletal muscle, a TRα-predominant tissue. These data further demonstrate the marked differences in these two synthetic thyromimetics.
This highlights the more unexpected results of this study. Two drugs designed to be agonists of the same receptor have quite different downstream effects on physiology. The likely explanation for the difference lies in the difference in receptor specificity of the two drugs, with the action of KB-2115 more restricted to TRβ-predominant tissues than the action of GC-1. Both agents are effective in reducing hepatic steatosis, but this metabolic gain came with unique costs. GC-1, by increasing glycerol turnover (possibly through actions in adipose tissue), increasing PEPCK activity, and impairing hepatic insulin sensitivity, led to increases in basal EGP and fasting hyperglycemia. However, there was no evidence of peripheral insulin resistance, possibly because of the compensatory increase in muscle GLUT4 expression leading to muscle with normal levels of GLUT4 protein. This pattern of increased EGP and hepatic insulin resistance is similar to the pattern seen in previous reports of human and canine hyperthyroid models (12, 25, 41, 56) and mirrors the pattern seen with chow-fed T4-induced thyrotoxic rats (22). In contrast, KB-2115 did not increase glycerol turnover and did not result in hepatic insulin resistance. However, via indirect mechanisms, KB-2115 led to a decrease in muscle GLUT4 abundance and decreased insulin-stimulated muscle glucose uptake.
Cardiovascular disease is still the leading cause of death globally, and new agents that can treat dyslipidemia are needed. Moreover, although NAFLD has become the most common chronic liver disease in developed countries, there are few effective therapies for NAFLD. Thus, the continued refinement and study of TRβ-specific agonists holds the promise for new and necessary therapies. The agonists studied here, GC-1 and KB-2115, have been successfully used in animal and human studies, respectively. While both potently lower hepatic lipid content and can correct dyslipidemia, we demonstrated that each compound had unique negative effects on insulin action in high-fat-fed rats, thereby questioning their therapeutic potential. These studies also suggest that attempts to selectively activate pathways in specific tissues that were intended to function as part of a coordinated whole body response may lead to surprising adverse effects, and underscores the importance of rigorous preclinical testing of novel agents, especially regarding the effects on insulin action and glucose metabolism.
GRANTS
This project was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-40936, R24-DK-085638, P30-DK-45735, R01-DK-092661, R01-DK-075772, and T32-DK-07058 (D. F. Vatner), a Manpei Suzuki Diabetes Foundation Fellowship (N. Kumashiro), from the American Diabetes Association, and a Veterans' Affairs Merit Grant, I01 BX000901 (V. T. Samuel).
DISCLOSURES
The authors have no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
Author contributions: D.F.V., D.W., G.J.G., P.W., K.J.P., R.E.W., J.S.B., J.B., G.I.S., and V.T.S. conception and design of research; D.F.V., D.W., S.A.B., N.K., D.M.E., X.-H.L., and V.T.S. performed experiments; D.F.V., D.W., and X.-H.L. analyzed data; D.F.V., D.W., R.E.W., J.S.B., G.I.S., and V.T.S. interpreted results of experiments; D.F.V. prepared figures; D.F.V. drafted manuscript; D.F.V., D.W., K.J.P., R.E.W., J.S.B., G.I.S., and V.T.S. edited and revised manuscript; D.F.V., D.W., S.A.B., N.K., D.M.E., X.-H.L., G.J.G., P.W., K.J.P., R.E.W., J.S.B., G.I.S., and V.T.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Yanna Kosover, Codruta Todeasa, Maria Batsu, and Aida Groszmann (all of the Yale University School of Medicine) for technical support.
Current addresses: D. Weismann, Medizinische Klinik und Poliklinik I, Universitätsklinikum Würzburg, Würzburg, Germany; and D. M. Erion, Pfizer, Cambridge, MA.
REFERENCES
- 1. Baxter JD, Webb P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug discovery 8: 308–320, 2009 [DOI] [PubMed] [Google Scholar]
- 2. Baxter JD, Webb P, Grover G, Scanlan TS. Selective activation of thyroid hormone signaling pathways by GC-1: a new approach to controlling cholesterol and body weight. Trends Endocrinol Metab 15: 154–157, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Berkenstam A, Kristensen J, Mellstrom K, Carlsson B, Malm J, Rehnmark S, Garg N, Andersson CM, Rudling M, Sjoberg F, Angelin B, Baxter JD. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci USA 105: 663–667, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bogan JS. Regulation of glucose transporter translocation in health and diabetes. Ann Rev Biochem 81: 507–532, 2012 [DOI] [PubMed] [Google Scholar]
- 5. Bogan JS, Hendon N, McKee AE, Tsao TS, Lodish HF. Functional cloning of TUG as a regulator of GLUT4 glucose transporter trafficking. Nature 425: 727–733, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Bogan JS, Kandror KV. Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr Opin Cell Biol 22: 506–512, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bogan JS, Rubin BR, Yu C, Loffler MG, Orme CM, Belman JP, McNally LJ, Hao M, Cresswell JA. Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J Biol Chem 287: 23932–23947, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bradley DJ, Young WS, 3rd, Weinberger C. Differential expression of alpha and beta thyroid hormone receptor genes in rat brain and pituitary. Proc Natl Acad Sci USA 86: 7250–7254, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 141: 1249–1253, 2011 [DOI] [PubMed] [Google Scholar]
- 10. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev 31: 139–170, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiellini G, Apriletti JW, Yoshihara HA, Baxter JD, Ribeiro RC, Scanlan TS. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem Biol 5: 299–306, 1998 [DOI] [PubMed] [Google Scholar]
- 12. Dimitriadis G, Baker B, Marsh H, Mandarino L, Rizza R, Bergman R, Haymond M, Gerich J. Effect of thyroid hormone excess on action, secretion, and metabolism of insulin in humans. Am J Physiol Endocrinol Metab 248: E593–E601, 1985 [DOI] [PubMed] [Google Scholar]
- 13. Dozin B, Magnuson MA, Nikodem VM. Tissue-specific regulation of two functional malic enzyme mRNAs by triiodothyronine. Biochemistry 24: 5581–5586, 1985 [DOI] [PubMed] [Google Scholar]
- 14. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology 147: 4036–4043, 2006 [DOI] [PubMed] [Google Scholar]
- 15. Erion MD, Cable EE, Ito BR, Jiang H, Fujitaki JM, Finn PD, Zhang BH, Hou J, Boyer SH, van Poelje PD, Linemeyer DL. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc Natl Acad Sci USA 104: 15490–15495, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng X, Jiang Y, Meltzer P, Yen PM. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol 14: 947–955, 2000 [DOI] [PubMed] [Google Scholar]
- 17. Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 48: 1270–1274, 1999 [DOI] [PubMed] [Google Scholar]
- 18. Hashimoto K, Yamada M, Matsumoto S, Monden T, Satoh T, Mori M. Mouse sterol response element binding protein-1c gene expression is negatively regulated by thyroid hormone. Endocrinology 147: 4292–4302, 2006 [DOI] [PubMed] [Google Scholar]
- 19. Hodin RA, Lazar MA, Chin WW. Differential and tissue-specific regulation of the multiple rat c-erbA messenger RNA species by thyroid hormone. J Clin Invest 85: 101–105, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johansson L, Rudling M, Scanlan TS, Lundasen T, Webb P, Baxter J, Angelin B, Parini P. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc Natl Acad Sci USA 102: 10297–10302, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim B. Thyroid hormone as a determinant of energy expenditure and the basal metabolic rate. Thyroid 18: 141–144, 2008 [DOI] [PubMed] [Google Scholar]
- 22. Klieverik LP, Sauerwein HP, Ackermans MT, Boelen A, Kalsbeek A, Fliers E. Effects of thyrotoxicosis and selective hepatic autonomic denervation on hepatic glucose metabolism in rats. Am J Physiol Endocrinol Metab 294: E513–E520, 2008 [DOI] [PubMed] [Google Scholar]
- 23. Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, Still CD, Gerhard GS, Han X, Dziura J, Petersen KF, Samuel VT, Shulman GI. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA 108: 16381–16385, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ladenson PW, Kristensen JD, Ridgway EC, Olsson AG, Carlsson B, Klein I, Baxter JD, Angelin B. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med 362: 906–916, 2010 [DOI] [PubMed] [Google Scholar]
- 25. Laville M, Riou JP, Bougneres PF, Canivet B, Beylot M, Cohen R, Serusclat P, Dumontet C, Berthezene F, Mornex R. Glucose metabolism in experimental hyperthyroidism: intact in vivo sensitivity to insulin with abnormal binding and increased glucose turnover. J Clin Endocrinol Metab 58: 960–965, 1984 [DOI] [PubMed] [Google Scholar]
- 26. Lazar MA. Thyroid hormone receptors: multiple forms, multiple possibilities. Endocr Rev 14: 184–193, 1993 [DOI] [PubMed] [Google Scholar]
- 27. Lazar MA, Hodin RA, Darling DS, Chin WW. Identification of a rat c-erbA alpha-related protein which binds deoxyribonucleic acid but does not bind thyroid hormone. Mol Endocrinol 2: 893–901, 1988 [DOI] [PubMed] [Google Scholar]
- 28. Liu YY, Brent GA. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol Metab 21: 166–173, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopez D, Abisambra Socarras JF, Bedi M, Ness GC. Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim Biophys Acta 1771: 1216–1225, 2007 [DOI] [PubMed] [Google Scholar]
- 30. O'Mara BA, Dittrich W, Lauterio TJ, St Germain DL. Pretranslational regulation of type I 5′-deiodinase by thyroid hormones and in fasted and diabetic rats. Endocrinology 133: 1715–1723, 1993 [DOI] [PubMed] [Google Scholar]
- 31. O'Shea PJ, Williams GR. Insight into the physiological actions of thyroid hormone receptors from genetically modified mice. J Endocrinol 175: 553–570, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Payne J, Morris JG. Pyruvate carboxylase in Rhodopseudomonas spheroides. J Gen Microbiol 59: 97–101, 1969 [DOI] [PubMed] [Google Scholar]
- 33. Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, Cocco MT, Ledda-Columbano GM, Columbano A. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J 22: 2981–2989, 2008 [DOI] [PubMed] [Google Scholar]
- 34. Petersen KF, Blair JB, Shulman GI. Triiodothyronine treatment increases substrate cycling between pyruvate carboxylase and malic enzyme in perfused rat liver. Metab Clin Exp 44: 1380–1383, 1995 [DOI] [PubMed] [Google Scholar]
- 35. Petersen KF, Cline GW, Blair JB, Shulman GI. Substrate cycling between pyruvate and oxaloacetate in awake normal and 3,3′-5-triiodo-l-thyronine-treated rats. Am J Physiol Endocrinol Metab 267: E273–E277, 1994 [DOI] [PubMed] [Google Scholar]
- 36. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54: 603–608, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, Shulman GI. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 109: 1345–1350, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR (Abstract). Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pohlenz J, Maqueem A, Cua K, Weiss RE, Van Sande J, Refetoff S. Improved radioimmunoassay for measurement of mouse thyrotropin in serum: strain differences in thyrotropin concentration and thyrotroph sensitivity to thyroid hormone. Thyroid 9: 1265–1271, 1999 [DOI] [PubMed] [Google Scholar]
- 40. Previs SF, Cline GW, Shulman GI. A critical evaluation of mass isotopomer distribution analysis of gluconeogenesis in vivo. Am J Physiol Endocrinol Metab 277: E154–E160, 1999 [DOI] [PubMed] [Google Scholar]
- 41. Raboudi N, Arem R, Jones RH, Chap Z, Pena J, Chou J, Field JB. Fasting and postabsorptive hepatic glucose and insulin metabolism in hyperthyroidism. Am J Physiol Endocrinol Metab 256: E159–E166, 1989 [DOI] [PubMed] [Google Scholar]
- 42. Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117: 739–745, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santalucia T, Moreno H, Palacin M, Yacoub MH, Brand NJ, Zorzano A. A novel functional co-operation between MyoD, MEF2 and TRalpha1 is sufficient for the induction of GLUT4 gene transcription. J Mol Biol 314: 195–204, 2001 [DOI] [PubMed] [Google Scholar]
- 45. Shin DJ, Osborne TF. Thyroid hormone regulation and cholesterol metabolism are connected through Sterol Regulatory Element-Binding Protein-2 (SREBP-2). J Biol Chem 278: 34114–34118, 2003 [DOI] [PubMed] [Google Scholar]
- 46. Shulman GI, Ladenson PW, Wolfe MH, Ridgway EC, Wolfe RR. Substrate cycling between gluconeogenesis and glycolysis in euthyroid, hypothyroid, and hyperthyroid man. J Clin Invest 76: 757–764, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sloop KW, Showalter AD, Cox AL, Cao JX, Siesky AM, Zhang HY, Irizarry AR, Murray SF, Booten SL, Finger EA, McKay RA, Monia BP, Bhanot S, Michael MD. Specific reduction of hepatic glucose 6-phosphate transporter-1 ameliorates diabetes while avoiding complications of glycogen storage disease. J Biol Chem 282: 19113–19121, 2007 [DOI] [PubMed] [Google Scholar]
- 48. Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, Shulman GI, Kibbey RG. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J Biol Chem 284: 26578–26590, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson CC. Thyroid hormone-responsive genes in developing cerebellum include a novel synaptotagmin and a hairless homolog. J Neurosci 16: 7832–7840, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Torrance CJ, Devente JE, Jones JP, Dohm GL. Effects of thyroid hormone on GLUT4 glucose transporter gene expression and NIDDM in rats. Endocrinology 138: 1204–1214, 1997 [DOI] [PubMed] [Google Scholar]
- 51. Trost SU, Swanson E, Gloss B, Wang-Iverson DB, Zhang H, Volodarsky T, Grover GJ, Baxter JD, Chiellini G, Scanlan TS, Dillmann WH. The thyroid hormone receptor-beta-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology 141: 3057–3064, 2000 [DOI] [PubMed] [Google Scholar]
- 52. van der Linden CG, Simonides WS, Muller A, van der Laarse WJ, Vermeulen JL, Zuidwijk MJ, Moorman AF, van Hardeveld C. Fiber-specific regulation of Ca(2+)-ATPase isoform expression by thyroid hormone in rat skeletal muscle. The Am J Physiol Cell Physiol 271: C1908–C1919, 1996 [DOI] [PubMed] [Google Scholar]
- 53. Wagner RL, Huber BR, Shiau AK, Kelly A, Cunha Lima ST, Scanlan TS, Apriletti JW, Baxter JD, West BL, Fletterick RJ. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol 15: 398–410, 2001 [DOI] [PubMed] [Google Scholar]
- 54. Webb P. Selective activators of thyroid hormone receptors. Exp Opinion Invest Drugs 13: 489–500, 2004 [DOI] [PubMed] [Google Scholar]
- 55. Weiss RE, Chassande O, Koo EK, Macchia PE, Cua K, Samarut J, Refetoff S. Thyroid function and effect of aging in combined hetero/homozygous mice deficient in thyroid hormone receptors alpha and beta genes. J Endocrinol 172: 177–185, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Wennlund A, Felig P, Hagenfeldt L, Wahren J. Hepatic glucose production and splanchnic glucose exchange in hyperthyroidism. J Clin Endocrinol Metab 62: 174–180, 1986 [DOI] [PubMed] [Google Scholar]
- 57. Yu C, Cresswell J, Loffler MG, Bogan JS. The glucose transporter 4-regulating protein TUG is essential for highly insulin-responsive glucose uptake in 3T3–L1 adipocytes. J Biol Chem 282: 7710–7722, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]