

Abstract
Chroman aldehydes bearing an acetyl group plus alkoxyl or hydroxyl groups inhibit HIV infectivity in HeLa37 cells.
Keywords: antiviral, chroman, aldehyde, HIV
As a result of a concerted effort to identify natural products that inhibit HIV, a wealth of useful natural product leads have been reported.1 Several small molecular weight natural products have been discovered that inhibit viruses.2 In 2003, Lee and coworkers isolated aldehyde 1, shown in Figure 1, from Desmos dumosus and evaluated it for inhibition of HIV replication in H9 lymphocyte cells.3 Remarkably, 1 demonstrated potent anti-HIV activity with an ability to inhibit 50% of HIV replication (IC50) at 0.022 µg/mL and with a large therapeutic index (TI) of 489. Lawinal (2) is a related antiviral aldehyde.3 As part of an effort to identify useful antiviral agents,4, 5 we report herein the synthesis of chroman aldehydes related to 1 and 2.
Figure 1.

Structures of small molecule antiviral agents
Chroman 3 was available in one pot from phloroglucinol and isoprene in 69% yield after modification of a literature procedure.6 As shown in Scheme 1, acetylation of 3 with acetic anhydride afforded a mixture of 4, 5 and 67 in 86% yield. Chromatography yielded a mixture of 4 and 5 in 82% yield and diketone 6 in 4% yield. Ketone 4 could be readily separated from 5 by recrystallization. Formylation of 4 with dichloromethyl methyl ether and TiCl48 produced keto aldehyde 7 in 62% yield.
Scheme 1.

Synthesis of analogs.
To better understand the effects of structure on activity, we prepared dialdehyde 9 from 3 and ether 10 from 8. As shown in Scheme 2, oxidation of 7 with DDQ9 in benzene produced chromene 11. Benzoylation followed by rearrangement to a beta-diketone generated diketone 12.
Scheme 2.

Derivatives of aldehyde 7.
Assessment of antiviral activity of the compounds. Compounds 6–12 were evaluated for their ability to inhibit HIV infectivity in HeLa37 cells as previously described.10 The concentration of compounds that inhibited 50 and 90% of HIV infectivity (IC50 and IC90) are shown in Table 1. In parallel, the cytotoxicity of each compound at those same concentrations was appraised. At concentrations as high as 100 µg/mL, little cytotoxicity was observed with any of the compounds except compound 12 which had significant cytotoxicity at the higher concentrations tested. The fourteen carbon compounds 7, 8 and 11 gave the lowest IC50 values between 30 and 40 µM.
Table 1.
Anti-HIV activity and cytotoxicity of synthesized phlorogluconols.
| Compound | Cytotoxicity (µM) | HIV-1 Infection (µM) | ||
|---|---|---|---|---|
| LC50 | LC90 | IC50 | IC90 | |
| 7 | 145 | 258 | 30.2 | 85 |
| 6 | >100 | N/D | >100 | >100 |
| 8 | >100 | N/D | 39.8 | 85.6 |
| 9 | >100 | N/D | 77.3 | >100 |
| 10 | >100 | N/D | 55.3 | >100 |
| 11 | >100 | N/D | 34.6 | 98.2 |
| 12 | 84.2 | N/D | 40.7 | 86.6 |
We investigated the HeLa37 cell cytotoxicity associated with compound 7 in further detail to obtain a therapeutic index (TI) for the fourteen carbon compound that gave the lowest IC50 values. We found that the concentration of compound that killed 50% or 90% of cells (LC50 or LC90) of compound 7 was 145 and 258 µM, respectively (Figure 2).
Figure 2.

A dose response curve of the ability of compound 7 to inhibit HIV-1 NL4-3 infectivity. Cytotoxicity in HeLa37 cells of compound 7 is also shown. All studies were performed in triplicate three independent times. Shown are the means and standard errors of the means.
Discussion
We found that all compounds had inhibitory activity against HIV. The fourteen carbon compounds, compounds 7, 8 and 11, were most efficacious with a range of IC50 values from 30 to 40 µM. Compound 7 that had the lowest IC50 values was also analyzed in detail for cytotoxicity, giving a LC50 of 145 µM, resulting in a TI of about 5. Compounds 8 and 11 that had slightly poorer IC50 may have similar TIs as compound 7. The TI of our lead compound was significantly smaller than that reported by Wu et al. for Compound 1,3 due to apparently reduced antiviral activity of Compound 7. However, it should be noted that in our studies we assessed the antiviral viral activity during a single round of infection, whereas Compound 1 was assess for the ability to inhibit multiple rounds of HIV infectivity.
Our other compounds had either higher IC50 values for HIV inhibition or LC50 values for cytotoxicity below 100 µM. The diketone 6 was the least active, indicating that an aldehyde moiety is necessary for good activity. Interestingly, the O-methylated derivative of aldehyde 8 showed good activity. Diketone 12, the closest analog to 1, exhibited good activity but had the highest cytotoxicity.
In summary, we synthesized a series of aromatic aldehydes that are related to naturally derived aldehydes 1 and 2. We evaluated the ability of these compounds to inhibit HIV infectivity. Although several aldehydes showed inhibitory activity, none of the compounds showed an IC50 value or a therapeutic index comparable to compound 1.
Acknowledgment
Studies were supported by funds provided by NIH P50 AT004155.
References
- 1.Gambari R, Lampronti I. Advances in Phytomedicine. 2006;2:299. (Lead Molecules from Natural Products) [Google Scholar]
- 2.Liu T, Li AX, Miao YP, Wu KZ, Ma Y. Chinese Chem. Lett. 2009;20(11):1386. [Google Scholar]
- 3.Wu J-H, Wang X-H, Yi Y-H, Lee K-H. Bioorg. Med. Chem. Lett. 2003;13(10):1813. doi: 10.1016/s0960-894x(03)00197-5. [DOI] [PubMed] [Google Scholar]
- 4.Maury W, Price JP, Brindley MA, Oh CS, Neighbors JD, Wiemer DF, Wills N, Carpenter S, Hauck C, Murphy P, Widrlechner MP, Delate K, Kumar G, Kraus GA, Rizshsky L, Nikolau B. Virology Journal. 2009;6:101. doi: 10.1186/1743-422X-6-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraus GA, Yuan Y, Kempema A. Molecules. 2009;14(2):807. doi: 10.3390/molecules14020807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalena GP, Jain A, Banerji A. Molecules. 1997;2:100–105. [Google Scholar]
- 7.Donnelly WJ, Shannon PVR. J. Chem. Soc. Perkin Trans. 1972;1:25. [Google Scholar]
- 8.Nakagawa-Goto K, Lee K-H. Tetrahedron Lett. 2006;47(47):8263–8266. [Google Scholar]
- 9.Ahluwalia VK, Jolly RS. Synlett. 1982:74. [Google Scholar]
- 10.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. J. Virology. 1998;72:2855. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reed-Inderbitzin E, Maury W. Virology. 2003;314:680. doi: 10.1016/s0042-6822(03)00508-7. [DOI] [PubMed] [Google Scholar]
- 12.Ahluwalia VK, Arora KK, Jolly RS. J. Chem. Soc. Perkin Trans. 1982;1:335–338. [Google Scholar]
-
13.Experimental2,2-Dimethyl-5,7-dihydroxychroman (3). A 100-mL round bottom flask equipped with a stir bar was charged with phloroglucinol dihydrate (500.9 mg, 3.97 mmol) followed by 1,4-dioxane (10 mL) and Amberlyst 15 (1.5 g). The resulting heterogeneous mixture was cooled to 0 °C, and then isoprene (600 mL, 6.0 mmol) was added over 5 min. The reaction vessel was next equipped with a reflux condenser and heated to 101 °C. After 17 h, the reaction mixture was cooled to rt, filtered through Celite, and the catalyst was washed with hot acetone. The filtrate was concentrated in vacuo to afford a light yellow solid, which was purified by column chromatography (hexanes:EtOAc = 9:1 → 1:1) to first give a mixture of two compounds, which were separated on a silica gel column eluted with toluene. The first fraction gave 2,2,8,8-tetramethyl-2,3,4,8,9,10-hexahydropyrano[2,3-f]chromen-5-ol 13 (54 mg). The second fraction gave 2,2,8,8-tetramethyl-2,3,4,6,7,8-hexahydropyrano[3,2-g]chromen-5-ol 14 (50 mg), spectral data matched the lit.12
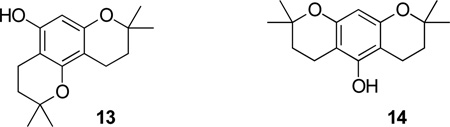 2,2-Dimethyl-5,7-dihydroxychroman 3 was isolated (536 mg, 69%) as colorless needles from benzene, mp 163–164 °C (lit.,12 163–164 °C). The final fraction contained recovered phloroglucinol (85.6 mg).Synthesis of 4, 5, and 6 by the acetylation of 3. A 50-mL round bottom flask equipped with a stir bar was charged with 2,2-dimethyl-5,7-dihydroxychroman (3) (1.70 g, 8.77 mmol) followed by AcOH (18.4 mL) and Ac2O (0.91 mL, 9.65 mmol). The resulting mixture was stirred vigorously and warmed to 40 °C until it was homogeneous, and then BF3·OEt2 (1.17 mL, 9.21 mmol) was added over 1 min. The resulting red mixture was warmed to 100 °C. After 10 hours at that temperature, the mixture was cooled to 0 °C, quenched with water (10 mL) and poured into a separatory funnel containing EtOAc (25 mL). The phases were separated and the aqueous phase was extracted with EtOAc (3×25 mL) and 5% MeOH/EtOAc (5×20 mL). The combined organic fractions were washed with water (2×25 mL) and brine (1×25 mL), dried over MgSO4, filtered and concentrated in vacuo. The pale orange solid was purified by chromatography. Elution with hexanes:EtOAc (50:1) gave 6,8-diacetyl-2,2-dimethylchroman-5,7-diol (6) (97.6 mg, 4%) as a pale yellow needles, mp 129–130 °C (lit.7 131–132.5 °C); 1H NMR (400 MHz, CDCl3) δ 2.71 (s, 3H), 2.64 (s, 3H), 2.60 (t, J = 6.8 Hz, 2H), 1.82 (t, J = 6.8 Hz, 2H), 1.42 (s, 3H). Elution with hexanes:EtOAc (5:1) gave a 1:1 mixture (by 1H NMR) of 6-acetyl-2,2-dimethylchroman-5,7-diol 4 and 8-acetyl-2,2-dimethylchroman (5) (1.68 g, 81.4%). Crystallization from benzene first gave 4 as a yellow solid, mp 230 – 230.5 °C (lit.7 230 °C); 1H NMR (400 MHz, acetone-d6) δ 5.87 (s, 1H), 2.61 (s, 3H), 2.53 (t, J = 6.8 Hz, 2H), 1.78 (t, J = 6.8 Hz, 2H), 1.30 (s, 6H). The mother liquor was concentrated and the yellow residue afforded 5 as a yellow solid from benzene.8-Acetyl-2,2-dimethylchroman (5). mp 150–151 °C (lit.,7 150 °C) 1H NMR (400 MHz, acetone-d6) δ 5.94 (s, 1H), 2.57 (t, partially obscured, J = 6.8 Hz, 2H), 2.55 (s, 3H), 1.76 (t, J = 6.8 Hz, 2H), 1.37 (s, 6H).6-Acetyl-2,2-dimethyl-8-formylchroman-5,7-diol (7). A 100-mL round bottom flask equipped with a stir bar was charged with 4 (620.9 mg, 2.63 mmol) followed by anhydrous CH2Cl2 (50 mL). The resulting mixture was cooled to −78 °C and then TiCl4 (1.5 mL, 13.4 mmol) was added. This mixture was treated with Cl2CHOMe (2.26 mL, 25.0 mmol). The resulting red solution was spontaneously warmed to rt and then stirred for 10 h. Then it was cooled to 0 °C, slowly quenched with ice-cold water (10 mL), stirred for 1 h and poured into a separatory funnel. The organic phase was washed with water (3×25 mL) and brine (1×25 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography. Elution with 2% EtOAc and 98% hexanes gave 7 (427.2 mg, 62%) as yellow prisms, mp 102–104 °C. 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 2.69 (s, 3H), 2.56 (t, J = 6.8 Hz, 2H) 1.82 (t, J = 6.8 Hz, 2H), 1.39 (s, 6H). %). 13C NMR (100 MHz, CDCl3) δ 203.7, 191.8, 171.0, 168.3, 161.8, 103.9, 103.4, 100.3, 77.8, 32.6, 31.5, 26.6, 15.5. HRMS, Calcd for C14H16O5: 264.0998, Found 264.1004.8-Acetyl-5,7-dihydroxy-2,2-dimethylchroman-6-carboxaldehyde (8). The reaction of 5 under conditions identical to those used for the formylation of 4 provided product 8. Pale yellow needles, mp 131–134 °C. 1H NMR (400 MHz, CDCl3) δ 13.23 (s, 1H), 10.18 (s, 1H), 3.63 (s, 3H), 2.60 (t, partially obscured, J = 6.8 Hz, 2H), 1.83 (t, J = 6.8 Hz, 2H), 1.44 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 203.4, 192.3, 169.7, 167.3, 163.0, 104.4, 104.2, 99.6, 78.4, 33.2, 31.1, 26.9, 15.3.5,7-Dihydroxy-2,2-dimethylchroman-6,8-dicarbaldehyde (9). A 100-mL round bottom flask equipped with a stir bar was charged with 3 (57 mg, 0.295 mmol) followed by anhydrous CH2Cl2 (10 mL). The mixture was gently warmed and vigorously stirred to fully dissolve the starting material. Next, Cl2CHOMe (160 µL, 1.77 mmol) was added to the reaction mixture at rt. After cooling to −78 °C, TiCl4 (98 µL, 0.885 mmol) was added in drops over 5 min. The resulting red solution was spontaneously warmed to rt and then stirred for 10h. It was then cooled to 0 °C, slowly quenched with ice-cold water (5 mL), and stirred for 1 h. CH2Cl2 was then distilled off using a rotary evaporator. The purplish slurry was dissolved in EtOAc (20 mL) and poured into a separatory funnel. The organic phase was washed with water (3×20 mL) and brine (1×20 mL) then dried over MgSO4, filtered and concentrated to give a dark purplish-brown residue. It was purified by column chromatography (EtOAc-hexanes) to give 9 (23 mg, 31%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 10.16 (s, 1H), 10.04 (s, 1H), 3.59 (t, J = 6.4 Hz, 2H), 1.82 (t, J = 6.4 Hz, 2H), 1.42 (s, 6H). 13C (100 MHz, CDCl3) δ 192.1, 168.9, 168.1, 163.3, 104.1, 103.7, 100.0, 78.3, 31.4, 26.7, 12.1. HSMS, Calcd for C13H14O5: 250.08412, Found 250.08458.8-Acetyl-5,7-dimethoxy-2,2-dimethylchroman-6-carbaldehyde (10). A 50-mL round bottom flask equipped with a stir bar was charged with 8 (55.3 mg, 0.209 mmol) followed by THF (2 mL) and t-BuOH (0.5 mL). The resulting mixture was cooled to 0 °C and then t-BuOK (56.4 mg, 0.502 mmol) was added all at once. MeI (65 µL, 1.05 mmol) was added in drops over 1 min. The ice bath was removed to allow the resulting yellow slurry to warm to rt; then it was warmed to 40 °C for 2 h. After cooling to rt, the mixture was quenched with water and acidified with 1 N HCl. The mixture was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4 and concentrated. The residue was purified by column chromatography on SiO2 (EtOAc-hexanes) to obtain 10 (6.3 mg, 10%). 1H NMR (400 MHz, CDCl3) δ 10.23 (s, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 2.73 (t, J = 6.4 Hz, 2H), 1.82 (t, J = 6.4 Hz, 2H), 1.36 (s, 6H).6-Acetyl-2,2-dimethyl-8-formylchromen-5,7-diol (11). A slurry of 7 (75.9 mg, 0.287 mmol) and DDQ (71.7 mg, 0.316 mmol) in benzene (3 mL) was heated at 100°C in a sealed tube for 20 h. Then the mixture was filtered through Celite. Benzene was used to rinse the reaction flask and wash the filter pad. The filtrate was concentrated, and then the residue was purified by column chromatography on SiO2 to obtain 11 (1.6 mg, 2%). 1H NMR (400 MHz, CDCl3) δ 13.15 (s, 1H), 10.20 (s, 1H), 6.61 (d, J = 10 Hz, 1H), 5.51 (d, J = 10 Hz, 1H), 6.68 (s, 3H), 1.55 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 203.6, 192.7, 178.3, 170.9, 164.6, 124.8, 115.4, 104.8, 104.3, 101.1, 80.7, 33.2, 28.8. HRMS Calcd for C14H14O5: 262.0841, Found 262.0846.6-(1,3-Dioxo-3-phenylpropyl)-2,2-dimethyl-8-formylchroman-5,7-diol (12). Potassium tert-butoxide (100 mg, 0.82 mmol) was suspended in t-BuOH (1.5 mL) and anhydrous THF (5 mL), and then cooled to −10°C. A solution of 7 (58.1 mg, 0.22 mmol) in anhydrous THF (5 mL) was added. After stirring at −10°C for 15 min, benzoyl chloride (0.512 mL, 0.44 mmol) was added. The ice bath was removed, and the mixture warmed to rt. Then the mixture was heated at reflux for 5 h. After cooling to rt, the mixture was quenched with water and acidified with 1N HCl. The mixture was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4 and concentrated. The residue was purified by column chromatography on SiO2 (EtOAc-hexanes 1:19) to obtain 12 (17 mg, 21%) as a mixture of tautomers. 1H NMR (300 MHz, CDCl3) δ 10.03 (s, 1H, CHO, for enol form), 9.95 (s, 0.73H, CHO, for 1,3-diketo form), 7.97–7.92 (m, ArH, for both forms), 7.65 (s, olefin for enol form), 7.53–7.44 (m, ArH, for both forms), 4.68 (s, 2H, C(O)CH2C(O) for 1,3-diketo form), 2.63–2.57 (m, CH2, for both forms), 1.86–1.81 (m, CH2, for both forms), 1.40 (s, 6H, 2xCH3, for enol form) 1.39 (s, 6H, 2xCH3, CH3 for 1,3-diketo form); HRMS Calcd for C21H20O6: 368.126, Found 368.126.Virus infectivity studies. Single round infectivity studies in HeLa37 cells were used to assess antiviral activity of the compounds. Approximately 250 infectious particles of a X4-tropic HIV strain that is lab adapted (NL4-3) were combined with serial dilutions of the compounds. Increasing amounts of vehicle, DMSO, were observed to result in cell cytotoxicity with 0.5% DMSO resulting in about 5% reduction in cell viability. Thus, in these studies, we never exceeded 0.5% DMSO in the cultures and a dose response curve from 0.05 to 0.5% DMSO was performed independently. The effect on cell viability of the DMSO concentrations was subtracted from the inhibition observed in the presence of each compound. The extract and virus mix was added to 5×104 cells/well of HeLa37 cells in a 48-well format resulting in a multiplicity of infection (MOI) of ~0.005 as previously described.11 The infections were maintained for 40 h. Cells were fixed with 75% acetone/25% water and immunostained for HIV antigens using human anti-HIV antisera. HIV antigen positive cells were enumerated in the cultures. The findings for each compound were normalized to control levels at that same concentration of DMSO. IC50 and IC90 concentrations were determined using TableCurve software (Systat Academic).Cell viability studies. Cells were plated and treated with compounds as described above for the infectivity assays. Forty hours following treatment, cell viability was monitored by ATPLite Assay (Packard Biosciences) per manufacturer’s instructions.
2,2-Dimethyl-5,7-dihydroxychroman 3 was isolated (536 mg, 69%) as colorless needles from benzene, mp 163–164 °C (lit.,12 163–164 °C). The final fraction contained recovered phloroglucinol (85.6 mg).Synthesis of 4, 5, and 6 by the acetylation of 3. A 50-mL round bottom flask equipped with a stir bar was charged with 2,2-dimethyl-5,7-dihydroxychroman (3) (1.70 g, 8.77 mmol) followed by AcOH (18.4 mL) and Ac2O (0.91 mL, 9.65 mmol). The resulting mixture was stirred vigorously and warmed to 40 °C until it was homogeneous, and then BF3·OEt2 (1.17 mL, 9.21 mmol) was added over 1 min. The resulting red mixture was warmed to 100 °C. After 10 hours at that temperature, the mixture was cooled to 0 °C, quenched with water (10 mL) and poured into a separatory funnel containing EtOAc (25 mL). The phases were separated and the aqueous phase was extracted with EtOAc (3×25 mL) and 5% MeOH/EtOAc (5×20 mL). The combined organic fractions were washed with water (2×25 mL) and brine (1×25 mL), dried over MgSO4, filtered and concentrated in vacuo. The pale orange solid was purified by chromatography. Elution with hexanes:EtOAc (50:1) gave 6,8-diacetyl-2,2-dimethylchroman-5,7-diol (6) (97.6 mg, 4%) as a pale yellow needles, mp 129–130 °C (lit.7 131–132.5 °C); 1H NMR (400 MHz, CDCl3) δ 2.71 (s, 3H), 2.64 (s, 3H), 2.60 (t, J = 6.8 Hz, 2H), 1.82 (t, J = 6.8 Hz, 2H), 1.42 (s, 3H). Elution with hexanes:EtOAc (5:1) gave a 1:1 mixture (by 1H NMR) of 6-acetyl-2,2-dimethylchroman-5,7-diol 4 and 8-acetyl-2,2-dimethylchroman (5) (1.68 g, 81.4%). Crystallization from benzene first gave 4 as a yellow solid, mp 230 – 230.5 °C (lit.7 230 °C); 1H NMR (400 MHz, acetone-d6) δ 5.87 (s, 1H), 2.61 (s, 3H), 2.53 (t, J = 6.8 Hz, 2H), 1.78 (t, J = 6.8 Hz, 2H), 1.30 (s, 6H). The mother liquor was concentrated and the yellow residue afforded 5 as a yellow solid from benzene.8-Acetyl-2,2-dimethylchroman (5). mp 150–151 °C (lit.,7 150 °C) 1H NMR (400 MHz, acetone-d6) δ 5.94 (s, 1H), 2.57 (t, partially obscured, J = 6.8 Hz, 2H), 2.55 (s, 3H), 1.76 (t, J = 6.8 Hz, 2H), 1.37 (s, 6H).6-Acetyl-2,2-dimethyl-8-formylchroman-5,7-diol (7). A 100-mL round bottom flask equipped with a stir bar was charged with 4 (620.9 mg, 2.63 mmol) followed by anhydrous CH2Cl2 (50 mL). The resulting mixture was cooled to −78 °C and then TiCl4 (1.5 mL, 13.4 mmol) was added. This mixture was treated with Cl2CHOMe (2.26 mL, 25.0 mmol). The resulting red solution was spontaneously warmed to rt and then stirred for 10 h. Then it was cooled to 0 °C, slowly quenched with ice-cold water (10 mL), stirred for 1 h and poured into a separatory funnel. The organic phase was washed with water (3×25 mL) and brine (1×25 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography. Elution with 2% EtOAc and 98% hexanes gave 7 (427.2 mg, 62%) as yellow prisms, mp 102–104 °C. 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 2.69 (s, 3H), 2.56 (t, J = 6.8 Hz, 2H) 1.82 (t, J = 6.8 Hz, 2H), 1.39 (s, 6H). %). 13C NMR (100 MHz, CDCl3) δ 203.7, 191.8, 171.0, 168.3, 161.8, 103.9, 103.4, 100.3, 77.8, 32.6, 31.5, 26.6, 15.5. HRMS, Calcd for C14H16O5: 264.0998, Found 264.1004.8-Acetyl-5,7-dihydroxy-2,2-dimethylchroman-6-carboxaldehyde (8). The reaction of 5 under conditions identical to those used for the formylation of 4 provided product 8. Pale yellow needles, mp 131–134 °C. 1H NMR (400 MHz, CDCl3) δ 13.23 (s, 1H), 10.18 (s, 1H), 3.63 (s, 3H), 2.60 (t, partially obscured, J = 6.8 Hz, 2H), 1.83 (t, J = 6.8 Hz, 2H), 1.44 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 203.4, 192.3, 169.7, 167.3, 163.0, 104.4, 104.2, 99.6, 78.4, 33.2, 31.1, 26.9, 15.3.5,7-Dihydroxy-2,2-dimethylchroman-6,8-dicarbaldehyde (9). A 100-mL round bottom flask equipped with a stir bar was charged with 3 (57 mg, 0.295 mmol) followed by anhydrous CH2Cl2 (10 mL). The mixture was gently warmed and vigorously stirred to fully dissolve the starting material. Next, Cl2CHOMe (160 µL, 1.77 mmol) was added to the reaction mixture at rt. After cooling to −78 °C, TiCl4 (98 µL, 0.885 mmol) was added in drops over 5 min. The resulting red solution was spontaneously warmed to rt and then stirred for 10h. It was then cooled to 0 °C, slowly quenched with ice-cold water (5 mL), and stirred for 1 h. CH2Cl2 was then distilled off using a rotary evaporator. The purplish slurry was dissolved in EtOAc (20 mL) and poured into a separatory funnel. The organic phase was washed with water (3×20 mL) and brine (1×20 mL) then dried over MgSO4, filtered and concentrated to give a dark purplish-brown residue. It was purified by column chromatography (EtOAc-hexanes) to give 9 (23 mg, 31%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 10.16 (s, 1H), 10.04 (s, 1H), 3.59 (t, J = 6.4 Hz, 2H), 1.82 (t, J = 6.4 Hz, 2H), 1.42 (s, 6H). 13C (100 MHz, CDCl3) δ 192.1, 168.9, 168.1, 163.3, 104.1, 103.7, 100.0, 78.3, 31.4, 26.7, 12.1. HSMS, Calcd for C13H14O5: 250.08412, Found 250.08458.8-Acetyl-5,7-dimethoxy-2,2-dimethylchroman-6-carbaldehyde (10). A 50-mL round bottom flask equipped with a stir bar was charged with 8 (55.3 mg, 0.209 mmol) followed by THF (2 mL) and t-BuOH (0.5 mL). The resulting mixture was cooled to 0 °C and then t-BuOK (56.4 mg, 0.502 mmol) was added all at once. MeI (65 µL, 1.05 mmol) was added in drops over 1 min. The ice bath was removed to allow the resulting yellow slurry to warm to rt; then it was warmed to 40 °C for 2 h. After cooling to rt, the mixture was quenched with water and acidified with 1 N HCl. The mixture was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4 and concentrated. The residue was purified by column chromatography on SiO2 (EtOAc-hexanes) to obtain 10 (6.3 mg, 10%). 1H NMR (400 MHz, CDCl3) δ 10.23 (s, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 2.73 (t, J = 6.4 Hz, 2H), 1.82 (t, J = 6.4 Hz, 2H), 1.36 (s, 6H).6-Acetyl-2,2-dimethyl-8-formylchromen-5,7-diol (11). A slurry of 7 (75.9 mg, 0.287 mmol) and DDQ (71.7 mg, 0.316 mmol) in benzene (3 mL) was heated at 100°C in a sealed tube for 20 h. Then the mixture was filtered through Celite. Benzene was used to rinse the reaction flask and wash the filter pad. The filtrate was concentrated, and then the residue was purified by column chromatography on SiO2 to obtain 11 (1.6 mg, 2%). 1H NMR (400 MHz, CDCl3) δ 13.15 (s, 1H), 10.20 (s, 1H), 6.61 (d, J = 10 Hz, 1H), 5.51 (d, J = 10 Hz, 1H), 6.68 (s, 3H), 1.55 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 203.6, 192.7, 178.3, 170.9, 164.6, 124.8, 115.4, 104.8, 104.3, 101.1, 80.7, 33.2, 28.8. HRMS Calcd for C14H14O5: 262.0841, Found 262.0846.6-(1,3-Dioxo-3-phenylpropyl)-2,2-dimethyl-8-formylchroman-5,7-diol (12). Potassium tert-butoxide (100 mg, 0.82 mmol) was suspended in t-BuOH (1.5 mL) and anhydrous THF (5 mL), and then cooled to −10°C. A solution of 7 (58.1 mg, 0.22 mmol) in anhydrous THF (5 mL) was added. After stirring at −10°C for 15 min, benzoyl chloride (0.512 mL, 0.44 mmol) was added. The ice bath was removed, and the mixture warmed to rt. Then the mixture was heated at reflux for 5 h. After cooling to rt, the mixture was quenched with water and acidified with 1N HCl. The mixture was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4 and concentrated. The residue was purified by column chromatography on SiO2 (EtOAc-hexanes 1:19) to obtain 12 (17 mg, 21%) as a mixture of tautomers. 1H NMR (300 MHz, CDCl3) δ 10.03 (s, 1H, CHO, for enol form), 9.95 (s, 0.73H, CHO, for 1,3-diketo form), 7.97–7.92 (m, ArH, for both forms), 7.65 (s, olefin for enol form), 7.53–7.44 (m, ArH, for both forms), 4.68 (s, 2H, C(O)CH2C(O) for 1,3-diketo form), 2.63–2.57 (m, CH2, for both forms), 1.86–1.81 (m, CH2, for both forms), 1.40 (s, 6H, 2xCH3, for enol form) 1.39 (s, 6H, 2xCH3, CH3 for 1,3-diketo form); HRMS Calcd for C21H20O6: 368.126, Found 368.126.Virus infectivity studies. Single round infectivity studies in HeLa37 cells were used to assess antiviral activity of the compounds. Approximately 250 infectious particles of a X4-tropic HIV strain that is lab adapted (NL4-3) were combined with serial dilutions of the compounds. Increasing amounts of vehicle, DMSO, were observed to result in cell cytotoxicity with 0.5% DMSO resulting in about 5% reduction in cell viability. Thus, in these studies, we never exceeded 0.5% DMSO in the cultures and a dose response curve from 0.05 to 0.5% DMSO was performed independently. The effect on cell viability of the DMSO concentrations was subtracted from the inhibition observed in the presence of each compound. The extract and virus mix was added to 5×104 cells/well of HeLa37 cells in a 48-well format resulting in a multiplicity of infection (MOI) of ~0.005 as previously described.11 The infections were maintained for 40 h. Cells were fixed with 75% acetone/25% water and immunostained for HIV antigens using human anti-HIV antisera. HIV antigen positive cells were enumerated in the cultures. The findings for each compound were normalized to control levels at that same concentration of DMSO. IC50 and IC90 concentrations were determined using TableCurve software (Systat Academic).Cell viability studies. Cells were plated and treated with compounds as described above for the infectivity assays. Forty hours following treatment, cell viability was monitored by ATPLite Assay (Packard Biosciences) per manufacturer’s instructions.