

Abstract
As members of the glycosaminoglycan (GAG) family, heparin and heparan sulfate (HS) are responsible for mediation of a wide range of essential biological actions, most of which are mediated by specific patterns of modifications of regions of these polysaccharides. To fully understand the regulation of HS modification and the biological function of HS through its interactions with protein ligands, it is essential to know the specific HS sequences present. However, the sequencing of mixtures of HS oligosaccharides presents major challenges due to the lability of the sulfate modifications, as well as difficulties in separating isomeric HS chains. Here, we apply a sequential chemical derivatization strategy involving permethylation, desulfation and trideuteroperacetylation to label original sulfation sites with stable and hydrophobic trideuteroacetyl groups. The derivatization chemistry differentiates between all possible heparin/HS sequences solely by glycosidic bond cleavages, without the need to generate cross-ring cleavages. This derivatization strategy combined with LC-MS/MS analysis has been used to separate and sequence five synthetic HS-like oligosaccharides of sizes up to dodecasaccharide, as well as a highly-sulfated Arixtra-like heptamer. This strategy offers a unique capability for the sequencing of microgram quantities of HS oligosaccharide mixtures by LC-MS/MS.
INTRODUCTION
Heparan sulfate (HS) and heparin are linear, highly negatively charged polysaccharides that belong to the glycosaminoglycan family, with molecular weights ranging from 5 to 70kDa1,2. Through specific binding to a variety of proteins, HS and heparin have been recognized as key factors for mediation of a wide range of biological actions, such as cell growth control, cell signaling, cell adhesion and migration, inflammation, anticoagulation, neural development and regeneration3-7. Their biological significance makes HS and heparin important targets for drug discovery. One of the most studied heparin protein binding motifs is the pentasaccharide responsible for binding antithrombin III and inhibiting the coagulation cascade8, which has been formulated into an anticoagulant drug (Arixtra). Ongoing efforts are identifying numerous other examples of protein-binding motifs in HS, which could be potential drug candidates9,10. Most of the interactions between HS motifs and proteins are structurally specific, requiring a specific sequence of modifications across an oligosaccharide of moderate length. Changes to the HS biosynthesis pathway, whether by regulation or disease state, can alter such interactions leading to change or loss of function. Thus, detailed information on these heparin/HS oligosaccharide sequences is required for a better understanding of their structure-function relationship, as well as for the development of HS/heparin-based drugs11.
The diversity of heparin/HS modification is what drives the biology, and what makes sequencing of these polysaccharides so challenging. HS and heparin polysaccharide chains are composed of glucosamine (GlcN) and uronic acid (UA) disaccharide repeat units with various types of modifications including acetylation, sulfation and epimerization of the C-5 position of the UA. In the GlcN residue, the amine group can either be a free amine, and acetylated amine, or a sulfated amine. The 6-O position of the GlcN residue can be sulfated, and in uncommon but biologically important instances, GlcNS can be additionally sulfated at the 3-O position. The uronic acid can be either glucuronic acid (GlcA) or iduronic acid (IdoA) differing by the stereochemistry at the C-5 position, and the uronic acid can be sulfated at the 2-O position2. The synthesis of these polysaccharides are not template-driven like DNA, but rather are driven by untemplated enzymatic modification of the sugar backbone, which is mediated by a complex and dynamic suite of modification enzymes12. The result is a mixture of heparin/HS sequences that are both polydisperse and heterogeneously modified, often resulting in isomeric sequences that differ widely in biological functions.
The high degree of heterogeneity and negative charge in these polysaccharides caused by the variety of chain lengths and diverse sulfation patterns makes their structural determination a very challenging task. Rapid progresses in mass spectrometry (MS) instrumentation and chromatography separation technique have led to an increasing use of these methodologies in GAG structural studies13. Several MS techniques have been used for structural analyses of GAGs including fast-atom bombardment (FAB)14,15, matrix-assisted laser desorption-ionization (MALDI)16-18, and electrospray ionization (ESI)19-21. ESI is the most commonly used ionization method for its gentle ionization giving minimum in-source fragmentation and sulfate loss. Most methods for the detailed structural analysis of heparin/HS involves either complete or partial depolymerization by either enzymatic or chemical means to obtain disaccharides mixtures for compositional analysis or a range of oligosaccharide fractions with different lengths for oligosaccharide analysis22,23. These methods always involve separation of HS/heparin oligosaccharides for either off-line purification or on-line liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, using capillary electrophoresis (CE)24 or high-performance liquid chromatography (HPLC)25 including strong anion-exchange chromatography (SAX)26,27, size-exclusion chromatography (SEC)21,28, hydrophilic interaction chromatography (HILIC)29,30, porous graphitized carbon separation (PGC)31,32, and ion-pairing reversed-phase (IPRP)33,34 chromatography.
Disaccharide compositional profiling of HS/heparin derivatized disaccharides is useful and allows for quantitative analysis by using commercially available disaccharides as standards. For differentiation of isomeric disaccharides, collision induced dissociation (CID) MS/MS or multi-stages tandem mass spectrometry (MSn) is used to generate diagnostic ions for identifying disaccharides, and recent development in IPRP-HPLC or IPRP-UPLC technique enabled separation of 12 common commercially available HS disaccharides allowing the use of retention time for disaccharide identification20,33-35. While disaccharide composition analysis uses relatively little sample and has short analysis times, significant sample preparation is still required. Certain regions of heparin/HS have been shown to be resistant to digestion down to disaccharides, biasing the composition results36. Another compositional analysis method was developed by Zaia’s group using a HILIC LC-MS platform for on-line separation and MS analysis of oligosaccharides with different lengths derivatized from enzyme digestion29,30,37. Compositional information like numbers of hexuronic acid, N-acetylglucosamine, sulfate groups and acetyl groups can be obtained for each observed oligosaccahride based on their accurate mass, and heparin/HS glycomics profiles can be obtained and compared for different samples. While both composition analysis methods are useful for studying patterns and trends in heparin/HS biology, they do not reveal the detailed sequence information of the oligosaccharide units required for most HS-protein interactions.
A major impediment to use tandem mass spectrometry for structural sequencing of heparin/HS oligosaccharides is sulfate loss during fragmentation. As heparin/HS is collisionally activated, one of the most common fragmentation pathways is loss of the sulfate modification, resulting in a loss of sequence information regarding the original site of sulfation. It has been demonstrated that the loss of sulfate groups can be minimized using a combination of charge state manipulation and metal ion adduction19,38,39. However, delicate optimization of buffer and ionization conditions is required for each oligosaccharide and on-line separation of isomeric sequences is highly limited, narrowing the applicability of such approach. Saad and Leary introduced a program called heparin oligosaccharide sequencing tool (HOST) for automated sequencing using the results of tandem mass spectrometry for disaccharides produced by enzyme digestion from the target oligosaccharides40. The use of such method is limited in application to structurally homogeneous sample because of its incompatibility with online LC separation. Besides the traditional CID, another tandem mass spectrometry technique, electron detachment dissociation (EDD) has recently been applied for GAG structural studies, with the capability of distinguishing GlcA from IdoA41. Like the HOST sequencing method, additional purification step for oligosaccharides or samples with very few contaminants is required in order to generate promising structural information.
While chemical derivatizations, for example, permethylation and acetylation, are not as commonly applied for structural analysis of GAG oligosaccharides as for other carbohydrates (such as N- and O-linked glycans), there were studies reported in 1980s using sequential chemical derivatization for structural sequencing of heparin oligosaccharides with fine structure by FAB MS42, and combined with chemical depolymerization for monosaccharide analysis of heparin polysaccharides by chemical ionization MS coupled with gas-liquid chromatography43. We modified this chemical derivatization scheme to improve versatility and yield, and used it to successfully separate and sequence mixtures of another class of GAGs, chondroitin sulfate, by LC-MSn 44. We present here a related approach involving sequential permethylation, desulfation and pertrideuteroacetylation to modify native HS oligosaccharides for on-line separation and structural analysis of mixtures of HS oligosaccharides, preserving the information regarding the original sites of sulfation and allowing for clear sequencing of heparin/HS sequences based solely on glycosidic bond cleavages. By replacing the labile and strongly polar sulfate groups with much more stable and hydrophobic trideuteroacetyl groups, the oligosaccharides can be separated well by reverse-phase capillary HPLC and fragmented by MS/MS without losing information regarding the sites of modification.
EXPERIMENTAL SECTION
Materials
HS disaccharide standards were purchased from Dextra (Reading, UK). Chemoenzymatically synthesized Arixtra-like heptamer and five HS-like oligosaccharides (two decamers, two dodecamers and one undecamer) were synthesized following the procedures in previous publications45,46. Unless otherwise noted, all chemical reagents used in the chemical derivatization scheme were purchased from Sigma-Aldrich Inc. (St. Louis, MO).
Chemical Derivatization of HS Oligosaccharides
A series of chemical derivatizations were performed to replace the labile and strongly polar sulfate groups with much more stable and hydrophobic trideuteroacetyl groups, which enable the HS oligosaccharides to be retained well and separated by RPLC and fragmented by MS/MS without losing the information about the sulfation modifications. Detailed procedures for the chemical derivatization have been described in our previous work for structural analysis of CS oligosaccharides44. To adjust the method for HS oligosaccahride analysis, some modifications have been made. Briefly, HS oligosaccharides were converted to triethylamine (TEA) salts before being permethylated, in order to increase their solubility in dimethyl sulfoxide (DMSO)47. For permethylation, the dried TEA salts (10-50 μg) were re-suspended in 200 μL DMSO and 200 μL anhydrous suspension of sodium hydroxide in DMSO (150 μg/μL) followed by addition of 100 μL iodomethane. After 5 min vortexing and 10 min sonicating, the reaction was stopped by adding 2 mL water and sparged with nitrogen to remove iodomethane, followed by desalting using a C18 Sep-Pak cartridge (Waters Co.). The dried permethylated products were then converted to their pyridinium salts for solvolytic desulfation by dissolving in 20μL DMSO containing 10% methanol and incubated for 4h at 95°C to remove the sulfate groups48. The solvents were lyophilized and the dried products were re-suspended in 175μL pyridine, 25μL D6-acetic anhydride and incubated at 50°C overnight to label the original sites of sulfation with trideuteroacetyl groups49 . The solvents were then removed by using a Speed-Vac concentrator, and the samples were re-suspended in 20% acetonitrile/water at a concentration of 0.2μg/μl for later analysis. An estimate of yield of fully derivatized product based on UV analysis of a dp4 mixture of heparan sulfate was 29.5% (data not shown).
LC-MS/MS Analysis
For structural analysis of each HS oligosaccharides, a RPLC-MS/MS method was used. Buffer A was prepared as water with 1mM sodium acetate, and buffer B was 80% acetonitrile, 20% water with 1mM sodium acetate. Online HPLC was performed on a regular porous capillary C18 column (0.2×50mm, 3μm, 200 Å, Michrom Bioresources, Aubum, CA), using a linear gradient of buffer B from 25%- 100% over 60min, with a flow rate of 4μL/min and a 10μL injection at a sample concentration of 0.2μg/μL. Mass spectrometry was performed on either a Thermo LTQ-FT instrument or Waters Synapt G2 Q-TOF mass spectrometer. Full MS and CID-MS/MS spectra were acquired in positive ion mode, with spray voltage of 2-3kV and capillary temperature 250°C for LTQ-FT and 80°C for Q-TOF. The collision energy was set between 40V and 50V.
By mixing the five synthesized HS-like oligomers together, online RPLC separation of HS oligosaccharides using different C18 packing material was also compared. In addition to the regular porous C18 column as we mentioned above, we also used a Halo C18 column with core-shell packing materials (0.2×50mm, 2.6μm, 160 Å, Advanced Material Technology, Wilmington, DE) for online separation. For the porous C18 column, a 70min gradient was used from 40-100% buffer B with flow rate of 4μL/min. For the Halo C18 column, an 18min gradient was used from 35%-80% buffer B with flow rate of 9μL/min. A10μL injection at a sample concentration of 0.2μg/μL for each of the five oligosaccharides was used for analysis. Mass spectrometry setup was as same as we mentioned above.
RESULTS AND DISCUSSION
Chemical Derivatizations of HS Disaccharide Standards
As listed in Figure 1, the 12 common HS disaccharide standards contain the basic structure ΔUA-GlcN, where the amine group can either be a free amine, acetylated amine, or a sulfated amine. O-sulfation can occur at the 6-O position of the GlcN residue and/or the 2-O position of the ΔUA residue. Five HS disaccharides are selected to illustrate how all possible modifications can be differentiated from each other after derivatization by MS and/or MS/MS analysis. Structures of these disaccharides are presented in Figure 1, as well as the structure for each derivatized product (with MS and MS/MS spectra of selected ones shown in Figure S-1). Comparison of the derivatized products for ΔUA2S-GlcNAc6S and ΔUA2S-GlcNS6S (Figure 1, I-A and I-S) illustrate the reason for the use of D6-acetic anhydride, as the use of trideuteroacetylation allows for the differentiation between native N-acetylation and peracetylation during the derivatization by mass only (Figure S-1A and B). For sulfation positional isomers, there are three common sulfation sites as we mentioned, including N-sulfation, 2-O-sulfation and 6-O-sulfation. By comparing two isomeric dissacharides ΔUA2S-GlcNS and ΔUA2S-GlcN6S (Figure 1, disaccharides III-S and I-H), the differentiation between N-sulfation and O-sulfation isomers can be easily achieved only by their mass difference after derivatization(Figure S-1C and D). As it has been reported in other literature about the permethylation of different types of amine groups50, our result also showed that the free amine group in GlcN residue was converted to trimethyl quaternary amine that could not be further modified, while a sulfated amine was converted to monomethylated sulfoamine that was desulfated and trideuteroactylated afterwards. This resulted in a diagnostic mass difference of 14Da between derivatized ΔUA2S-GlcN6S (detected as M+) and ΔUA2S-GlcNS (detected as [M+H]+), which in their underivatized forms are isomers.
Figure 1.

Structures of 12 commercially available common HS disaccharides before and after the chemical derivatizations, with each corresponding molecular weight labeled. In the structures, the symbol Me, Ac and Ac’ represents for methyl group, acetyl group and trideuteroacetyl group respectively. GlcN, GlcNAc and GlcNS represents for free glucosamine, acetylated glucosamine and N-sulfated glucosamine respectively. 2S and 6S indicate the position of the sulfate group. ΔUA represents for unsaturated uronic acid.
Differentiation of 2-O-sulfation and 6-O-sulfation of the glucosamine by MS analysis is more difficult. For this isomeric pair, ΔUA-GlcNS6S and ΔUA2S-GlcNS (Figure 1, disaccharides II-S and III-S), the derivatized products were still isomers with exact same mass which require MS/MS analysis for differentiation. For the derivatized ΔUA-GlcNS6S, there was a trideuteroacetyl group on the 6-O position of the GlcNS residue and a methyl group on the 2-O position of the ΔUA residue. On the opposite side, the derivatized ΔUA2S-GlcNS had a methyl group on the 6-O position of the GlcNS residue and a trideuteroacetyl group on the 2-O position of the ΔUA residue. Compared to the derivatized ΔUA2S-GlcNS, the Y1 and Z1 ions of the derivatized ΔUA-GlcNS6S would be 31Da heavier (the mass difference between a methyl group and a trideuteroacetyl group), and the B1 and C1 ions would be 31Da lighter (Figure S-1E and F). One of the major benefits of our derivatization strategy is the greatly increased stability of the trideuteroacetylations as compared to the sulfation, with negligible trideuteroacetyl losses from the fragment ions upon CID. This stability preserves the information of the site of original sulfation, allowing the site of sulfation to be easily determined. The differentiation of this isomer pair is achieved by simply comparing the glycosidic bond cleavage fragments generated from the MS/MS, without requiring for the generation of cross-ring cleavage fragments as required for chondroitin sulfate44.
With these initial results from the HS disaccharide analyses, we demonstrated that the chemical derivatization strategy enable the differentiation between all common sulfation patterns based on mass or solely on glycosidic bond cleavages, and therefore we can accurately sequence HS oligosaccharides consisted with these repeating disaccharide units up to any length amenable to glycosidic bond cleavage. However, no disaccharides containing the rare, but biologically important, sulfation at the 3-O position of the GlcN are commercially available. The derivatization products and characteristics of 3-O sulfated GlcN are described below as studied using an Arixtra-like heptamer.
Structural Sequencing and LC Separation of Five Synthesized HS-like oligosaccharides
To evaluate the application of our method to longer HS oligosaccharides, we performed sequential chemical derivatizations and LC-MS/MS analysis of five chemoezymatically synthesized HS-like oligosaccharides with known sequences (Table 1). Full derivatization was achieved for all five oligosaccharides with byproducts caused primarily by the β–elimination reaction between the carbon-4 and -5 of the GlcA during the permethylation, with a smaller amount of byproducts apparently formed during the desulfation procedure. As mentioned in our previous work on CS oligosaccharides44, these byproducts are notable for not introducing species that would lead to the false identification of sites of sulfation; their major effect is reducing the apparent sensitivity of the technique and result in shortened oligomer sequences (direct infusion MS spectrum shown in Figure S-2). From the MS/MS spectra of the parent ion [M+2Na]2+ for each oligosaccharides (with two decamers and two dodecamers shown in Figure 2 and the undecamer shown in Figure S-3), we observed sufficient sequential glycosidic bond cleavage fragments to enable accurate, full structural sequencing of the modifications. Comparing the series of Y ions for NS-decamer and NS6S-decamer (Figure 2A and B), the Y ions of the NS6S-decamer gave additional n×31Da mass than the Y ions of the NS-decamer, where n is the number of the basic GlcN residue the Y ion has. For example, Y2 ions with no GlcN residue shared the same m/z of 447 for each decamer, and Y4 ions with only one GlcN residue had 31Da mass difference, and Y6 ions with two GlcN residues had 62Da mass difference, and so on. Together with the similar results obtained for other three longer oligosaccharides, the chemical derivatization strategy was demonstrated to enable successful structural sequencing of HS-like oligosaccharides up to dodecamer (the longest we attempted to sequence) with a single MS/MS experiment using glycosidic bond cleavages only. We did notice detectable losses of MeOH and AcOH from product ions, with these losses correlating strongly with the derivatization state at the 6O-position of each precursor. These neutral mass losses from the glycosidic bond cleavage products are always paired with the intact glycosidic bond cleavage product, making assignment of the product ion straightforward. Additionally, the neutral loss product cannot be mistaken for a different sequence, as the loss of MeOH or AcOH results in a mass not consistent with a fully derivatized glycosidic bond cleavage of another related sequence.
Table 1.
Sequences for five chemoenzymatic synthesized HS-like oligosaccharides.
| NS-decamer | GlcA-GlcNS-(GlcA-GlcNS)3-GlcA-AnMan |
| NS6S-decamer | GlcA-GlcNS6S-(GlcA-GlcNS6S)3-GlcA-AnMan |
| NS-undecamer | GlcNS-(GlcA-GlcNS)4-GlcA-AnMan |
| NS-dodecamer | GlcA-GlcNS-(GlcA-GlcNS)4-GlcA-AnMan |
| NS6S-dodecamer | GlcA-GlcNS6S-(GlcA-GlcNS6S)4-GlcA-AnMan |
Figure 2.

MS/MS spectra from the LTQ analyzer of an LTQ-FT of [M+2Na]2+ for NS-decamer (A), NS6S-decamer (B), NS-dodecamer (C) and NS6S-dodecamer (D) after the chemical derivatizations. Sufficient sequential glycosidic bond cleavage fragments were observed for each synthesized HS-like oligosaccharides, which enable the accurate and complete structural sequencing.
MS/MS of the derivatized HS results primarily in glycosidic bond cleavages. Due to the unique mass differences for each monosaccharide unit after derivatization, interpretation of the MS/MS spectra is similar to interpretation of peptide MS/MS spectra. For example, we refer to the compound analyzed in Figure 2A. Starting at the non-reducing end, if the monosaccharide is GlcA, we should observe m/z of 1058.7 for Y9 ion; while if it is GlcA2S, an m/z of 1043.2 should be observed. The presence of 1058.7 ion and absence of 1042.7 ion indicate that it is a GlcA residue at the non-reducing end. Following with GlcA, the 2nd residue could be GlcNS, GlcNAc, GlcNS6S, GlcNAc6S, GlcNS3S, GlcNAc3S, GlcNS3S6S, GlcNAc3S6S; and the corresponding B2 ion for each possibility should give m/z of 503.3, 500.3, 534.3, 531.3, 520.3, 517.3, 551.3 and 548.3. From the MS/MS spectra, we only observed the 503.3 ion without clear signs for other ions, which indicate the second residue is GlcNS. For the 3rd residue, we observed C3 ion with m/z of 739.5 but not 770.5, indicating it is a GlcA not GlcA2S. For the 4th residue, we observed B4 ion with m/z of 969.5 but no other possibilities (966.5, 1000.5, 997.5, 986.5, 983.5, 1017.5 and 1014.5), indicating it is a GlcNS. Sequencing for the rest of residues is very similar. The C5 ion with m/z of 1205.5 but not 1236.5 indicates that it is a GlcA at the 5th residue. The B6 ion with m/z of 1435.7 but no other possibilities (1432.7, 1466.7, 1463.7, 1452.7, 1449.7, 1483.7 and 1480.7) indicates that it is a GlcNS at the 6th residue. The C7 ion with m/z of 1671.9 but not 1702.9 indicates that it is a GlcA at the 7th residue. The B8 ion (doubly charged) with m/z of 962.5 but not others (961.0, 978.0, 976.5, 971.0, 969.5, 986.5 and 985.0) indicates that it is a GlcNS at the 8th residue. The last two residues –GlcA-AnMan at the reducing end are fixed according to the synthesis protocol, and is proven by the presences of Y2 ion with m/z of 447.3 for all five oligosaccharides. The absence of 478.3 for Y2 ion also confirmed that the GlcA is not sulfated (see Figure S-4 for zoomed in views of the appropriate regions in the MS/MS spectrum).
Initial attempts at separation of the mixture of derivatized HS oligosaccharides was performed on a standard C18 column, by mixing the five HS-like oligosaccharides together for LC-MS-MS analysis. As shown in Figure 3A, with the optimized LC gradient, 4 of the 5 oligosaccharides were separated from each other, while NS-undecamer and NS-decamer were overlapped. To seek better separation, we moved to a fused core, porous shell Halo C18 column. As shown in Figure 3B, 5 oligosaccharides were separated, with better peak shapes and narrower elution times. The core-shell design of the Halo column significantly reduced the back pressure and allowed for a higher flow rate (9μL/min compare to 4μL/min for standard C18 column) to be used under the pressure limit of 400 bar. With their own optimized conditions for these two types of C18 column, the Halo column of the same length and similar particle size was able to generate narrower peak widths and better peak shapes in a shorter time scale. Therefore, for on-line separation of derivatized HS oligosaccharides, the core-shell Halo column is preferred for its higher separation efficiency.
Figure 3.

LC separation of derivatized products of five synthesized HS-like oligosaccharides by reverse-phase C18 column packed with either traditional porous material (A) or fused core-porous shell material (B). Same colors in two chromatograms represent the same oligasaccharide as indicated on the right side. For porous C18 column (50mm, 200Å, 3μm): 4 of the 5 oligosaccharides were separated, with NS-hendecamer and NS-decamer co-eluted at 45.29min. For core-shell Halo C18 column (50mm, ~160Å, ~2.6μm): 5 oligosaccharides were able to be separated from each other or at least partially separated.
Structural Analysis of Synthesized Arixtra-like heptamer containing 3-O-sulfation
Compared to N-sulfation and 6-O-sulfation on GlcN residue, 3-O-sulfation is a relatively rare modification for HS oligosaccharides that usually occurs only on N-sulfated GlcN. 3-O sulfation is known to be essential for many HS-protein interactions, including antithrombin, herpes simplex virus 1 glycoprotein D, and growth factor receptor and fibroblast growth factor 7, indicating the important roles of this rare modification in diverse biological functions51. For structural analysis of samples with 3-O sulfation present, there is one more isomer pair to be considered for structural sequencing of HS oligosaccharides, which involved the differentiation between 3-O-sulfated GlcNS and 6-O-sulfated GlcNS. Unfortunately, no commercially-available 3-O sulfated disaccharide is available, and we were unable to purify or synthesize such a disaccharide in any usable quantity. An Arixtra-like heptamer containing a GlcNS6S3S residue (Figure 4A) was analyzed to study the derivatization properties of 3-O sulfated GlcNS, and the 3-O-sulfated residue was found to have a curious derivatization product resulting from our standard chemical derivatization procedure. After performing complete derivatization for the heptamer, we observed the derivatization product m/z of 901.48 with 14Da less than the theoretical calculated m/z for complete derivatized heptamer (Figure S-5). This underpermethylation was consistent across three attempts, suggesting that the unexpected underpermethylation was not due to improper sample handling. Subsequent derivatization efforts with other heparan sulfate oligosaccharides not containing 3-O sulfation did not exhibit underpermethylation. To determine if the underpermethylation was site-specific, and to locate the position of underpermethylation, we ran a MS/MS experiment on the parent ion with m/z of 901.48 and interpreted the spectra by comparing the observed m/z value with the theoretical m/z value for glycosidic bond cleavage fragments of the fully derivatized product (Figure 4C). Compared to the theoretical m/z value, the reduction of 14Da started from the Y5 and B3 ions, with Y2, Y4 and B2 ions sharing the same m/z with their theoretical values. With these results, we located the under-methylated position to the sulfated amine group on the GlcNS6S3S residue, which contains only one possible position for methylation, the sulfated amine. This also explained why the under-permethylated site was not further trideuteroacetylated, because the free amine group generated after desulfation will only be trideuteroacetylated to a mono-trideuteroacetylated amine group but not to a di-trideuteroacetylated amine group. From the structure of the derivatized heptamer (Figure 4B), we found that the sulfated amine group only became inert when the 3-O position of the same GlcN residue were sulfated, based on the fact that GlcNS6S could be fully methylated while GlcNS6S3S could not. This finding is consistent with previously reported NMR studies showing that the proton on the sulfated amine group of a 3-O sulfated GlcNS is deactivated, exhibiting a much slower exchange rate compared to the amine proton on GlcNS with no 3-O sulfation52. With these data, as illustrated in Figure 5, we conclude that the derivatized 3-O-sulfated and 6-O-sulfated isomers should be differentiated solely by their 14Da mass difference from the underpermethylation of the 3-O sulfated GlcNS.
Figure 4.

Structures of the synthesized Arixtra-like heptamer before (A) and after (B) the derivatizations. Two black arrows indicated the different behavior during the derivatization between GlcNS6S and GlcNS6S3S residues, which was interpreted from the MS/MS spectra and the comparison between theoretical and observed m/z values for each glycosidic bond cleavage fragment as obtained from a Synapt G2 HDMS Q-TOF (C).
Figure 5.
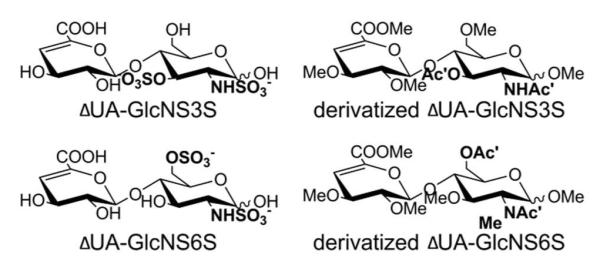
Prediction for the structure of the derivatized product of 3-O-sulfated HS disaccharide ΔUA-GlcNS3S, and there is a 14Da mass difference between 3-O-sulfated and 6-O-sulfated isomeric disaccharides after derivatizations.
CONCLUSIONS
In this work, we have demonstrated that the sequential chemical derivatization strategy enabled differentiation between all sulfation patterns possible based solely on glycosidic bond cleavages from single-stage MS/MS. Therefore, we can accurately sequence all sulfation and acetylation modifications of oligosaccharides up to any length amenable to glycosidic bond cleavage. We have successfully sequenced synthetic HS oligosaccharides up to dodecamers with a single MS/MS experiment solely using glycosidic bond cleavages, which is the longest oligomer we have attempted to sequence so far. In addition, by replacing the labile and strongly polar sulfate groups with much more stable and hydrophobic trideuteroacetyl groups, the oligosaccharides can be retained well and separated by reverse-phase capillary HPLC, allowing for analysis of mixtures of HS oligosaccharides by automated online LC-MS/MS. The fused core, porous shell Halo C18 columns have been demonstrated to give better separation than traditional C18 columns using the same gradient but at higher flow rates.
At present, the chemical derivatization protocol combined with LC-MS/MS analysis clearly enables the separation and sequencing of HS oligosaccharides in microgram quantities, but further improvements are possible. One of the limitations is the sample loss and beta-elimination byproducts generated during the permethylation, which requires homogeneous length for oligosaccharides being analyzed. One strategy that is currently being pursued by our group to reduce sample loss is the reduction of the carboxylic acid of the uronic acid residues to alcohols to eliminate the beta-elimination during the permethylation step. Additionally, our current technology does not purport to identify C-5 epimerization of the uronic acid. Preliminary chemometric analysis results suggest that epimerization of the C-5 position of uronic acid may be differentiated by our derivatization and LC-MS/MS protocol, but a broader sample set of analytes is required to ensure robustness of the chemometric analysis.
In applications where a complex mixture of depolymerized heparin/HS is to be analyzed, our methodology provides great promise, even with the issues presented by beta-elimination byproducts unresolved. The beta-elimination byproducts do not shift the sequences of modifications in the byproduct; the remaining sequence is a real sequence that existed in the original mixture, merely truncated from its original size. This truncated sequence can still provide valuable information regarding changes in the sequence. If accurate analysis of full length is required (e.g. affinity purification analyses), then all fragments smaller than the dp fraction size isolated from the size exclusion column can be safely ignored, leaving only full-length sequence data if the researcher so desires.
One notable advantage to our LC-MS/MS protocol is the fact that baseline LC resolution of isomers is not required to address mixtures of isomeric sequences. Partial separation of isomeric sequences by as little as one MS scan in the LC-MS/MS run can successfully identify both sequences by plotting the selection product ion chromatogram specific to each isomeric sequence, which is possible given that our derivatization strategy only relies upon the more abundant glycosidic bond fragments to differentiate between all isomers. While the separation of non-isomeric HS-like oligosaccharides has been achieved in regular HPLC system, and we have previously achieved resolution of isomeric chemically-derived GAG sequences of chondroitin sulfate, the improved resolution granted by a nano-UPLC system would be useful for separations of isomeric oligosaccharides mixtures where much higher resolution is required.
In summary, we have successfully developed and demonstrated a strategy for chemical derivatization combined with standard reverse phase LC-MS/MS analysis using CID fragmentation that enables the structural sequencing for heparin/HS oligosaccharides with different modification patterns. We have used this strategy to analyze samples with as little as 5 μg of total starting material for simple mixtures. This strategy shows tremendous promise for sequencing of mixtures of heparin/HS oligosaccharides obtained from complex biological systems or partial purification strategies, when detailed information about sulfation and acetylation modifications is needed for mixtures of oligosaccharides with certain biological activity or consequence. We anticipate that this methodology will provide a practical strategy for sequencing of the “heparanome” from heparin/HS oligosaccharide mixtures from various systems of biological interest, and represents a valuable follow-up to compositional analysis studies.
Supplementary Material
ACKNOWLEDGEMENTS
This research is supported in part by the National Institute of General Medical Sciences-funded “Research Resource for Integrated Glycotechnology” (P41 GM103390) from the National Institutes of Health.
Footnotes
SUPPORTING INFORMATION Supporting material including full MS and MS/MS spectra for derivatized HS disaccharide standards; MS/MS spectra for derivatized NS-undecamer and MS spectra for derivatized Arixtra-like heptamer. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1).Gandhi NS, Mancera RL. Chem. Biol. Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- (2).Rabenstein DL. Nat. Prol. Rep. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- (3).Tumova S, Woods A, Couchman JR. Int. J. Biochem. Cell Biol. 2000;32:269–288. doi: 10.1016/s1357-2725(99)00116-8. [DOI] [PubMed] [Google Scholar]
- (4).Kresse H, Schönherr E. J. Cell. Physiol. 2001;189:266–274. doi: 10.1002/jcp.10030. [DOI] [PubMed] [Google Scholar]
- (5).Lyon M, Gallagher JT. Matrix Biol. 1998;17:485–493. doi: 10.1016/s0945-053x(98)90096-8. [DOI] [PubMed] [Google Scholar]
- (6).Li J.-p., Vlodavsk I. Thromb. Haemost. 2009;102:823–828. doi: 10.1160/TH09-02-0091. [DOI] [PubMed] [Google Scholar]
- (7).Dityatev A, Schachner M. Nat. Rev. Neurosci. 2003;4:456–468. doi: 10.1038/nrn1115. [DOI] [PubMed] [Google Scholar]
- (8).Capila I, Linhardt RJ. Angew. Chem. Int. Ed. 2002;41:390–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- (9).Lever R, Page CP. Nature Reviews Drug Discovery. 2002;1:140–148. doi: 10.1038/nrd724. [DOI] [PubMed] [Google Scholar]
- (10).Theocharis AD, Skandalis SS, Tzanakakis GN, Karamanos NK. FEBS Journal. 2010;19:3904–3923. doi: 10.1111/j.1742-4658.2010.07800.x. [DOI] [PubMed] [Google Scholar]
- (11).Guerrini M, Beccati D, Shriver Z, Naggi A, Viswanathan K. Nat. Biltechnol. 2008;26:669–675. doi: 10.1038/nbt1407. al., e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Carlsson P, Kjellén L. Handb. Exp. Pharmacol. 2012;207:23–41. doi: 10.1007/978-3-642-23056-1_2. [DOI] [PubMed] [Google Scholar]
- (13).Chi LL, Amster J, Linhardt RJ. Curr. Anal. Chem. 2005;1:223–240. doi: 10.2174/157341105774573929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mallis LM, Wang HM, Loganathan D, Linhardt RJ. Anal. Chem. 1989;61:1453–1458. doi: 10.1021/ac00188a030. [DOI] [PubMed] [Google Scholar]
- (15).Linhardt RJ, Wang HM, Loganathan D, Lamb DJ, Mallis LM. Carbohydr. Res. 1992;225:137–145. doi: 10.1016/0008-6215(92)80045-3. [DOI] [PubMed] [Google Scholar]
- (16).Tissot B, Gasiunas N, Powell AK, Ahmed Y, Zhi Z-I, Haslam SM, Morris HR, Turnbull JE, Gallagher JT, Dell A. Glycobiology. 2007;17:972–987. doi: 10.1093/glycob/cwm072. [DOI] [PubMed] [Google Scholar]
- (17).Laremore TN, Linhardt RJ. Rapid Commun. Mass Spectrom. 2007;21:1315–1320. doi: 10.1002/rcm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Minamisawa T, Hirabayashi J. Trends Glycosci. Glyc. 2006;18:293–312. [Google Scholar]
- (19).Zaia J, Costello CE. Anal. Chem. 2003;75:2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- (20).Saad OM, Leary JA. Anal. Chem. 2003;75:2985–2995. doi: 10.1021/ac0340455. [DOI] [PubMed] [Google Scholar]
- (21).Zaia J, Costello CE. Anal. Chem. 2001;73:233–239. doi: 10.1021/ac000777a. [DOI] [PubMed] [Google Scholar]
- (22).Jandik KA, Gu KA, Linhardt RJ. Glycobiology. 1994;4:289–296. doi: 10.1093/glycob/4.3.289. [DOI] [PubMed] [Google Scholar]
- (23).Shively JE, Conrad HE. Biochemistry. 1976;15:3932–3942. doi: 10.1021/bi00663a005. [DOI] [PubMed] [Google Scholar]
- (24).Volpi N, Maccari F, Linhardt RJ. Electrophoresis. 2008;29:3095–3106. doi: 10.1002/elps.200800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zaia J. Mass Spectrom. Rev. 2009;28:254–272. doi: 10.1002/mas.20200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vives RR, Goodger S, Pye DA. Biochem. J. 2001;354:141–147. doi: 10.1042/0264-6021:3540141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bruggink C, Maurer R, Herrmann H, Cavalli S, Hoefler F. J. Chromatogr. A. 2005;1085:104–109. doi: 10.1016/j.chroma.2005.03.108. [DOI] [PubMed] [Google Scholar]
- (28).Hitchcock AM, Costello CE, Zaia J. Biochemistry. 2006;45:2350–2361. doi: 10.1021/bi052100t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Staples GO, Bowman MJ, Costello CE, Hitchcock AM, Lau JM, Leymarie N, Miller C, Naimy H, Shi X, Zaia J. Proteomics. 2009;9:686–695. doi: 10.1002/pmic.200701008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Naimy H, Leymarie N, Bowman MJ, Costello CE, Zaia J. Biochemistry. 2008;47:31553161. doi: 10.1021/bi702043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Karlsson NG, Schulz BL, Pacher NH, Whitelock JM. J. Chromatogr. B. 2005;824:139–147. doi: 10.1016/j.jchromb.2005.07.014. [DOI] [PubMed] [Google Scholar]
- (32).Estrella RP, Whitelock JM, pacher NH, Karlsson NG. Anal. Biochem. 2007;79:3597–3606. doi: 10.1021/ac0622227. [DOI] [PubMed] [Google Scholar]
- (33).Yang B, Chang Y, Weyers AM, Sterner ES, Linhardt RJ. J.Chromatogr. A. 2012;1225:91–98. doi: 10.1016/j.chroma.2011.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yang B, Weyers AM, Baik JY, Sterner ES, Sharfstein S, Mousa SA, Zhang F, Dordick JS, Linhardt RJ. Anal. Biochem. 2011;415:59–66. doi: 10.1016/j.ab.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wei W, Niñonuevo MR, Sharma A, Danan-Leon LM, Leary JA. Anal. Chem. 2011;83:3703–3708. doi: 10.1021/ac2001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yamada S, Yoshida K, Sugiura M, Sugahara K. J. Biol. Chem. 1993;268:4780–4787. [PubMed] [Google Scholar]
- (37).Leymarie N, McComb ME, Naimy H, Staples GO, Zaia J. Int. J. Mass Spectrom. 2012;312:144–154. doi: 10.1016/j.ijms.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ly M, Leach F. E. r., Laremore TN, Toida T, Amster IJ, Linhardt RJ. Nat. Chem. Biol. 2011;11:827–833. doi: 10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kailemia MJ, Li L, Ly M, Linhardt RJ, Amster IJ. Anal. Chem. 2012;84:5475–5478. doi: 10.1021/ac3015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Saad OM, Leary JA. Anal. Chem. 2005;77:5902–5911. doi: 10.1021/ac050793d. [DOI] [PubMed] [Google Scholar]
- (41).Wolff JJ, Chi LL, Linhardt RJ, Amster J. Anal. Chem. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dell A, Rogers ME, Thomas-Oates JE. Carbohydr. Res. 1988;179:7–19. [Google Scholar]
- (43).Barker SA, Hurst RE, Settine J, Fish FP, Settine RL. Carbohydr. Res. 1984;125:291–300. doi: 10.1016/0008-6215(84)85164-2. [DOI] [PubMed] [Google Scholar]
- (44).Huang R, Pomin VH, Sharp JS. J. Am. Soc. Mass Spectrom. 2011;22:1577–1587. doi: 10.1007/s13361-011-0174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Liu R, Xu Y, Chen M, Weiwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J. J. Biol. Chem. 2010;285:34240–34249. doi: 10.1074/jbc.M110.159152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa SA, Linhardt RJ, Liu J. Science. 2011;334:498–501. doi: 10.1126/science.1207478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Heiss C, Wang Z, Azadi P. Rapid Commun. Mass Spectrom. 2011;25:774–778. doi: 10.1002/rcm.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nagasawa K, Inoue Y, Kamata T. Carbohydr. Res. 1977;58:47–55. doi: 10.1016/s0008-6215(00)83402-3. [DOI] [PubMed] [Google Scholar]
- (49).Bendiak B, Fang TT, Jones DNM. Can. J. Chem. 2002;80:1032–1050. [Google Scholar]
- (50).Baldwin MA, Stahl N, Reinders LG, Gibson BW, Prusiner SB, Burlingame AL. Anal. Biochem. 1990;191:174–182. doi: 10.1016/0003-2697(90)90405-x. [DOI] [PubMed] [Google Scholar]
- (51).Lindahl U, Li JP. Int. Rev. Cell Mol. Biol. 2009;276:105–159. doi: 10.1016/S1937-6448(09)76003-4. [DOI] [PubMed] [Google Scholar]
- (52).Guerrini M, Elli SS, Mourier P, Rudd TR, Gaudesi D, Casu B, Boudier C, Torri G, Viskov C. Biochem. J. 2012 doi: 10.1042/BJ20121309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.