

Abstract
Background
Epidermal growth factor receptor (EGFR) is co-activated by the μ-opioid receptor (MOR), expressed on non-small cell lung cancer (NSCLC) cells and human lung cancer. We hypothesized that clinically used opioid analgesics that are MOR agonists co-activate EGFR, resulting in growth- and survival-promoting signaling.
Methods
We used H2009, a human adenocarcinoma NSCLC cell line, with constitutive EGFR phosphorylation, which showed increased expression of MOR and delta opioid receptor (DOR) by reverse transcriptase polymerase chain reaction. We used Western immunoblotting, magnetic bead-based Bio-plex cytokine assay, immunofluorescent staining, BrdU incorporation ELISA and BioCoat™ Matrigel™ invasion assay, to examine cell signaling, cytokine expression, co-localization of MOR and EGFR in human lung cancer, cell proliferation and invasion, respectively.
Results
Like epidermal growth factor (EGF), morphine stimulated phosphorylation of EGFR, Akt/protein kinase B (Akt), and mitogen-activated protein kinase/extracellular signal regulated kinase (MAPK/ERK) signaling in H2009 cells. Opioid receptor (OR) antagonist, naloxone, EGFR tyrosine kinase inhibitor, erlotinib, and silencing of MOR and DOR abrogated morphine- and EGF-induced phosphrylation of signaling, suggestive of OR mediated co-activation of EGFR. H2009 cells secreted significantly higher levels of cytokines as compared to control Beas2B epithelial cells. H2009 conditioned medium stimulated MOR expression in Beas2B cells, suggesting that cytokines secreted by H2009 may be associated with increased OR expression in H2009. We observed co-localization of EGFR and MOR, in human NSCLC tissue. Functionally, morphine and EGF-induced proliferation and invasion of H2009 cells was ameliorated by naloxone as well as erlotinib.
Conclusion
Morphine-induced phosphorylation of EGFR occurs via ORs, leading to downstream MAPK/ERK, Akt phosphorylation, cell proliferation and increased invasion. Notably, ORs are also associated with EGF-induced phosphorylation of EGFR. Increased co-expression of MOR and EGFR in human lung cancer suggests that morphine may have a growth-promoting effect in lung cancer.
INTRODUCTION
Lung cancer is the most common cause of cancer deaths worldwide.1,2 Non-small cell lung cancer (NSCLC) comprises approximately 80% of cases; of those, adenocarcinoma is the most common histology.3 The vast majority are diagnosed at an advanced stage, and median survival ranges from 8 to 11 months, indicating a desperate need to further elucidate the molecular pathways driving these tumors and develop new treatments.
Epidermal growth factor receptor (EGFR, also known as erbB-1) is a receptor tyrosine kinase (RTK), which has been shown to correlate with poor outcomes in both resected and advanced NSCLC.4-7 The EGFR tyrosine kinase inhibitors (TKIs) erlotinib and Gefitinib and the anti-EGFR monoclonal antibody cetuximab are used for the treatment of advanced NSCLC,8-11 and mutations imparting significant sensitivity12-14 or resistance15-16 to EGFR TKI therapy are predictive and prognostic biomarkers in NSCLC. Unfortunately, none of these agents is curative, indicating a need to further elucidate mechanisms of resistance to anti-EGFR therapy.
Mu opioid receptors (MORs) are G-protein coupled receptors (GPCRs) that mediate the analgesic activity of morphine and its congeners to treat pain. In addition to analgesia, morphine/MOR activation stimulates signaling pathways involved in cell proliferation, survival, and migration in a number of cell types.17-24 We showed that morphine stimulates angiogenesis by activating mitogen-activated protein kinase/extracellular signal regulated kinase (MAPK/ERK) and Akt/protein kinase B (Akt) phosphorylation in human dermal microvascular endothelial cells (HDMEC) and breast cancer progression in mice.22 Morphine activates MAPK/ERK directly and also co-activates vascular endothelial growth factor 2 (VEGFR2) on endothelium.19,20,25 In breast cancer, the growth- and survival-promoting activity of morphine translates into tumor growth, metastasis, and decreased survival in murine models of breast cancer.22,26 Complementary to MOR agonist-induced promotion of tumor growth, the non-selective opioid receptor (OR) antagonist naloxone inhibits human MCF-7 breast cancer cell proliferation and tumor growth in rodents.22,27 The MOR-specific antagonist methylnaltrexone (MNTX) inhibits proliferation and migration of endothelial cells,28 enhances the antitumor effects of the chemotherapeutic agent 5-fluorouracil (5-FU) in breast, lung, and colon cancer cell lines, and synergizes with bevacizumab and 5-FU to inhibit VEGF-induced angiogenesis.29,30
A recent demonstration of inhibition of Lewis lung carcinoma (LLC) in MOR knockout mice as compared to wild type mice further exemplified the significance of MOR in lung cancer.23 Expression of the immunoreactive opioid peptides β-endorphin, enkephalin and dynorphin, and the presence of high affinity membrane receptors for mu-, delta-, and kappa-opioid receptors (MOR, DOR, and KOR) on diverse small cell lung carcinoma (SCLC) and NSCLC cell lines was demonstrated on the basis of ligand binding studies31,32 two decades ago. Subsequent studies showed that methadone inhibited lung cancer cell growth by promoting apoptosis via stimulation of MAPK-phosphatase, inactivation of MAPK, and suppression of bcl-2, in low-concentration bombesin secreting SCLC and NSCLC cells but not in cells secreting higher concentrations of bombesin.33 Importantly, in the same study, morphine and the MOR-specific agonist [D-Ala2, N-MePhe4, Glu-ol]-enkephalin (DAMGO) stimulated MAPK/ERK phosphorylation while methadone inhibited MAPK/ERK phosphorylation. The authors suggested that methadone acted via a non-OR mediated mechanism, but did not provide an explanation for morphine- and DAMGO-induced MAPK/ERK phosphorylation. The presence of MOR and DOR has been shown in human lung cancers in vivo using positron emission tomography (PET) scanning.34 These authors showed the presence of binding sites for the DOR-selective antagonist 11C-methylnaltrindole (11C-MeNTI) and the MOR-specific agonist 11C-carfentanil (11C-CFN) in patients with small cell, squamous cell, and adenocarcinoma. Increased binding of 11C-MeNTI and 11C-CFN was observed in all lung tumors compared to non-cancerous lung. These studies clearly demonstrate the presence of ORs and highlight their significance in lung cancer growth. However, the mechanism(s) of OR-mediated lung cancer progression are unclear.
MOR agonists also transactivate receptor RTKs, including EGFR35-37 but this has not been examined in NSCLC. Because MOR is activated by clinically relevant concentrations of opioids and EGFR signaling is critical in the malignant phenotype of lung adenocarcinomas, we hypothesize that ORs in NSCLC co-activate EGFR and downstream signaling pathways that promote proliferation and survival. We used NCI-H2009 human adenocarcinoma lung cancer cells due to their resistance to therapy and increased expression of MOR compared to other NSCLC cell lines. We show that morphine stimulates the phosphorylation of EGFR, MAPK/ERK and Akt via MOR, and the effect is attenuated by the OR antagonist naloxone as well as by the EGFR inhibitor erlotinib.
Methods
Cell culture
The human NCI lung carcinoma cell lines H2009 and H460 were obtained from ATCC (Manassus, VA) and maintained in RPMI1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum. Beas2B, an adenovirus-12 SV40 immortalized human bronchoepithelial cell line, was obtained from ATCC and maintained in BEBM (Invitrogen, Carlsbad CA) supplemented with SingleQuots (Cambrex Bio Science, Walkersville, MD). HDMEC were cultured from neonatal human foreskin, maintained in media containing 20% male human serum as described previously,38 and used between passages 4-8. The human epidermal keratinocyte cell line, HaCAT, was obtained from Carol A. Lange, PhD, at the University of Minnesota. All cells were incubated at 37°C and 5% CO2.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
RNA was isolated from H460, H2009, and HDMEC using Trizol (Invitrogen, Carlsbad, CA) and RT-PCR was done using Taq DNA polymerase (Continental Laboratory Products, San Diego, CA). The following Primers were used:
| MOR(440bp): | forward: GGT ACT GGG AAA ACC TGC TGA AGA TCT GTG. |
| reverse: GGT CTC TAG TGT TCT GAC GAT TCG AGT GG. | |
| DOR (365 bp): | forward: ATC TTC ACC CTC ACC ATG ATG. |
| reverse: CGG TCC TTC TCC TTG GAA CC. | |
| GAPDH(452bp): | forward: GAA GGT GAA GGT CGG AGT C. |
| reverse: GAA GAT GGT GAT GGG ATT TC. |
DNA samples were visualized by 2% agarose gel electrophoresis.
Western Immunoblotting
H2009 cells were grown in regular growth media for 24 hours, incubated in 0.5% reduced serum media overnight, and whole cell lysates were prepared after the treatment incubations (Figures 1 through 6). Lysates (100 micro-g of protein) resolved on a 3-15% SDS-PAGE gel were transferred to a polyvinylidine fluoride membrane (Immobilon, Millipore, Bedford, MA), and probed for mouse anti-human phospho-EGFRTyr1173 (Millipore) at 1:2000 dilution or total EGFR, rabbit anti-human phospho-p42/44 MAPK (ERK1/2)Thr202/Tyr204 or total MAPK/ERK (Cell Signaling) at 1:500, and rabbit anti-human phospho AktThr308 or total Akt (Cell Signaling) at 1:500. After washing, membranes were incubated in the appropriate species-specific secondary antibody linked to alkaline phosphatase for one hour at room temperature. Chemiluminescent signals were detected using the ECF System (Amersham Biosciences), and images were acquired using a Storm 860 Phosphoimager (Molecular Dynamics, Sunnyvale, CA).
Figure 1. Increased expression of MOR and morphine-induced phosphorylation of EGFR in H2009 NSCLC cells.
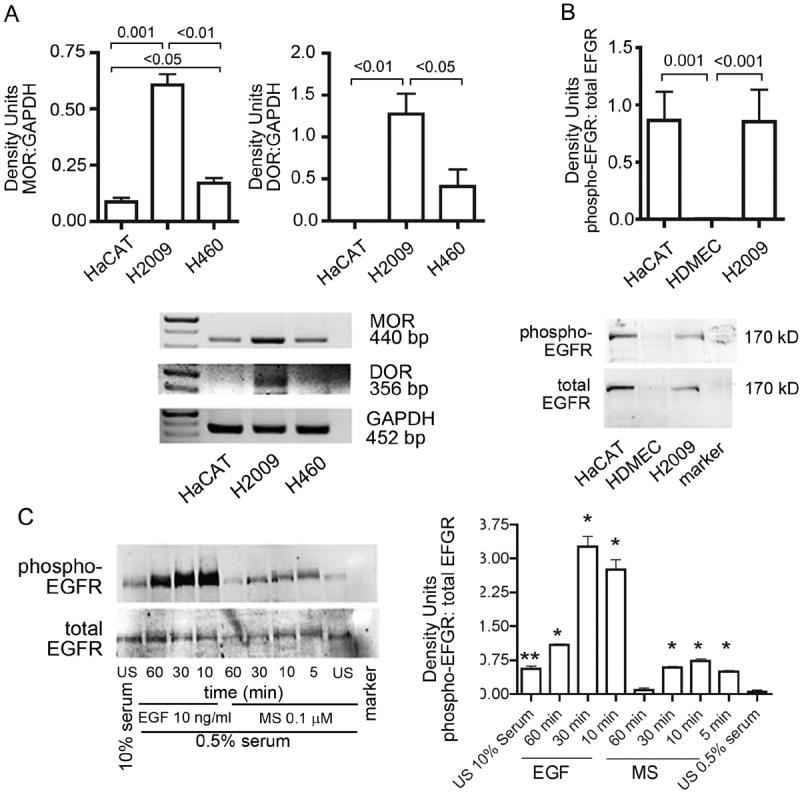
(A) RT-PCR analysis showing MOR and DOR expression on human lung cancer cell lines H2009 and H460. HaCaT, human keratinocyte cell line known to express high levels of EGFR also shows strong expression of MOR. Loading control for GAPDH is shown in the bottom row. Density Units relative to GAPDH are shown. (B) Western immunoblotting showing constitutive phosphorylation of EGFR on H2009 cells. HaCaT, were used as a positive control and HDMEC as a negative control. Density Units for phospho-EGFR relative to total-EGFR are shown. (C) Morphine and EGF-induced time-dependent phosphorylation of EGFR on H2009 grown overnight in serum-reduced conditions. Density Units for phosph-EGFR relative to total-EGFR for each treatment are shown. *P <0.001 and **P < 0.01, as compared to US 0.5% serum. Each figure represents three separate, reproducible experiments. Abbreviations: EGFR: epidermal growth factor receptor; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; HaCAT, human keratinocyte cell line; HDMEC: human dermal microvascular endothelial cells; MOR: mu opioid receptor; NSCLC: non-small cell lung cancer; US: unstimulated; DOR: delta opioid receptor; RT-PCR: Reverse transcriptase-polymerase chain reaction
Figure 6. Morphine stimulates the proliferation and invasiveness of H2009 cells.
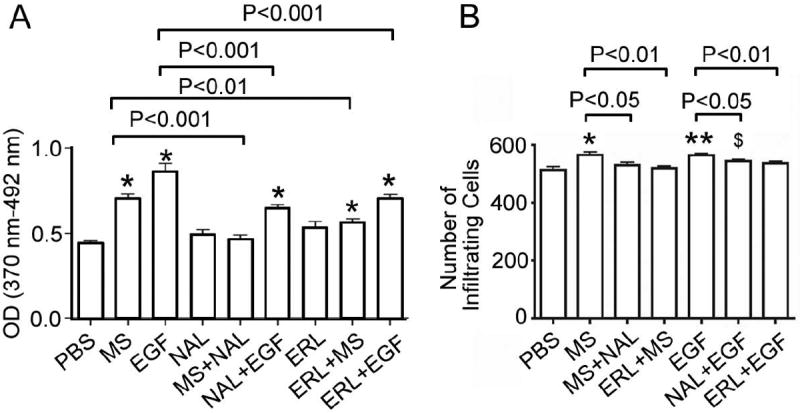
(A) H2009 cells were plated at a density of 2 × 103 per well in 96 a well plate and incubated in regular growth media overnight followed by serum starvation for another night. Cells were then incubated with 0.1 μM morphine or 20 ng/mL EGF, with or without 0.1 μM naloxone or 1 μM erlotinib for 48 hours. Cell proliferation was measured using BrdU incorporation ELISA. Data are expressed as mean ± SEM from 3 experiments. *P < 0.001 as compared to PBS. (B) H2009 cells (1 × 105 per chamber) were allowed to migrate in Matrigel-coated invasion chambers for 24 hours in the treatment conditions described for A. Cells migrated per field of view (average of 4 fields per well) are expressed as a mean ± SEM from 3 separate experiments. $P <0.05, *P <0.01 and **P <0.001, as compared to PBS. Morphine-induced proliferation and invasion were blocked by naloxone as well as erlotinib. Likewise, EGF-induced proliferation and invasion were blocked by either naloxone or erlotinib, indicative of the dependence of EGFR on ORs and vice versa. Abbreviations: MS: morphine sulfate; EGF: epidermal growth factor; NAL: naloxone; ERL: erlotinib; PBS, phosphate buffered saline.
Densitometry
Quantification of immunoblotting and RT-PCR bands were obtained by densitometric analysis using ImageJ Software (National Institutes of Health, Bethesda MD).
siRNA transfection
H2009 cells were grown in regular growth media overnight, then transfected with 100 nM of human MOR siRNA, DOR siRNA, or scramble siRNA (all from Santa Cruz Biotechnology, Santa Cruz, California) in Optimem (Invitrogen, Carlsbad, California) using siPORT lipid transfection agent (Ambion, Austin, Texas). Regular growth media was added after 8 hours of transfection and cells were incubated overnight. Both siRNAs comprised a pool of three target-specific siRNAs. RNA was isolated as described above and RT-PCR was done to assess knockdown. DNA samples were visualized by 2% agarose gel electrophoresis to ensure gene knockdown using the primers described above. For immunoblotting experiments, transfected cells were serum-starved for 6 hours and then stimulated with morphine or epidermal growth factor (EGF) as described below.
Cytokine analysis
Sub-confluent cultures of H2009 and Beas2B cells were incubated for 48 hours in their respective media, and the conditioned media was snap frozen and stored at −80°C until assayed. Control media was prepared in parallel by incubating media without cells in identical flasks. Magnetic bead based Bio-Plex assays (BioRad Laboratories, Inc., Hercules, CA) were used to determine the concentration of the angiogenic factors as described previously.39 Briefly, color-coded magnetic beads are coupled to an antibody unique to the marker analyzed and dyed with 2 fluorophores (classification dyes), which absorb maximally at 635 nm and emit at 2 distinct wavelengths. The reporter dye is a third fluorophore, phycoerythrin (PE), which emits at a third distinct wavelength and has a maximal absorption at 532 nM. The detector unit consists of a flow cell that enables the magnetic beads to travel in a single file (laminar flow) through a region illuminated by a pair of lasers. Values shown are the mean ± SEM of 6 separate experiments. P-values were determined by ANOVA.
Beas2B cell treatment with H2009 conditioned medium
H2009 cells were incubated with serum-free H2009 medium for 48h, to prepare H2009 conditioned medium. Beas2B cells plated overnight in complete Beas2B culture medium overnight were growth factor-starved and incubated for an additional night. The following day Beas2B cells were incubated for 48h in growth factor-free medium or in 50% growth factor-free medium + 50% H2009 serum-free medium or in 50% growth factor-free medium + H2009 conditioned medium. RNA was isolated for MOR and DOR expression, and RT-PCR was performed as described above. Cell lysates were examined for autophosphorylation of Beas2B cells by Western immunoblotting as described above.
Immunofluorescent microscopy of H2009 cells
H2009 cells were grown on glass slides for 24 hours in regular growth media and serum-starved overnight. Cells were treated with 0.1 μM morphine or 100 ng/mL EGF for 10 minutes with or without preincubation with 10 nM naloxone for 10 minutes. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 on ice. Cells were then blocked with 3% bovine serum albumin and incubated with mouse anti-human phospho EGFRTyr1068 (Cell Signaling, Danvers, MA) primary antibody at 1:50 dilution, followed by incubation with Rhodamine (TRITC) AffiniPure donkey anti-mouse immunoglobulin (Ig)G (Jackson Immunoresearch, Westgrove, PA) secondary antibody at 1:100, and 4’,6-diamidino-2-phenylindole (DAPI, Molecular Probes, Eugene, OR) at 1:50,000 to co-stain the nucleus during the last 10 minutes of incubation. An isotype IgG antibody (Jackson Immunoresearch) was used in parallel as a control. Fluorescent images were obtained using an Olympus 1×70 microscope fitted with an Olympus DP70 digital camera at X600.
Immunofluorescent staining of human adenocarcinoma tissue
Archived, paraffin-embedded human lung adenocarcinomas were obtained from the VA Medical Center, Minneapolis, MN. Six micron sections were co-stained with the following primary antibodies: goat anti-human CD31 (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:50 dilution to label blood vessels, mouse anti-human total EGFR (Abcam, Cambridge, MA) at 1:100, and rabbit anti-human MOR (Chemicon International, Temecula, CA) at 1:100, followed by staining with species-specific secondary antibodies labeled with Cy2 (CD31), Cy3 (EGFR), and Cy5 (MOR) (Jackson Immunoresearch, Westgrove, PA). Images were obtained using an Olympus BX50 upright epifluorescent microscope at X200. MOR was pseudo-colored green, EGFR red, and CD31 blue using Adobe Photoshop. Images were overlaid using Adobe Photoshop.
Proliferation Assay
Cell proliferation was measured using BrdU cell proliferation ELISA (Roche, Indianapolis, IN) according to the manufacturer’s instructions, as described previously24. Briefly, 2,000 cells/well were plated on a 96-well plate in complete culture medium for H2009 cells and incubated overnight. The following day cells were incubated with serum-free medium overnight, followed by incubation with different agonists and antagonists for 48h (Figure 6) (morphine sulfate 0.1 microM; naloxone, 0.1 microM; erlotinib, 1 microM; EGF, 20 ng/ml). BrdU ELISA was performed using peroxidase-conjugated anti-BrdU antibodies. Color developed was measured at 370 nm with 492 nm as the reference wavelengths.
Invasion Assay
H2009 cell invasion was assessed using 24-well BioCoat™ Matrigel™ invasion chambers (Becton Dickenson Biosciences, Bedford, MA) with 8 μm pore size according to the manufacturer’s instructions. Serum-free medium supplemented with morphine or EGF, with and without naloxone or erlotinib, was added to the bottom chamber of the well in a 24-well cluster. Serum-free medium containing 1 × 105 H2009 cells was added to the top chamber of the inserts. Cells were allowed to migrate for 24 hours. Cells remaining on the upper side of the membrane were removed with a cotton-tipped applicator. Cells that had migrated through the membrane were fixed in methanol and stained with crystal violet. Cells were counted in a blinded fashion in 4 random areas/membrane at a magnification of X100 using an Olympus CK2 inverted microscope (Olympus America, Melville, NY).
Statistical analysis
All statistical analyses were performed using Prism software (GraphPad Prism Inc., San Diego, CA). Un-paired Student’s t-tests or ANOVA followed by Tukey’s post hoc test were performed to determine the significance between different conditions. A P < 0.05 was considered to be statistically significant.
Results
MOR and EGFR are upregulated in NSCLC cell lines, and morphine co-activates EGFR
H2009, an adenocarcinoma cell line resistant to therapy, showed significantly higher expression of MOR and DOR as compared to H460, a large cell lung carcinoma cell line (Fig 1A). Therefore, H2009 was selected for further experiments. Increased MOR expression was accompanied by significantly higher constitutive phosphorylation of EGFR similar to that of HaCaT cells, a human keratinocyte cell line, as compared to HDMEC (Fig 1B). Morphine as well as EGF stimulated the phosphorylation of EGFR in a time-dependent manner (Fig 1C). These data show correlative expression of increased MOR and constitutively phosphorylated EGFR, and morphine-induced phosphorylation of EGFR, suggestive of MOR – EGFR cross-talk.
Morphine and EGF induced EGFR co-activation and downstream signaling are abrogated by naloxone and erlotinib
Western immunoblotting and densitometric analysis of protein bands showed that morphine-induced EGFR phosphorylation in H2009 cells was accompanied by a significant increase in phosphorylation of MAPK/ERK and Akt (Fig 2A). Naloxone, a nonselective OR antagonist, significantly inhibited morphine- as well as EGF-induced phosphorylation of EGFR, MAPK/ERK and Akt (Fig 2A). Activation of these pathways by morphine as well as EGF was significantly ameliorated by erlotinib, an inhibitor of EGFR. Immunofluorescent analysis of the phosphorylation of EGFR by EGF and morphine also showed increased staining of membrane bound phospho-EGFR after 10 min of incubation (Fig 2B). Dense red punctate staining of phospho-EGFR was seen over the entire cell body after 10 min treatment with EGF (inset in the box), which was reduced in cells pretreated with naloxone. Morphine-induced staining was more densely associated with the cell membrane and sparsely with the nuclei and cytoplasm, and was inhibited by naloxone. These data suggest OR and EFGR cross-talk that influences downstream signaling pathways, MAPK/ERK and Akt upon stimulation with morphine or EGF.
Figure 2. Naloxone and erlotinib ameliorate morphine- and EGF-induced EGFR, MAPK/ERK, and Akt phosphorylation in H2009 cells.

(A) Serum starved H2009 cells were treated with 0.1 microM morphine MS or 20 ng/ml EGF for 10 minutes in the presence or absence of 100 nM naloxone or 1 microM erlotinib. MS- and EGF-induced EGFR, MAPK/ERK and Akt phosphorylation were abrogated by both naloxone and erlotinib. Respective total protein levels did not change. Since erlotinib was dissolved in DMSO, H2009 cells were stimulated with MS and EGF containing the same concentration of DMSO as erlotinib. Ratios of Density Units for each phospho- to total-protein are shown. $P <0.05, *P <0.01 and **P <0.001, as compared to PBS in each graph. (B) H2009 cells were grown on glass slides for 24 hours in regular growth media, serum starved overnight, and treated with morphine or EGF in the presence or absence of naloxone for 10 minutes (similar to A). Cells were incubated with mouse anti-human phospho- EGFRTyr1068 at 1:50 dilution, and then incubated with Rhodamine (TRITC) AffiniPure donkey anti-mouse IgG at 1:100. An isotype IgG antibody was used as a control. DAPI was used to co-stain the nuclei. Please note the inset in EGF 10’ showing intense granular staining of phospho-EGFR over the entire cell. In contrast EGF + Nal 10’ shows red staining of phospho-EGFR confined to one end and overall weak staining for phospho-EGFR. Magnification X600. Each figure represents three separate, reproducible experiments. MS: morphine sulfate; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; DAPI: 4’,6-diamidino-2-phenylindole; SF: serum free; GM: growth medium; Nal: naloxone; Erl: erlotinib; DMSO: dimethyl sulfoxide.
Morphine-induced co-activation of EGFR occurs via MOR and DOR
Silencing of MOR and DOR was achieved by transfecting H2009 cells with a cocktail of three different siRNA sequences for MOR or DOR (Fig 3A). siMOR and siDOR silenced the expression of MOR and DOR, respectively on H2009 cells but scramble sequence did not alter the MOR and DOR expression. Western immunoblotting and densitometric analysis of protein bands showed that silencing of MOR and DOR significantly abrogated morphine- and EGF-induced phosphorylation of EGFR, MAPK/ERK and Akt as compared to H2009 cells treated with scramble siRNA (Figure 3B). These data suggest that activation of EGFR and downstream MAPK/ERK and Akt pathways is dependent upon activation of MOR and DOR.
Figure 3. Silencing of MOR or DOR inhibits morphine- and EGF-induced signaling.
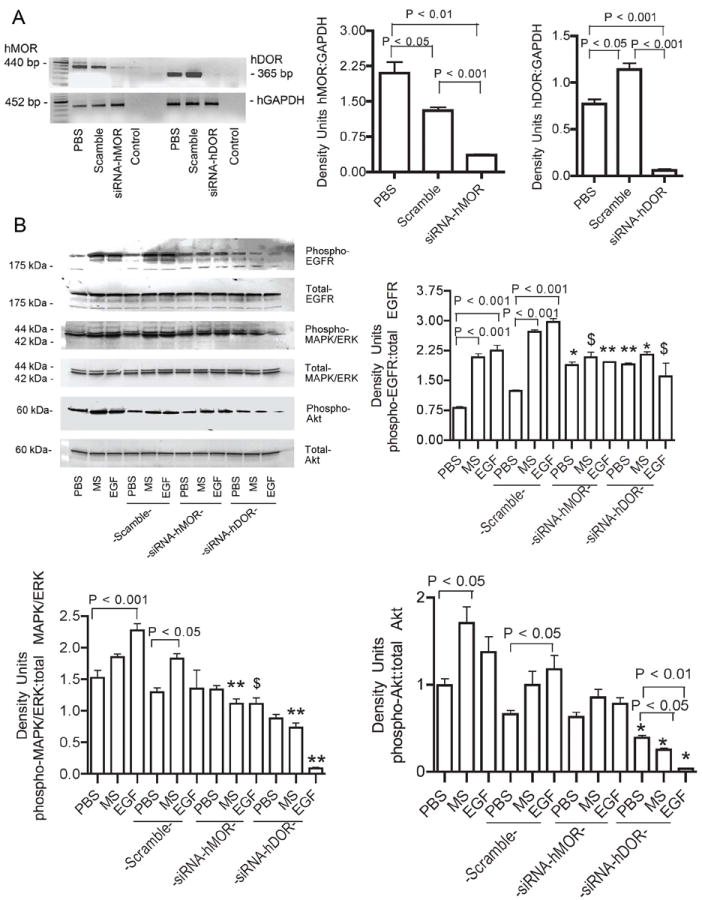
(A) H2009 cells were grown in complete media for 24 hours prior to transfection with human MOR or DOR siRNA (100 nM). RNA was isolated and silencing assessed by RT-PCR. Both MOR and DOR silencing was achieved. Density units for the ratios of MOR or DOR to GAPDH are expressed as mean ± SEM from 3 separate experiments. (B) H2009 cells transfected with MOR or DOR siRNA were serum starved for six hours followed by stimulation with MS or EGF for 10 minutes. MS- and EGF-induced phosphorylation of EGFR, MAPK/ERK and Akt were abrogated in siRNA treated cells compared to untransfected cells. Scramble siRNA was used as a negative control and had no effect. Density units of phospho- to total-protein for each signaling pathway are shown as mean ± SEM of 3 different experiments. $P <0.05, *P <0.01 and **P <0.001, as compared to scramble with the same treatment in each graph. These data indicate that MS-induced co-activation of EGFR occurs via opioid receptors, and indicate the necessity of opioid receptors in EGF-induced phosphorylation of EGFR and its downstream pathways. Abbreviations: MOR: mu opioid receptor; DOR: delta opioid receptor; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; MS: morphine sulfate; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; MOR: mu opioid receptor.
NSLCL lung cancer cells secrete pro-survival and pro-angiogenic factors
We next examined cytokines secreted by H2009 cells compared to benign Beas2B epithelial cells (Table 1) to determine whether the increased MOR and DOR expression and EGFR auto-phosphorylation is due to the secretion of cytokines by H2009 cells. Hepatocyte growth factor (HGF), the only known ligand for mesenchymal-epithelial transition factor (c-MET), a tyrosine kinase overexpressed in the majority of NSCLC involved in cell proliferation, survival, migration, and carcinogenesis40-42 was significantly higher in H2009 conditioned media (p=0.0023). c-MET crosstalk with EGFR has been demonstrated,41 and indeed, EGFR inhibition has been shown to lead to abrogation of HGF-induced c-MET activation, thus mitigating HGF-induced migration.43 Additionally, cMET amplification has been shown to impart resistance to the EGFR TKI gefitinib in vitro.44 Interleukin-8 and VEGF were significantly higher in H2009 cells as compared to Beas2B conditioned medium (p<0.001). VEGF upregulates MOR expression in endothelial cells;25 this is a plausible explanation of the MOR upregulation observed in H2009 cells (Fig 1) and in NSCLC tissue (Fig 5). Anti-angiogenic therapy in lung cancer has not been therapeutic, despite its success in animal models45-47, perhaps due to a self-supportive and pro-angiogenic environment created by the lung cancer cells. Granulocyte-colony stimulating factor (G-CSF) is also over-expressed in H2009 medium as compared to Beas2B medium (p=0.0004). G-CSF appears to play a role in angiogenesis, and imparts resistance to bevacizumab when given exogenously in a mouse model.48 PECAM-1 Platelet endothelial cell adhesion molecule-1 (PECAM-), felt to be involved in angiogenesis and endothelial cell migration48 and follistatin, which is overexpressed in patients with NSCLC,49 were also markedly higher in H2009 cell supernatants (p=0.0001). Together, these data support the hypothesis that these adenocarcinoma cells possess the ability to self-create a pro-angiogenic, growth-promoting and pro-survival environment, by inducing the expression of ORs and constitutive phosphorylation of EGFR.
Table 1.
H2009 cells secrete growth-promoting cytokines.
| H2009 | H2009 control | Beas2B | Beas2B control | p-value
|
|||
|---|---|---|---|---|---|---|---|
| H2009 vs Beas2B | H2009 vs control | Beas2B vs control | |||||
|
| |||||||
| follistatin | 298 ± 10 | 1.7 ± 0.3 | 566 ± 11.0 | 9.7 ± 0.9 | <0.0001 | <0.0001 | <0.0001 |
| HGF | 45 ± 6 | 3.0 ± 1.0 | 5.7 ± 1.5 | 5.3 ± 2.3 | 0.0023 | 0.002 | 0.91 |
| IL-8 | 4217 ± 265 | 0.21 ± 0.02 | 112 ± 2.3 | 0.7 ± 0.3 | <0.0001 | <0.0001 | <0.0001 |
| G-CSF | 411 ± 37 | 1.7 ± 0.3 | 28.3 ± 1.0 | 2.7 ± 0.3 | 0.0005 | 0.0004 | <0.0001 |
| PDGF-BB | 11.3 ± 0.9 | 3.3 ± 0.3 | 8.0 ± 0.6 | 7.0 ± 0.0 | 0.035 | 0.001 | 0.11 |
| VEGF | 7253 ± 179 | 4.0 ± 0.6 | 650 ± 6.5 | 5.0 ± 0.6 | <0.0001 | <0.0001 | <0.0001 |
| leptin | 9.0 ± 3.5 | 1.7 ± 1.7 | 0.3 ± 0.3 | 12.0 ± 1.6 | 0.07 | 0.13 | 0.0006 |
| PECAM-1 | 157 ± 5.8 | 12.7 ± 4.5 | 66.3 ± 1.7 | 51.7 ± 6.6 | 0.0001 | <0.0001 | 0.1 |
H2009 and Beas2B (control) cells were grown in regular growth conditions for 48 hours, and the conditioned medium was assayed for pro-angiogenic cytokines using magnetic bead based BioPlex assays. All factors except leptin were significantly higher in H2009 conditioned media compared to that obtained from Beas2B epithelial cells, indicating H2009 cells possess the ability to create a self-promoting environment. The markedly increased levels are also a plausible explanation of the mu opioid receptor (MOR) and epidermal growth factor (EGF) upregulation observed. All results are reported as means ± SEM and significance was calculated by ANOVA/Tukey’s post-hoc correction using Graphpad Prizm. HGF: hepatocyte growth factor; IL-8: interleukin 8; G-CSF: granulocyte colony stimulating factor; PDGF-BB: platelet derived growth factor-BB; VEGF: vascular endothelial growth factor; PECAM-1: platelet endothelial cell adhesion molecule
Figure 5. MOR, EGFR, and CD31 co-expression in human NSCLC tumors.

Six micron sections of archived human adenocarcinoma (Stage I-III) were co-stained for EGFR (red), MOR (green), and CD31 (blue) as shown in the top row. Images were pseudocolored and overlaid in Adobe Photoshop. Magnification X400. MOR expression is shown by the green arrows. EGFR expression is indicated by the red arrows in the area outlined. EGFR and MOR are co-expressed in tumor tissue but not normal lung tissue as shown in the bottom left panel. CD31 likewise co-localizes with MOR as shown in the bottom middle panel. Most significantly, tumors contained areas where all three markers were co-expressed, as shown in the bottom right panel, indicating endothelial cells are important to the micro-environment of NSCLC tumors and may correlate with MOR and EGFR expression. The H&E image is a histologically representative area of the immunofluorescent images. Images shown represent 8 tumors stained. The images above were obtained from a Stage IB adenocarcinoma. Abbreviations: MOR: mu opioid receptor; EGFR: epidermal growth factor receptor; H&E: hematoxylin & eosin.
Furthermore, incubating Beas2B cells in H2009-conditioned medium for 48h resulted in significantly increased MOR expression (Figure 4), but did not stimulate DOR expression or autophosphorylation of EGFR (data not shown). Of note, Beas2B cells did not show any constitutive expression of DOR or autophosphorylation of EGFR. Therefore, the tumor microenvironment appears to be more conducive to MOR expression in non-malignant cells.
Figure 4. H2009 conditioned medium increases MOR expression in benign Beas2B bronchoepithelial cells.

Beas2B epithelial cells incubated with H2009 conditioned medium for 48h as compared to H2009 blank medium show significantly increased expression of MOR. Density units of MOR:GAPDH are expressed as mean ± SEM of 3 separate experiments. No DOR expression was observed on Beas2B cells incubated with or without H2009 conditioned medium (data not shown). X-axis shows different medium used as follows: Beas2B, Complete Beas2B culture medium BEBM with Singlequotes; H2009 Blank, H2009 culture medium without serum incubated for 48h in a T-75 flask without any cells; H2009 conditioned, H2009 medium incubated with H2009 cells for 48h. Abbreviations: GAPDH: glyceraldehyde 3-phosphate dehydrogenase; MOR: mu opioid receptor
MOR, EGFR, and CD31 are co-expressed by human NSCLC tumors
Correlative to our in vitro observations of increased MOR and EGFR expression on H2009 cells and increased levels of pro-angiogenic and survival-promoting cytokines, we observed increased co-expression of EGFR and MOR in the tumor region of resected human lung adenocarcinomas compared to normal lung tissue (Fig 5). EGFR and MOR expression co-localized with CD31-positive as well as CD31-negative cells, suggesting that MOR and EGFR expression increases on vascular endothelial cells as well as on tumor cells. Green staining for vascular endothelium (top left hand panel) shows MOR co-expression (bottom left hand panel) in turquoise color. Merged images of CD31, MOR and EGFR show white staining suggestive of EGFR and MOR co-expression on CD31 positive endothelial cells (bottom right panel); magenta staining shows the co-expression of MOR with EGFR on nonvascular cells. A tumor microenvironment replete with increased cytokines and growth factors may be involved in the increased vascularity that correlates with increased expression of MOR and EGFR in human lung tumors.
Morphine- and EGF-induced proliferation and invasion of H2009 is mediated by OR(s) and EGFR
EGF as well as morphine significantly stimulated the proliferation and invasiveness of H2009 cells as compared to phosphate buffered saline (PBS)-treated cells (Figures 6A and B; P <0.001 and 0.05, respectively). These functional effects of morphine and EGF were significantly inhibited by pretreatment of cells with OR antagonist, naloxone or EGFR inhibitor, erlotinib. These data demonstrate the functional dependence of EGFR on ORs and vice versa. Correlatively, these observations provide the functional significance of morphine-induced activation of growth-promoting signaling pathways described above.
Discussion
The influence of opioids on tumor biology has remained elusive, and limited data argue for both tumor-inhibitory and tumor-promoting effects of opioids.22,23,32,33 However, emerging literature strongly suggests that MOR agonist opioids at clinically used doses induce endothelial and tumor cell proliferation, enhance survival, and migration both in vitro and in vivo.18-19,27,28 Our data demonstrate that, [i] activation of ORs by analgesic morphine co-activates EGFR in highly resistant NSCLC cells, [ii] ORs are involved with EGF-induced EGFR, MAPK/ERK- and Aktphosphorylation, proliferation and invasiveness and [iii] the self-supportive tumor microenvironment created by NSCLC cells may be associated with increased expression of MOR on tumor cells. Therefore, our data provide an insight into the complex mechanism(s) that may impart resistance to therapy in difficult to treat lung cancer.
EGFR is a critical target in the treatment of advanced NSCLC; however, EGFR-targeted therapies are not curative, indicating a need to identify additional targets to block cancer progression. In this regard, we identified ORs as potential contributing factors to EGFR TKI resistance. We show that ORs are expressed on NSCLC human lung cancer cells both in vitro and in vivo. This is in agreement with previous work demonstrating increased MOR expression in human lung cancers as detected by PET scanning34, opioid agonist and antagonist binding,32 and by immunohistochemistry23. Our observations are in agreement with a recent study showing overexpression of MOR on several human NSCLC adenocarcinoma and bronchioalveolar carcinoma cell lines and human lung adenocarcinoma and bronchioalveolar carcinoma tissue23. However, herein, the finding of increased co-expression of EGFR with overexpression of MOR in human lung adenocarcinoma tissue specimens is a novel finding. Correlative to EGFR and MOR overexpression in the cancerous lung adenocarcinoma, we observed auto-phosphorylation of EGFR without external stimulation in H2009 cells. We believe that the tumor microenvironment replete with proinflammatory and growth- and survival-promoting cytokines stimulates the expression of MOR in lung cancer cells as well as in the cancerous tissue, observed by us. This is further supported by the markedly increased expression of cytokines in the culture medium of H2009 cells herein. In earlier studies we observed that VEGF increased MOR gene and protein expression several-fold in mouse retinal microvascular endothelial cells25. It is therefore conceivable that the highly enriched cytokine microenvironment in the tumor and that observed by us in H2009 cell culture medium orchestrates MOR expression in the tumor and H2009 cells, respectively.
Indeed, incubation of benign Beas2B lung epithelial cells with H2009-conditioned medium, which led to increased MOR expression on Beas2B cells, supports our hypothesis that increased secretion of cytokines by NSCLC enhance MOR expression on H2009 and in lung tumors. Whether this increased MOR expression is transformative remains to be investigated. Beas2B cells did not show constitutive DOR expression or autophosphorylation of EGFR, nor were they stimulated by H2009-conditioned medium. It is likely that long-term stimulation with cytokines such as those secreted by H2009 may be required to cause EGFR autophosphorylation in benign cells or transformation to tumor cell phenotype may be a prerequisite to cytokine-induced DOR and/or EGFR autophosphorylation. Thus, NSCLC cells are able to maintain a self-promoting and self-sustaining environment, perhaps via overexpression of MOR and constitutive activation of EGFR observed in H2009 cells and in human lung adenocarcinoma.
The constitutive phosphorylation of EGFR is further increased upon stimulation with morphine in H2009 cells, which is accompanied by phosphorylation of downstream MAPK/ERK and Akt signaling pathways analogous to EGF-induced signaling. Our observations are in agreement with morphine- and highly selective MOR agonist DAMGO-induced MAPK/ERK phosphorylation in NSCLC cells shown more than a decade ago33. Activation of these pathways by morphine translates into increased proliferation and invasiveness of H2009 cells, suggestive of a growth promoting effect of morphine in lung cancer. These signaling and functional activities of morphine in H2009 cells are reminiscent of morphine-induced phosphorylation of MAPK/ERK, Akt and VEGFR2, and proliferation, migration and survival of endothelial cells observed by us, and others19-22, 25. Our observations on morphine-induced proliferation of H2009 NSCLC cells are further supported by a recent study showing significantly increased proliferation of LLC cells in response to morphine and a highly selective MOR agonist peptide, DAMGO23. Together, these observations demonstrate that morphine stimulates lung cancer cell proliferation and invasion by activating EGFR, MAPK/ERK and Akt signaling pathways.
Inhibition of morphine-induced EGFR, Akt and MAPK/ERK phosphorylation by naloxone implicates ORs in this phenomenon. Silencing of MOR as well as DOR on H2009 cells results in abrogation of morphine as well as EGF-induced signaling, suggesting an integral role of MOR and DOR in lung cancer progression. These observations on H2009 cells complement the findings on inhibition of invasion and colony formation in MOR-silenced LLC cells and a significant inhibition in tumor burden and metastases in MOR knockout mice as compared to wild type mice with subcutaneously growing LLC23. Invasion of LLC was also inhibited by a highly specific MOR antagonist, MNTX, in this study. Moreover, inhibition of morphine-induced EGFR, Akt and MAPK/ERK phosphorylation by erlotinib and in the MOR/DOR-silenced H2009 noted by us, supports a mechanism whereby morphine-induced stimulation of growth- and survival-promoting signaling requires cross-talk between MOR/DOR and EGFR. The observations on MOR overexpression and inhibition of its activity pharmacologically by MNTX or erlotinib may be critical in resistant, difficult-to-treat lung cancer. Taken together, these data suggest that the addition of morphine to a NSCLC microenvironment with upregulated MOR may increase growth-promoting and pro-survival signaling directly via ORs and by co-activating EGFR.
The observed inhibition of morphine- and EGF-induced signaling by silencing DOR may be due to the formation of MOR and DOR heterodimers50,51, but this requires further investigation. Due to severe pain accompanying advanced stage lung cancer, MOR appears to be highly critical, because of the use of opioid analgesics that act via MOR. However, our observations on the abrogation of morphine- and EGF-induced signaling by silencing of DOR may be important in developing DOR antagonists to inhibit the unwanted tumor growth-promoting activity of opioids without compromising analgesia. Naltrindole, a classical DOR antagonist, was shown to inhibit the Akt survival-promoting pathway and modulation of several Akt-dependent genes and the downstream effectors, glycogen synthase kinase-3 beta and Forkhead transcription factors AFX and FKHR, in SCLC cells.52 In this study, naltrindole also induced apoptosis in SCLC cells by inhibiting Akt signaling. Thus, DOR may provide additional targets to abrogate the unwanted effect of morphine on lung cancer progression.
Although the exact mechanism by which OR inhibition abrogates morphine- and EGF-induced EGFR, Akt, and MAPK/ERK phosphorylation has not yet been elucidated in NSCLC, our model suggests that EGFR co-activation by MOR occurs intracellularly as it does in a number of other cell types. EGFR transactivation occurs via intracellular, zinc dependent ADAM (a disintegrin and metalloprotease) family protease cleavage of membrane-bound EGF-like ligands such as heparin binding-EGF, amphiregulin, and transforming growth factor-alpha, in a variety of cell types.37,51,53 Moreover, EGFR and MAPK can be transactivated by MOR stimulation via a calmodulin-dependent mechanism in human embryonal kidney (HEK) cells.35 Conversely, EGF has been shown to phosphorylate MOR via GRK2 (G protein receptor kinase 2) in HEK cells.54 Cross-talk between EGFR and GPCRs has been suggested to be src and ERK dependent.55 In this study, inhibition of GPCR or EGFR inhibited the activity of the other. This is a plausible explanation for our observation that naloxone and OR silencing or erlotinib attenuated EGF- or morphine-induced EGFR phosphorylation. Therefore it is likely that MOR-specific antagonists will have an inhibitory effect on lung cancer as suggested by Biji et al.23 These data provide insight into a potential means to overcome resistance to anti-EGFR therapy, which is made particularly attractive with the recent development of peripherally only acting MOR antagonists such as MNTX which do not antagonize analgesia by morphine.
Recently, Singleton et al reported synergy between MNTX and the mTOR inhibitors rapamycin and temsirolimus in inhibiting VEGF-induced human pulmonary vein endothelial cell proliferation, migration and angiogenesis.56 Similar to therapies for other cancers, multitargeted therapies addressing both the endothelium and tumor cells appear to be more effective than monotherapy.57 These recent observations are further supported by earlier observations that MNTX can have an inhibitory effect on VEGFR2 and EGFR activity on endothelial and lung cancer cells, respectively,28,29,58 and by our data showing co-localization of EGFR and MOR on CD31-positive endothelial cells in addition to tumor cells.
Based on our observations, we speculate important clinical implications for the involvement of ORs in NSCLC: (1) upregulation of MOR may have adverse effects on the promotion of tumor growth, (2) opioid analgesics that are MOR agonists such as morphine may inadvertently promote cancer progression when used for analgesia, and (3) peripherally acting opioid receptor antagonism can be targeted to develop adjunctive therapy to treat lung cancer.
Acknowledgments
We thank Ms. Carol Taubert for preparation of the document for publication.
Funding: This work is funded by National Institute of Health Grants RO1 HL68802 and CA109582 to K.G. and NHLBI Training Grant 5T32HL007062-34 (NF).
Footnotes
DISCLOSURES:
Name: Naomi Fujioka, MD
Contribution: This author helped conduct the study and write the manuscript.
Attestation: Naomi Fujioka has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Julia Nguyen
Contribution: This author helped conduct the study.
Attestation: Julia Nguyen has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Chunsheng Chen
Contribution: This author helped conduct the study and analyze the data.
Attestation: Chunsheng Chen has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Yunfang Li
Contribution: This author helped conduct the study and analyze the data.
Attestation: Yunfang Li has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Teena Pasrija
Contribution: This author helped conduct the study and analyze the data.
Attestation: Teena Pasrija has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Gloria Niehans
Contribution: This author helped conduct the study and analyze the data.
Attestation: Gloria Niehans has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Kathryn N Johnson
Contribution: This author helped conduct the study.
Attestation: Kathryn N Johnson has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Vinita Gupta, PhD
Contribution: This author helped conduct the study.
Attestation: Vinita Gupta has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Robert A Kratzke, MD
Contribution: This author helped design the study, conduct the study, and analyze the data.
Attestation: Robert A Kratzke has seen the original study data, reviewed the analysis of the data, and approved the final manuscript.
Name: Kalpna Gupta, PhD
Contribution: This author helped design the study, conduct the study, analyze the data, and write the manuscript.
Attestation: Kalpna Gupta has seen the original study data, reviewed the analysis of the data, approved the final manuscript, and is the author responsible for archiving the study files.
The authors declare no conflicts of interest.
Contributor Information
Naomi Fujioka, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Julia Nguyen, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Chunsheng Chen, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Yunfang Li, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Teena Pasrija, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Gloria Niehans, Department of Lab Medicine/Pathology, VA Medical Center, Minneapolis, MN.
Kathryn N Johnson, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Vinita Gupta, Bio-Rad Laboratories, Hercules, CA.
Robert A Kratzke, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
Kalpna Gupta, Department of Medicine, Division of Hematology, Oncology, Transplantation, University of Minnesota, Minneapolis, MN.
References
- 1.American Cancer Society. Cancer Facts & Figures 2007. Atlanta: American Cancer Society; 2007. [Google Scholar]
- 2.Garcia M, Jemal A, Ward EM, Center MM, Hao Y, Siegel RL, Thun MJ. Global Cancer Facts & Figures 2007. Atlanta, GA: American Cancer Society; 2007. [Google Scholar]
- 3.Altekruse SF, Kosary CL, Krapcho M, Neyman N, Aminou R, Waldron W, Ruhl J, Howlader N, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Cronin K, Chen HS, Feuer EJ, Stinchcomb DG, Edwards BK, editors. SEER Cancer Statistics Review, 1975-2007. National Cancer Institute; Bethesda, MD: 2010. http://seer.cancer.gov/csr/1975_2007/, based on November 2009 SEER data submission, posted to the SEER web site. [Google Scholar]
- 4.Grandis JR, Sok JC. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharm Ther. 2004;102:37–46. doi: 10.1016/j.pharmthera.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Brabender J, Danenberg KD, Metzger R, Schneider PM, Park JM, Salonga D, Holscher AH, Danenberg PV. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin Can Res. 2001;7:1850–55. [PubMed] [Google Scholar]
- 6.Veale D, Kerr N, Gibson GJ, Kelly PJ, Harris AL. The relationship of quantitative epidermal growth factor receptor expression in non small cell lung cancer to long term survival. Br J Cancer. 1993;68:162–5. doi: 10.1038/bjc.1993.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selvaggi G, Novello S, Torri V, Leonardo E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P, Scagliotti GV. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann Oncol. 2004;15:28–32. doi: 10.1093/annonc/mdh011. [DOI] [PubMed] [Google Scholar]
- 8.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabárbara P, Seymour L National Cancer Institute of Canada Clinical Trials Group. Erlotinib in previously treated non small cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch F, Varella-Garcia M, Bunn PA, Jr, Franklin WA, Dziadziuszko R, Thatcher N, Chang A, Parikh P, Pereira JR, Ciuleanu T, von Pawel J, Watkins C, Flannery A, Ellison G, Donald E, Knight L, Parums D, Botwood N, Holloway B. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:5024–34. doi: 10.1200/JCO.2006.06.3958. [DOI] [PubMed] [Google Scholar]
- 10.Douillard J, Shepherd FA, Hirsh V, Mok T, Socinski MA, Gervais R, Liao ML, Bischoff H, Reck M, Sellers MV, Watkins CL, Speake G, Armour AA, Kim ES. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non small cell lung cancer: Data from the randomized phase III INTEREST trial. J Clin Oncol. 2010;28:744–52. doi: 10.1200/JCO.2009.24.3030. [DOI] [PubMed] [Google Scholar]
- 11.Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomized, placebo-controlled, multicentre study (Iressa survival evaluation in lung cancer) Lancet. 2005;366:1527–37. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 12.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 13.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 14.Jackman DM, Yeap BY, Sequist LV, Lindeman N, Holmes AJ, Joshi VA, Bell DW, Huberman MS, Halmos B, Rabin MS, Haber DA, Lynch TJ, Meyerson M, Johnson BE, Jänne PA. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small-cell lung cancer patients treated with gefitinib or erlotinib. Clin Can Res. 2006;12:3908–14. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;8:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 16.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Nat Acad Sci USA. 2008;105:2070–75. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tegeder I, Geisslinger G. Opioids as modulators of cell death and survival--unraveling mechanisms and revealing new indications. Pharmacol Rev. 2004;56:351–69. doi: 10.1124/pr.56.3.2. [DOI] [PubMed] [Google Scholar]
- 18.Gupta M, Li Y, Gupta K. Opioids as promoters and regulators of angiogenesis. In: Maragoudakis ME, Papadimitriou E, editors. Angiogenesis: Basic Science and Clinical Applications. Transworld Research Network; 2007. pp. 303–17. [Google Scholar]
- 19.Singleton PA, Lingen MW, Fekete MJ, Garcia JG, Moss J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: Role of receptor transactivation. Microvasc Res. 2006;72:3–11. doi: 10.1016/j.mvr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Singleton PA, Garcia JG, Moss J. Synergistic effects of methylnaltrexone with 5-fluorouracil and bevacizumab on inhibition of vascular endothelial growth factor–induced angiogenesis. Mol Cancer Ther. 2008;7:1669–79. doi: 10.1158/1535-7163.MCT-07-2217. [DOI] [PubMed] [Google Scholar]
- 21.Singleton PA, Mambetsariev N, Lennon FE, Mathew B, Siegler JH, Moreno-Vinasco L, Salgia R, Moss J, Garcia JG. Methylnaltrexone potentiates the anti-angiogenic effects of mTOR inhibitors. J Angiogenes Res. 2010;2:5. doi: 10.1186/2040-2384-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta K, Kshirsagar S, Chang L, Schwartz R, Law PY, Yee D, Hebbel RP. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002;62:4491–98. [PubMed] [Google Scholar]
- 23.Mathew B, Lennon F, Siegler J, Mirzapoiazova T, Mambetsariev N, Sammani S, Gerhold LM, Lariviere PJ, Chen CT, Garcia JG, Salgia R, Moss J, Singleton PA. The novel role of the mu opioid receptor in lung cancer progression: A laboratory Investigation. Anesth Analg. 2011;112:558–67. doi: 10.1213/ANE.0b013e31820568af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber ML, Farooqui M, Nguyen J, Ansonoff M, Pintar JE, Hebbel RP, Gupta K. Morphine induces mesangial cell proliferation and glomerulopathy via kappa-opioid receptor. Am J Physiol Renal Physiol. 2008;294:F1388–97. doi: 10.1152/ajprenal.00389.2007. [DOI] [PubMed] [Google Scholar]
- 25.Chen C, Farooqui M, Gupta K. Morphine stimulates vascular endothelial growth factor-like signaling in mouse retinal endothelial cells. Curr Neurovasc Res. 2006;3:171–180. doi: 10.2174/156720206778018767. [DOI] [PubMed] [Google Scholar]
- 26.Farooqui M, Li Y, Rogers T, Poonawala T, Griffin RJ, Song CW, Gupta K. COX-2 inhibitor celecoxib prevents chronic morphine-induced promotion of angiogenesis, tumor growth, metastasis and mortality, without compromising analgesia. Br J Cancer. 2007;97:1523–31. doi: 10.1038/sj.bjc.6604057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farooqui M, Jiang J, Stephenson EJ, Yee D, Gupta K. Naloxone acts as an antagonist of estrogen receptor in MCF7 cancer cells. Mol Cancer Ther. 2006;5:611–20. doi: 10.1158/1535-7163.MCT-05-0016. [DOI] [PubMed] [Google Scholar]
- 28.Singleton PA, Lingen MW, Fekete MJ, Garcia JG, Moss J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: role of receptor transactivation. Microvasc Res. 2006;72:3–11. doi: 10.1016/j.mvr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Singleton PA, Garcia JG, Moss J. Synergistic effects of methylnaltrexone with 5-fluorouracil and bevacizumab on inhibition of vascular endothelial growth factor-induced angiogenesis. Mol Can Ther. 2008;7:1669–79. doi: 10.1158/1535-7163.MCT-07-2217. [DOI] [PubMed] [Google Scholar]
- 30.Wang CZ, Li XL, Sun S, Xie JT, Aung HH, Tong R, McEntee E, Yuan CS. Methylnaltrexone, a peripherally acting opioid receptor antagonist, enhances tumoricidal effects of 5-Fu on human carcinoma cells. Anticancer Res. 2009;29:2927–32. [PubMed] [Google Scholar]
- 31.Roth KA, Barchas JD. Small cell carcinoma cell lines contain opioid peptides and receptors. Cancer. 1986;57:769–73. doi: 10.1002/1097-0142(19860215)57:4<769::aid-cncr2820570415>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 32.Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci USA. 1990;87:3294–98. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heusch WL, Maneckjee R. Effects of bombesin on methadone-induced apoptosis of human lung cancer cells. Cancer Lett. 1999;136:177–85. doi: 10.1016/s0304-3835(98)00335-8. [DOI] [PubMed] [Google Scholar]
- 34.Madar I, Bencherif B, Lever J, Heitmiller RF, Yang SC, Brock M, Brahmer J, Ravert H, Dannals R, Frost JJ. Imaging delta- and mu-opioid receptors by PET in lung carcinoma patients. J Nucl Med. 2007;48:207–13. [PubMed] [Google Scholar]
- 35.Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ. μ-opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:33847–53. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 36.Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. Mu opioid transactivation and down-regulation of the epidermal growth factor receptor in astrocytes: implications for mitogen-activated protein kinase signaling. Mol Pharmacol. 2003;64:1391–1401. doi: 10.1124/mol.64.6.1391. [DOI] [PubMed] [Google Scholar]
- 37.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 38.Gupta K, Ramakrishnan S, Browne PV, Solovey A, Hebbel RP. A novel technique for culture of human dermal microvascular endothelial cells under either serum-free or serum-supplemented conditions: isolation by panning and stimulation with vascular endothelial growth factor. Exp Cell Res. 1997;230:244–251. doi: 10.1006/excr.1996.3421. [DOI] [PubMed] [Google Scholar]
- 39.Kohli DR, Li Y, Khasabov SG, Gupta P, Kehl LJ, Ericson ME, Nguyen J, Gupta V, Hebbel RP, Simone DA, Gupta K. Pain related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids. Blood. 2010;116:456–465. doi: 10.1182/blood-2010-01-260372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 41.Puri N, Salgia R. Synergism of EGFR and c-met pathways, crosstalk, and inhibition, in non small cell lung cancer. J Carcinogenesis. 2008;7:1–8. doi: 10.4103/1477-3163.44372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, Laterra J. Transcription dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol Can Res. 2008;6:139–150. doi: 10.1158/1541-7786.MCR-07-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonine-Summers AR, Aakre ME, Brown KA, Arteaga CL, Pietenpol JA, Moses HL, Cheng N. Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological response in mammary epithelial cells. Cancer Biol Ther. 2007;6:561–570. doi: 10.4161/cbt.6.4.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engleman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. MET amplification leads to gefitinib resistance in lung cancer by activation ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 45.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. NEJM. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 46.Blumenschein GR, Jr, Gatzemeier U, Fossella F, Stewart DJ, Cupit L, Cihon F, O’Leary J, Reck M. Phase II, multicenter, uncontrolled trial of single-agent sorafenib in patients with relapsed or refractory, advanced non-small-cell lung cancer. J Clin Oncol. 2009;27:4274–80. doi: 10.1200/JCO.2009.22.0541. [DOI] [PubMed] [Google Scholar]
- 47.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, Meng YG, Ferrara N. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA. 2009;16:6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao G, O’Brien CD, Zhou Z, Sanders SM, Greenbaum JN, Makrigiannakis A, DeLisser HM. Involvement of human PECAM in angiogenesis and in vitro endothelial cell migration. Am J Physiol Cell Physiol. 2002;282:C1181–1190. doi: 10.1152/ajpcell.00524.2001. [DOI] [PubMed] [Google Scholar]
- 49.Planque C, Kulasingam V, Smith CR, Reckamp K, Goodglick L, Diamandis EP. Identification of five candidate lung cancer biomarkers by proteomics analysis of conditioned media of four lung cancer cell lines. Mol Cell Proteomics. 2009;8:2746–2758. doi: 10.1074/mcp.M900134-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomes I, Devi L. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. PNAS. 2004;14:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Transactions. 2003;6:1203–1208. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 52.Chen YL, Law PY, Loh HH. Inhibition of Akt/protein kinase B signaling by naltrindole in small cell lung cancer cells. Cancer Res. 2004;64:8723–30. doi: 10.1158/0008-5472.CAN-03-3091. [DOI] [PubMed] [Google Scholar]
- 53.Murphy G. The ADAMs: signaling scissors in the tumour microenvironment. Nat Rev Can. 2008;8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 54.Chen Y, Long H, Wu Z, Jiang X, Ma L. EGF transregulates opioid receptors through EGFR-mediated GRK2 phosphorylation and activation. Mol Biol Cell. 2008;19:2973–2983. doi: 10.1091/mbc.E07-10-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El Zein N, D’Hondt S, Sariban E. Crosstalks between the receptors tyrosine kinase EGFR and TrkA and the GPCR, FPR, in human monocytes are essential for receptors-mediated cell activation. Cell Signal. 2010;22:1437–47. doi: 10.1016/j.cellsig.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 56.Singleton PA, Mambetsariev N, Lennon FE, Mathew B, Siegler JH, Moreno-Vinasco L, Salgia R, Moss J, Garcia JG. Methylnaltrexone potentiates the anti-angiogenic effects of mTOR inhibitors. J Angiogenes Res. 2010;2:5–18. doi: 10.1186/2040-2384-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byers LA, Heymach JV. Dual targeting of the vascular endothelial growth factor and epidermal growth factor receptor pathways: rationale and clinical applications for non-small-cell lung cancer. Clin Lung Cancer. 2007;8(Suppl 2):S79–8. doi: 10.3816/clc.2007.s.006. [DOI] [PubMed] [Google Scholar]
- 58.Mathew B, Lennon FE, Siegler J. The mu opioid receptor regulates lewis lung carcinoma tumor growth and metastasis. Molecular Cancer Therapeutics. 2009;8:C79–79C. [Google Scholar]