

Abstract
Claudins constitute a family of tight junction transmembrane proteins whose first extracellular loop (ECL1) determines the paracellular permeability and ion selectivity in epithelia. There are two cysteines in the ECL1 that are conserved among all claudins. We hypothesized that these extracellular cysteines are linked by an intramolecular disulfide bond that is necessary for correct pore folding and function. To test this, we mutated C54 and C64 in claudin-2, either individually or together to alanine or serine, and generated stable Madin-Darby canine kidney (MDCK) I Tet-off cell lines. Immunoblotting showed a higher molecular mass band in the mutants with a single cysteine mutation, consistent with a claudin-2 dimer, suggesting that the two conserved cysteines normally form an intramolecular disulfide bond in wild-type claudin-2. By immunofluorescent staining, the alanine mutants were mislocalized intracellularly, while the serine mutants were expressed at the tight junction. Thus dimerization of both C54A and C64A did not require tight junction expression, suggesting that C54 and C64 are located near an intermolecular interface involved in cis-interaction. The conductance and Na+ permeability of the serine mutants were markedly lower than the wild type, but there was no difference between the single mutants and the double mutant. We conclude that the disulfide bond between the conserved extracellular cysteines in claudin-2 is necessary for pore formation, probably by stabilizing the ECL1 fold, but is not required for correct protein trafficking. We further speculate that this role is generalizable to other claudin family members.
Keywords: tight junction, paracellular transport, ion channel, cysteines, disulfide bond
the tight junction complex is the most apical junctional complex in epithelia and is the main determinant of paracellular permeability. Claudins constitute a family of tight junction transmembrane proteins whose first extracellular loop (ECL1) protrudes into the paracellular space and forms the lining of the paracellular pore or barrier. A claudin pore has an estimated diameter of 6.5–8 Å (14, 19), and its charge selectivity is determined by the electrostatic force exerted by charged amino acid(s) on the ECL1 (7, 10, 16, 19). However, precisely what determines correct folding of the ECL1 is incompletely understood.
The claudin family shares a highly conserved signature sequence GLW-[X]2-C-[X]8–10-C in the ECL1. The two cysteines in the ECL1 signature sequence are the only extracellular cysteines and are completely conserved in all claudins. They are believed to be important in the folding of ECL1. In claudin-5, a barrier claudin, mutating either one of the conserved cysteines resulted in lower transepithelial resistance (17). In cells expressing claudin-1, a coreceptor for cell entry of hepatitis C virus (HCV), mutating the cysteines caused the cells to become insensitive to HCV infection (4). These studies suggest that, in general, loss of the conserved cysteines impairs claudin function. In a previous study of claudin-2, the conserved cysteines C54 and C64 were not modifiable by thiol-reactive reagents (2). Thus we hypothesized that C54 and C64 are linked by an intramolecular disulfide bond that is necessary for correct pore folding and proper function of claudin-2.
To test this hypothesis, we mutated C54 and C64 in claudin-2, either individually or together, to alanine or serine, and generated stable Madin-Darby canine kidney (MDCK) I Tet-off cell lines expressing each mutant protein. Our data suggest that the two conserved cysteines in the ECL1 are disulfide bonded and that the disulfide bond is necessary for pore formation but not for claudin trafficking.
MATERIALS AND METHODS
Generation and screening of MDCK I Tet-off claudin-2 cell lines.
Six MDCK I Tet-off cell lines expressing claudin-2 mutants (C54A, C64A, C54A/C64A, C54S, C64S, and C54S/C64S) were generated by the methods described previously(18). In short, the mutants were generated by site-directed mutagenesis on the template plasmid pRevTREP-mClaudin2-wild type, using the QuikChange Kit (Agilent Technologies). The plasmids were lipofected into the viral packaging cell line PT67. Viral particles were collected from PT67 cell growth medium and used to transduce MDCK I Tet-off cells. After 7–10 days in a 0.3 mg/ml hygromycin-selective medium, independent clones were selected using cloning cylinders. To induce claudin-2 expression, doxycycline was omitted from the culture medium; otherwise, 50 ng/ml doxycycline was included to suppress its expression.
Immunoblotting.
Claudin-2 protein expression was tested by SDS-PAGE and immunoblotting. Confluent cells grown on tissue culture dishes were mechanically lysed by passing through a 25-gauge needle 10 times in a sucrose-histidine lysis buffer, containing 250 mM sucrose, 30 mM histidine, 1 mM EDTA (pH 7), and protease inhibitor (Complete Mini; Roche Diagnostics). Cell lysates were loaded in a reducing SDS-PAGE buffer [1% (vol/vol) 2-mercaptoethanol added] or a nonreducing SDS-PAGE buffer and heated at 75°C for 10 min. Twenty micrograms of protein samples were loaded on 12% polyacrylamide gel, transferred to a PVDF membrane, blotted with 1:500 mouse anti-claudin-2 antibody or 1:1,000 rabbit anti-claudin-2 antibody (Invitrogen) and appropriate horseradish peroxidase-conjugated secondary antibodies (GE), detected with the ECL chemiluminescent method (Pierce), and imaged in an ImageQuant LAS-4000 (GE Healthcare).
Immunofluorescent staining.
The MDCK I claudin-2 wild type and mutants were plated at a density of 105 cells/1.16 cm2 on 12-well Transwell plates and grown for 7 days. The cells were washed in ice-cold PBS, fixed with 4% paraformaldehyde at 4°C for 15 min, permeabilized, and blocked in a permeation buffer (0.3% Triton X-100, 1% BSA, and 5% goat serum in PBS) for 1 h. The filters were incubated in primary antibodies [rabbit anti-claudin-2, 1:200; mouse anti-zona occludens 1 (ZO-1), 1:400] for 2 h at room temperature, washed three times in PBS, and then incubated in secondary antibodies (Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 555-conjugated anti-mouse IgG, both 1:1,000, from Life Technologies) for 1 h, washed three times in PBS, and mounted in ProLong anti-fade mounting medium (Life Technologies). Slides were viewed with a Leica TCS SP2 multi-photon confocal microscope with a Z-section of 0.4-μm thick per layer.
Cysteine-specific biotinylation.
To test the accessibility of cysteine, cysteine-specific biotinylation was performed. Cells were plated at a density of 5 × 105 cells/well on six-well plates and grown for 6 days. Cells were pretreated with 2 mM Tris-2-carboxyethyl-phosphine (TCEP) with or without 0.01% Tween-20 or 0.005% NP-40 for 30 min, washed with PBS with 1 mM CaCl2 and 1 mM MgCl2 (PBS/CM), and treated with 0.5 ml/well freshly dissolved 2-[(biotinoyl)amino]ethyl methanethiosulfonate (MTSEA-biotin) in PBS/CM at a concentration of 0.5 mg/ml. The plate was incubated at room temperature for 10 min and washed three times with ice-cold PBS, and the cells were harvested in RIPA buffer [50 mM Tris·HCl pH 8, 150 mM NaCl, 0.1% (wt/vol) SDS, 0.5% (wt/vol) deoxycholic acid and 1% (vol/vol) NP-40]. The cell lysate was centrifuged at 16,000 g for 15 min. The supernatant was added to a 40-μl slurry of streptavidin-coated beads and rotated at 4°C for 2 h. The beads were then pelleted, and the supernatant was saved for analysis. The beads were washed three times in TBS (50 mM Tris·HCl and 150 mM NaCl), added to 20 μl 2× reducing SDS-PAGE loading buffer, and heated at 75°C for 10 min with occasional agitation. Both bead (biotinylated protein fraction) and supernatant (nonbiotinylated fraction) samples were then subjected to immunoblotting as described above.
Ussing chamber electrophysiological studies.
Cells were plated at a density of 105 cells/1.16 cm2 on Snapwell filters (Corning) and cultured for 7 days in the presence (Dox+) or absence (Dox−) of 50 ng/ml doxycycline. The Ussing chamber setup and liquid junction potential correction method was employed as previously described (18). In short, the standard Ringer solution used at baseline contained the following (in mM): 150 NaCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 Tris-HEPES, pH 7.4. To measure Na+ permeability, the solution of the basolateral chamber was changed to a 75-mM NaCl Ringer solution (osmolarity adjusted with mannitol). The ion permeability ratio, β = PCl−/PNa+, was calculated from the Goldman-Hodgkin-Katz voltage equation. The absolute Na+ permeability was estimated by the method of Kimizuka and Koketsu (8). The conductance and permeability attributable to claudin-2 pore was calculated by subtracting the values of the uninduced (Dox+) state from the values of the induced (Dox−) state.
Statistics.
The data are presented as means ± SE. Statistical significance was determined using unpaired two-tailed Student's t-test or one-way ANOVA test. The P value of the multiple comparisons was corrected using the Bonferroni correction. P < 0.05 was considered to be statistically significant.
RESULTS
Single mutants form a claudin-2 homodimer linked by an intermolecular disulfide bond.
C54 and C64 were mutated individually (single mutants) or together (double mutants) to either alanine or serine. The rationale for making both alanine and serine mutants is to avoid misinterpretation of data that is due to the artifact of introducing the mutation. Each mutant was transduced into MDCK I Tet-off cells using retroviral transduction, and stable clones were selected. Inducible protein expression was verified by immunoblotting (Fig. 1), in which all mutants showed a claudin-2 monomer band at ∼20 kDa in the absence of doxycycline. When normalized to β-actin, the protein expression of the single mutants was similar to wild type and the double mutants expressed approximately two- to threefold more protein than wild type. There were multiple smaller molecular mass bands present in the C64 single mutants and the double mutants, which were not seen in the claudin-2 blot of mouse kidney lysates using the same antibody (data not shown). These bands may be the products of proteolysis, which are commonly seen in overexpressing exogenous protein in cells. Also, the double mutants had multiple higher molecular mass bands, which might be related to posttranslational modification, but they have not been fully characterized yet. Interestingly, there was an additional higher molecular mass band clearly present in C64A and C64S and just visible in C54A and C54S. This higher molecular mass band was not seen in the wild type or in the double mutants. The band was ∼35 kDa, smaller than the predicted size of a claudin-2 dimer. However, since claudin-2 monomer migrates faster than its predicted size of 24 kDa, the dimer may migrate faster as well. In addition, although the putative dimer band was not exactly at the molecular mass of a claudin-2 dimer (40 kDa), this band had the same apparent molecular mass as D65C, which had previously been suggested to form a disulfide-bonded dimer (3). We hypothesized that in wild-type claudin-2, C64 is normally in an intramolecular disulfide bond with C54. Mutation of C64 to alanine or serine would free the thiol side chain of C54 to interact with C54 on an adjacent molecule, forming an intermolecular disulfide-bonded dimer. Likewise, mutating C54 could free up C64 to form an intermolecular disulfide bond with its counterpart on a neighboring molecule.
Fig. 1.
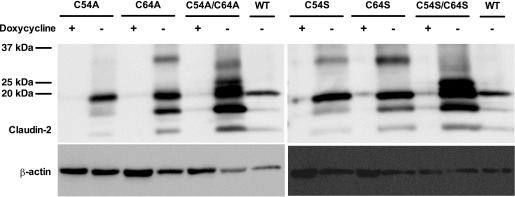
Immunoblot of claudin-2 expression in clones stably transduced with wild-type (WT) claudin-2 or the following claudin-2 mutants: C54A, C64A, C54A/C64A, C54S, C64S, and C54S/C64S. Cells were cultured in the presence (+) or absence (−) of doxycycline. Cell lysates were subjected to reducing SDS-PAGE and immunoblotted using rabbit claudin-2 antibody. Inducible expression of a band at the predicted molecular mass for claudin-2 monomer (∼20 kDa) was seen in all mutants. Note the appearance of a higher molecular mass band of ∼35 kDa most prominently in C64A and C64S in the absence of doxycycline.
To verify this hypothesis, protein samples were loaded with nonreducing SDS-PAGE buffer and reducing SDS-PAGE buffer. Under nonreducing conditions, the four single mutants (C54A, C64A, C54S, and C64S) exhibited a higher molecular mass band at the same level of the dimer band of D65C. Furthermore, under reducing conditions, the abundance of this band decreased concomitant with the increased abundance of the monomer band (Fig. 2), demonstrating that the band was a disulfide-bonded dimer and that it was composed of at least one claudin-2 molecule. There was a residual dimer band of C64S and C64A in reducing conditions either because the intermolecular disulfide bond formed by C54 is very stable and cannot be completed reduced or because its position within the protein fold makes it poorly accessible to reducing agents.
Fig. 2.

Effect of the reducing agent 2-mercaptoethanol on the higher molecular mass band in the extracellular conserved cysteine mutants. Cell lysates from Madin-Darby canine kidney (MDCK) I Tet-off cells expressing wild-type claudin-2 and the indicated mutants were loaded on an SDS-PAGE gel using either nonreducing (−) or reducing (+) gel loading buffer, and immunoblotted with anti-claudin-2. D65C was included as a positive control to show the expected size of a claudin-2 dimer. In mutants of a single cysteine, a higher molecular mass band consistent with a claudin-2 dimer was seen in nonreducing conditions, and the abundance of this band decreased under reducing conditions. This band was not present in wild-type or the double cysteine mutants.
To further confirm that the band was a claudin-2 homodimer, the membrane was blotted with antibodies against the other claudin isoforms known to be present in MDCK I cells: claudin-1, claudin-3, claudin-4, and claudin-7 (1, 6, 13, 19). No heterodimerization of claudin-2 with claudin-3, claudin-4, or claudin-7 was seen. In the claudin-1 blot, there was a faint band just above 25 kDa in all samples, consistent with a nonspecific band. Also, there was another faint band present at ∼35 kDa in C54A and C64A, but not in D65C or wild type, suggesting a possible heterodimerzation of claudin-1 and claudin-2 single mutant (Fig. 3). However, the abundance of this band was extremely low compared with the dimers in the claudin-2 blot. We conclude that the vast majority of the single mutant molecules form claudin-2 homodimers.
Fig. 3.
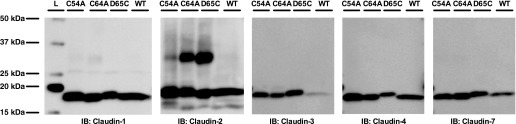
Claudin composition of the putative dimer band. Cell lysates from MDCK I Tet-off cells expressing C54A, C64A, D65C, and wild type were loaded on an SDS-PAGE gel using a nonreducing gel loading buffer and blotted using anti-claudin-1, anti-claudin-2, anti-claudin-3, anti-claudin-4, and anti-claudin-7 antibodies respectively. The signal of the other claudins was exposed to similar intensity as the claudin-2 signal. IB, immunoblot. L, ladder.
Extracellular conserved cysteines are not required for claudin-2 trafficking to tight junction.
Mutating conserved amino acids may cause protein misfolding and retention in the endothelium reticulum. The localization of the mutants was therefore examined by immunofluorescent staining of the tight junction marker ZO-1 and claudin-2, followed by confocal microscopy. Initially, we generated the alanine mutants and found that C54A and C54A/C64A were localized intracellularly, while C64A expressed both intracellularly and on the lateral membrane. This suggests that the conserved cysteines may be important for claudin-2 trafficking, but we could not exclude the possibility of an artifact due to introducing the alanine. Because serine mutations at this site were found to be well tolerated in claudin-5 (17), we then generated the serine mutants and found that the serine mutants colocalized with ZO-1 at the tight junction similar to wild type (Fig. 4). This suggests that the loss of the disulfide bond per se does not affect claudin-2 trafficking and, by inference, that it probably is not critical for global protein folding. However, the specific introduction of alanine at positions 54 or 64 may in some way disrupt normal protein trafficking.
Fig. 4.
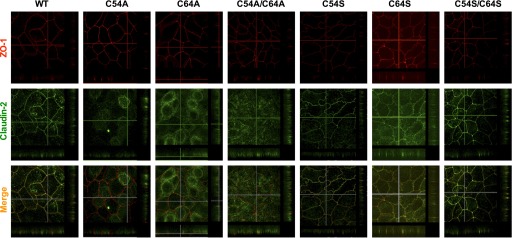
Localization of claudin-2 mutants by immunofluorescent staining and confocal microscopy examination. Cells were cultured on Transwells for 6 days and then immunostained with zona occludens 1 (ZO-1; red) and claudin-2 (green) antibodies. A series of Z-sections of 0.4-μm thickness per layer was collected for each mutant. The Z-stack along the y-axis was displayed at right of the image, and the Z-stack along the x-axis was displayed below the image. All serine mutants colocalized well with ZO-1 at tight junction. C54A and C54A/C64A were mislocalized intracellularly. C64A were localized both intracellularly and on the lateral membrane.
Single cysteine mutations makes the remaining extracellular cysteine accessible to MTSEA-biotin.
If C54 and C64 are normally disulfide bonded to each other in wild-type claudin-2, there should be no accessible free thiol groups on the extracellular surface of claudin-2. Consistent with this prediction, we previously demonstrated that wild-type claudin-2 could not be biotinylated by extracellular addition of the membrane-impermeant thiol-reactive reagent MTSEA-biotin (3). As a positive control in this experiment, MTSEA-biotin was shown to be able to biotinylate a claudin-2 mutant, Y35C, in which a cysteine was introduced at an extracellularly accessible site. It should be noted that some wild-type claudin-2 could be recovered in the streptavidin bead fraction even without MTSEA-biotin (Fig. 5A), probably due to nonspecific binding of claudin-2 molecules to the beads. We attempted to reduce the putative intramolecular disulfide bond with a strong reducing agent, TCEP, either on its own or together with weak detergents to partially unfold the protein. There was a small fraction of claudin-2 recovered in the bead fraction of both the wild-type without TCEP and the C54S/C64S samples, probably due to nonspecific binding. Nevertheless, we were unable to detect any enhancement of biotinylation in TCEP-treated groups (Fig. 5B). This indicates that the disulfide bond is buried inside the extracellular fold of the protein where it is inaccessible to TCEP.
Fig. 5.

Identification of accessible extracellular cysteines in claudin-2 constructs by MTSEA-biotin biotinylation. A: MDCK I Tet-off cells expressing wild-type claudin-2 were treated with or without MTSEA-biotin, followed by the biotinylation procedure, and were subjected to SDS-PAGE and blotted by anti-claudin-2 antibody. B: MDCK I Tet-off cells expressing wild-type claudin-2 were preincubated with 2 mM Tris-2-carboxyethyl-phosphine (TCEP) with or without detergent (0.01% Tween-20 or 0.005% NP-40) for 30 min and then were subjected to MTSEA biotinylation and streptavidin precipitation. Beads (B) and the supernatant (S) were subjected to SDS-PAGE and blotted by anti-claudin-2 antibody. MDCK I Tet-off claudin-2 Y35C and C54S/C64S were included as the positive and negative controls for the extracellular cysteine biotinylation, respectively. C: MDCK I Tet-off cells expressing serine mutants were cultured for six days, and then exposed to 0.5 mg/ml MTSEA-biotin. Cells were lysed in RIPA buffer and then precipitated with streptavidin beads. Beads and the supernatant were subjected to SDS-PAGE and immunoblotting with anti-claudin-2 antibody (both beads and supernatant samples) and anti-β-actin (supernatant samples) for loading control.
Our hypothesis further predicts that the mutation of either one of the extracellular cysteines will free up the other cysteine and make it biochemically accessible to covalent modification by MTSEA-biotin. Indeed, we found that both C54S and C64S could be biotinylated by MTSEA-biotin, whereas there was a very small amount of biotinylated fraction in wild type due to nonspecific binding and no biotinylated fraction in C54S/C64S (Fig. 5C). This provides additional support for the contention that C54 and C64 are normally disulfide bonded to each other in the native protein.
Cation pore function is lost in the serine mutants.
The functional consequence of losing the conserved cysteines on the electrophysiological characteristics of the claudin-2 pore was determined by the measurement of the transepithelial conductance and NaCl dilution potential in Ussing chambers. Induction of claudin-2 wild-type expression in MDCK I Tet-off cells increased the conductance and PNa+ by eight- and ninefold, respectively. By contrast, there was only very small increase in conductance and PNa+ in the serine mutants (Fig. 6A). By subtracting the baseline value in the Dox+ condition from values in Dox−, the conductance and PNa+ attributable to claudin-2 could be determined. From this, we found that the conductance and PNa+ of each mutant were almost completely abolished compared with wild type, whereas there was no statistical significance between either of the single mutants and the double mutant (Fig. 6B). The observation that mutating either one or both of the conserved cysteines (C54 and C64) impaired conductance and PNa+ to a similar extent strongly suggests that the loss of pore function is not an effect of any individual cysteine but rather is a consequence of disrupting the disulfide bond between them. We conclude that this disulfide bond is necessary for the formation of a functional cation pore.
Fig. 6.

Characterization of the electrophysiological properties of claudin-2 serine mutants by Ussing chamber measurements of the transepithelial conductance and NaCl dilution potential. A: transepithelial conductance (left) and the calculated Na+ permeability (right) without (Dox+) or with (Dox−) induction of expression of the indicated claudin-2 constructs. B: absolute conductance (left) and the absolute Na+ permeability (right) attributable to claudin-2 obtained by subtracting the baseline values of the noninduced cell from induced cells. Data points represent means of 3–4 filters ± SE. P value in A was obtained from unpaired Student's t-test between induced (Dox−) and uninduced (Dox+) state. P value in B was obtained from one-way ANOVA with multiple comparisons using Bonferroni correction. NS: P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
The ECL1 of claudins forms the lining of ion-selective paracellular pores at the tight junction. The ECL1 signature sequence GLW-[X]2-C-[X]8–10-C is likely to be important for correct folding of ECL1. In this study, we find that two conserved cysteines of ECL1 (C54 and C64 in claudin-2) form an intramolecular disulfide bond that is necessary for correct pore folding and function.
C54 and C64 in claudin-2 form an intramolecular disulfide bond.
A cysteine side chain in a protein generally exists in one of three main biochemical states: intermolecular disulfide-bonded, intramolecular disulfide-bonded, or as a reduced free thiol group. We do sometimes see a faint dimer band even in wild-type protein (as in Fig. 2), but it always constitutes an extremely small portion of total claudin-2 protein compared with the monomer band, and this band does not change in abundance in nonreducing conditions. We think it could be a result of noncovalent interaction, consistent with the findings of Van Itallie et al. (15), who used blue native gels to show that claudin-2 does undergo spontaneous dimerization mediated by the second transmembrane domain. On a nonreducing SDS-PAGE gel, the vast majority of wild-type claudin-2 migrates as a monomer. This excludes the idea that C54 or C64 forms an intermolecular disulfide bond. We have shown previously that there is no free thiol group accessible extracellularly in wild-type claudin-2 (2). Thus either C54 and C64 form an intramolecular disulfide bond or, if they exist as free cysteines, they must both be buried within the protein fold. The following evidence supports the intramolecular disulfide bond hypothesis. First, mutating either one of the two cysteines induces the remaining cysteine to form a claudin-2 homodimer linked by an intermolecular disulfide bond. Although it has been reported that C54S and C64S mutants of claudin-5 do not form a dimer (17), it is possible that this is because the dimers are reduced under the SDS-PAGE conditions or that C54 and C64 in claudin-5 happen not to be located near the intermolecular interface. Second, mutating either one of the two extracellular cysteines in claudin-2 makes the other one modifiable by the thiol-reactive reagent, MTSEA-biotin. It is unlikely that two free cysteines would have a mutual effect on each other such that mutating either one would make the other accessible. Collectively, our data support the proposition that the two cysteines of ECL1 normally form an intramolecular disulfide bond. Our inability to reduce the disulfide bond extracellularly in claudin-2 wild type indicates either that reducing the disulfide bond is energetically unfavorable or that the disulfide bond is buried inside the extracellular fold where it is inaccessible to the reducing agent.
Losing the disulfide bond does not affect claudin-2 trafficking.
In general, mutating conserved amino acids may cause protein misfolding and retention in the endoplasmic reticulum. In claudin-2, mutating the “GLW” of the signature sequence causes claudin-2 mislocalization (15). However, mutating the “C-C” of the signature sequence to serine does not affect claudin-2 trafficking to tight junction. This finding is consistent with observations by others in claudin-5 (17) and claudin-1 (5) and suggests that the disulfide bond is not critical for global claudin protein folding.
Disulfide bond is necessary for pore function.
In our studies, the serine mutants, C54S, C64S, and C54S/C64S expressed at similar levels to wild type at tight junction, yet the transepithelial conductance and PNa+ attributable to each mutant were almost abolished. Furthermore, all three mutants lost pore function to the same extent. This suggests that the impaired pore function is likely caused not by disruption of any one individual cysteine residue but by disruption of the disulfide bond between them. We conclude that the role of the extracellular disulfide bond between the two conserved cysteines is to stabilize ECL1 in its correctly folded conformation and that this is important for many of the important functions of claudins such as paracellular pore and barrier formation and extracellular interactions with other proteins. We believe this role is generalizable to most or all claudin family members.
C54 and C64 are localized at an intermolecular interface involved in cis-interaction.
Claudins are believed to polymerize within the tight junction and to be stabilized by side-by-side interactions between claudin protomers within the same cell and same tight junction strand (cis-interaction) and by head-to-head interactions between claudins from the lateral plasma membranes of two adjacent cells (trans-interaction). One way in which we have been able to identify residues that are located at or close to an intermolecular interface between claudin proteins is by showing that they form an intermolecular disulfide bond when they are mutated to cysteine, for example, D65 in claudin-2 (3). All four of our C54 and C64 single mutants were shown to form claudin-2 homodimers linked by intermolecular disulfide bonds, suggesting that C54 and C64 are located at or close to an intermolecular interface. In the case of C54A and C64A, the dimerization presumably occurs intracellularly since these mutants did not localize at the tight junction. Since therefore there is no possibility of trans-interaction, these results strongly suggest that the two conserved cysteines are localized at the cis-interaction interface. The cysteines at the cytoplasmic end of the second transmembrane domain of claudin-2 have also been shown to be in proximity to an intermolecular interface for cis-interaction (15). These two findings suggest that claudin-2 can homodimerize within the cell, presumably as early as in the endoplasmic reticulum, via both the transmembrane and extracellular domains. Whether C54 and C64 could in addition be close to a trans-interaction interface requires further study.
Mutation of the extracellular conserved cysteines leads to familial hypomagnesaemia with hypercalciuria and nephrocalcinosis.
Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) is a rare autosomal recessive renal tubule disorder characterized by renal magnesium and calcium wasting and nephrocalcinosis. It is the result of a defect in divalent cations reabsorption in the thick ascending limb of Henle, due to claudin-16 (11) or claudin-19 mutations (9). One such mutation, claudin-16 C120R, was reported in a patient with FHHNC (12). C120 is predicted to be an extracellular cysteine in claudin-16 and is homologous to C54 in claudin-2. The finding that mutation of this extracellular cysteine can cause clinical disease supports our conclusion that the conserved cysteines of ECL1 are critical for its pore function.
In conclusion, the extracellular conserved cysteines form an intramolecular disulfide bond that is necessary for pore function in claudin-2. This finding, along with observations about the critical role of these cysteines in claudin-1 (4, 5), claudin-5 (17), and claudin-16 (12), suggests that the correct folding and hence proper function of ECL1 requires the two conserved cysteines to form an intramolecular disulfide bond. We speculate that the role of these two cysteines is to stabilize an extracellular fold that is common to all claudins.
GRANTS
This work is supported by National Institutes of Health Grants R01-DK-062283 and U01-GM-094627 (to A. Yu). The Confocal Imaging Core at Kentucky University Medical Center is supported by National Institutes of Health Grant 9P20-GM-104936 (to D. Abrahamson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.L., S.A., and A.S.Y. conception and design of research; J.L., S.A., A.L., and M.Z. performed experiments; J.L., S.A., A.L., M.Z., and A.S.Y. analyzed data; J.L., S.A., M.Z., and A.S.Y. interpreted results of experiments; J.L. and A.S.Y. prepared figures; J.L. drafted manuscript; J.L. and A.S.Y. edited and revised manuscript; J.L., S.A., M.Z., and A.S.Y. approved final version of manuscript.
REFERENCES
- 1. Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, Fromm M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci 115: 4969–4976, 2002 [DOI] [PubMed] [Google Scholar]
- 2. Angelow S, Yu AS. Cysteine mutagenesis to study the structure of claudin-2 paracellular pores. Ann NY Acad Sci 1165: 143–147, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Angelow S, Yu AS. Structure-function studies of claudin extracellular domains by cysteine-scanning mutagenesis. J Biol Chem 284: 29205–29217, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cukierman L, Meertens L, Bertaux C, Kajumo F, Dragic T. Residues in a highly conserved claudin-1 motif are required for hepatitis C virus entry and mediate the formation of cell-cell contacts. J Virol 83: 5477–5484, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis C, Harris HJ, Hu K, Drummer HE, McKeating JA, Mullins JG, Balfe P. In silico directed mutagenesis identifies the CD81/claudin-1 hepatitis C virus receptor interface. Cell Microbiol 14: 1892–1903, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Furuse M, Furuse K, Sasaki H, Tsukita S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol 153: 263–272, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci USA 107: 18010–18015, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kimizuka H, Koketsu K. Ion transport through cell membrane. J Theor Biol 6: 290–305, 1964 [DOI] [PubMed] [Google Scholar]
- 9. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, Schlingmann KP, Schmid M, Rodriguez-Soriano J, Ariceta G, Cano F, Enriquez R, Juppner H, Bakkaloglu SA, Hediger MA, Gallati S, Neuhauss SC, Nurnberg P, Weber S. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79: 949–957, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krug SM, Gunzel D, Conrad MP, Rosenthal R, Fromm A, Amasheh S, Schulzke JD, Fromm M. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell Mol Life Sci 69: 2765–2778, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285: 103–106, 1999 [DOI] [PubMed] [Google Scholar]
- 12. Staiger K, Staiger H, Haas C, Thamer C, Risler T, Machicao F, Haring HU. Hypomagnesemia and nephrocalcinosis in a patient with two heterozygous mutations in the CLDN16 gene. J Nephrol 20: 107–110, 2007 [PubMed] [Google Scholar]
- 13. Tatum R, Zhang Y, Salleng K, Lu Z, Lin JJ, Lu Q, Jeansonne BG, Ding L, Chen YH. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am J Physiol Renal Physiol 298: F24–F34, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 121: 298–305, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Van Itallie CM, Mitic LL, Anderson JM. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J Biol Chem 286: 3442–3450, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol 291: F1288–F1299, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Wen H, Watry DD, Marcondes MC, Fox HS. Selective decrease in paracellular conductance of tight junctions: role of the first extracellular domain of claudin-5. Mol Cell Biol 24: 8408–8417, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yu AS. Electrophysiological characterization of claudin ion permeability using stably transfected epithelial cell lines. Methods Mol Biol 762: 27–41, 2011 [DOI] [PubMed] [Google Scholar]
- 19. Yu AS, Cheng MH, Angelow S, Gunzel D, Kanzawa SA, Schneeberger EE, Fromm M, Coalson RD. Molecular basis for cation selectivity in claudin-2-based paracellular pores: identification of an electrostatic interaction site. J Gen Physiol 133: 111–127, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]