

Abstract
The cancer stem cell marker, EpCAM, is an important indicator of wnt-β-catenin signaling activation and a functional component of hepatocellular tumor initiating cells. A high-throughput screening assay was developed to identify inhibitors of EpCAM-dependent growth of hepatocellular carcinoma cells. EpCAM(+) and EpCAM(−) HCC cell lines were assessed for differential sensitivity to a wnt-β-catenin pathway inhibitor. Libraries comprising 22,668 pure compounds and 107,741 crude or partially purified natural product extracts were tested and 12 pure compounds and 67 natural product extracts were identified for further study. Three active compounds and the positive control were further characterized in terms of effects on EpCAM expression. Treatment of EpCAM(+) Hep3B cells resulted in loss of EpCAM expression as assessed by flow cytometry. This reduction was incomplete (most cells continued to express EpCAM), but resulted in generation of cell populations expressing lower levels of EpCAM. Sublethal concentrations (~IC50) reduced median EpCAM expression to 28% of control after 1 d and 19% of control after 2 d. Reduction in EpCAM expression preceded growth inhibition suggesting that a threshold of EpCAM expression may be required for growth of EpCAM-dependent cells. The identification of compounds with a variety of possible molecular targets suggests a likelihood of multiple mechanisms for modulation of EpCAM-dependent cell growth.
Keywords: Hepatocellular carcinoma (HCC), EpCAM (epithelial cell adhesion molecule), wnt/β-catenin, high-throughput screening (HTS)
INTRODUCTION
Dysregulation of wnt/β-catenin signaling has been implicated in development of hepatocellular carcinoma (HCC) (1). Development of wnt pathway inhibitors has been a significant research focus for HCC and a variety of other cancer types (2-6). Recently, epithelial cell adhesion molecule (EpCAM), originally described in the 1970s as a colon carcinoma cell antigen (7), has been identified as a marker of HCC stem cells (or tumor initiating cells) (8-10). EpCAM has been shown to be a direct transcriptional target of wnt/β-catenin signaling in HCC (11), and thus a possible target for development of HCC therapeutics (12). Although the function of EpCAM remains uncertain, various lines of evidence suggest its involvement in cell adhesion, mitogenic signaling, cell migration, proliferation, and differentiation (9-14). EpCAM expression has been implicated in tumor development in a variety of contexts, including HCCs. In addition to having its expression regulated by wnt/β-catenin signaling, EpCAM itself is a signal transduction molecule (9). Experimentally, EpCAM(+) HCC cells have been shown to be much more efficient at forming highly invasive tumors in mice than are EpCAM(−) HCC cells and blocking EpCAM expression with siRNA reduced tumor formation by EpCAM(+) cells (8, 15). Similar results have been obtained with several other carcinomas (13). In culture, growth of EpCAM(+) HCC cell lines are dependent on maintenance of wnt/β-catenin signaling as well as on continued expression of EpCAM (8, 15). As a result, inhibition of either wnt/β-catenin signaling or of EpCAM expression or function may be expected to be growth inhibitory for EpCAM(+) cells but not for EpCAM(−) cells. This was confirmed by use of several inhibitors of wnt/β-catenin signaling (8). These observations now form the basis for development of a high-throughput assay for identification of potential inhibitors of EpCAM expression and/or function by assessing differential growth inhibition of EpCAM(+) and EpCAM(−) HCC cell lines. The assay has been optimized to screen both pure compound libraries and natural product extracts and has resulted in identification of a number of apparent EpCAM-dependent HCC growth inhibitors. Further evaluation suggests a correlation between growth inhibition and modulation of EpCAM expression.
MATERIALS AND METHODS
Materials
All cell culture components were from Invitrogen (Carlsbad, CA). Clear 384-well tissue culture-treated assay plates were from Perkin-Elmer (Waltham, MA). 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide (XTT) and natural products were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Frederick, MD). Natural product extracts were obtained from the Natural Products Support Group of the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Frederick, MD). Available confirmed active compounds were either obtained from the original supplier or were purchased from Sigma (St. Louis, MO). Maintenance and characteristics of HCC cell lines (Hep3B, HepG2, MHCC97, and SK-Hep1) are described in previous work (11).
Control compound
EpCAM(+) HCC cell lines have previously been shown to be more sensitive to growth inhibition by wnt pathway inhibitors than are EpCAM(−) HCC cells (11). A wnt pathway inhibitor, AV606 (kindly provided by Avalon Pharmaceuticals, Germantown, MD), was therefore used as a control in the development of the screening assay. In preliminary experiments this compound gave similar results when compared to inhibitors already cited (11) (data not shown). Based on these results and availability of sufficient quantities of material, this compound was used for assay development for screening. Further characterization with regard to EpCAM expression and EpCAM-dependent HCC growth is discussed in the present report.
Differential growth inhibition assay
EpCAM(−) and EpCAM(+) HCC cell lines were maintained in MEM supplemented with 10% fetal bovine serum. Subconfluent cells were harvested by trypsinization, resuspended in the same medium, counted, and transferred to 384-well tissue-culture treated plates (45 μl per well) using a μFill microplate liquid dispenser (BioTek, Winooski, VT). Cells were allowed to attach overnight after which test or control samples were added (prediluted to 10 x final concentration in PBS). After 1-4 days further incubation, cell numbers were estimated by XTT assay (16). For screening, cells were plated at 2000-3000 cells/well based on results of optimization and XTT response. Pure compound libraries (both synthetic and pure natural products) were assessed at 1 and 10 μM for their ability to inhibit growth of Hep3B and SK-Hep1 cells. In total, 22,668 samples from a variety of sources were screened. Natural product extracts and partially-purified extracts were tested at 10 μg/ml (107,741 total samples).
Flow cytometry
For flow cytometry applications, EpCAM(+) Hep3B cells were treated for 1 d or 2 d , harvested using PBS/5 mM EDTA and stained with APC-labeled anti-EpCAM (clone EBA-1, B-D Biosciences, San Jose, CA) and/or FITC-labeled anti-human epithelial antigen (clone Ber-EP4, Dako, Carpenteria, CA). Trypsin was avoided since results of preliminary experiments (not shown) suggested that trypsinization reduced EpCAM detection. Data were acquired on an accuri C6 flow cytometer and analyzed using the instrument’s CFlow software (Accuri Cytometers, Ann Arbor, MI).
Data analysis
All XTT-derived cell numbers were normalized to untreated controls on the same plates. IC50 values were calculated using SigmaPlot (SPSS, Inc., Chicago, IL) 4-parameter logistic nonlinear regression analysis. “Hit” identification was based on inhibition of EpCAM(+) cells and lack of inhibition of EpCAM(−) cells as discussed in the Results and Discussion sections. Confirmation of active samples was assessed by re-assaying each putative “hit” in quadruplicate followed by calculation of confidence intervals. A confirmed hit was defined as a sample that gave < 50% survival of EpCAM(+) Hep3B cells and > 50% survival of EpCAM(−) SK-Hep1 cells after 2 d treatment, each at a 95% confidence interval. Furthermore, hit confirmation required that the difference in effect between the two cell lines was > 50%. Upon confirmation active compounds were resupplied, whenever possible from new lots of material, and were assessed in a dose-response format using the XTT assay and/or an alternative assay, monitoring viable cell numbers using calcein AM (Invitrogen, Carlsbad, CA).
RESULTS
Choice of cell lines for screening
The suitability for HTS of several HCC cell lines previously shown to differ in their expression of EpCAM(11) was investigated. EpCAM(+) cell lines, Hep3B and HepG2, and EpCAM(−) cell lines, SK-Hep1 and MHCC97, were assessed for their growth characteristics and response to the positive control compound AV606. In order to simplify experimental design and data analysis in comparing multiple cell lines, it is desirable for the cells to have roughly comparable growth characteristics (e.g. doubling times, required cell densities for XTT analysis, etc.). Growth curves showed population doubling times to be similar for Hep3B, HepG2, and SK-Hep1 (16.0, 13.9, and 19.9 h respectively) whereas MHCC97 population doubling time was 36.1 h. Similarly, XTT signal for MHCC97 cells only reached 2 x background (no cells) after 2 d incubation. By contrast the other cell lines gave XTT signals of 15-25 x background, depending on cell density. By these criteria, the MHCC97 cell line was excluded from further analysis.
In a preliminary experiment with the remaining cell lines (Hep3B, HepG2, and SK-Hep1), the conditions under which they were previously shown to be differentially sensitive to wnt inhibitors(11) were recapitulated, albeit with a different inhibitor and in 384-well rather than 96-well plates. Cell densities were varied in this experiment in order to assess whether this would be relevant to the screening assay. Table 1 lists the IC50 values calculated from dose-response curves (not shown) for the three cell lines after 3 d incubation with compound (to match the time-course in the reference cited). Hep3B and SK-Hep1 gave the greatest differential sensitivity to the wnt pathway inhibitor and so, given the goals of the project, these cell lines were used for subsequent assay development and for screening. In contrast to results obtained with Hep3B cells, the other EpCAM(+) cell line tested (HepG2) showed a significant deterioration in response with time. At 2 d, IC50 for AV606 with HepG2 cells was 1.01 μM, but increased to 6.7 μM at 3 d (2000 cells, Table 1). Given that results with Hep3B cells were more robust with regard to cell density and incubation time, they were more suitable for HTS.
TABLE 1.
Differential sensitivity of 3 hepatocellular carcinoma cell lines to a wnt inhibitor
| Sensitivity to wnt inhibitor (IC50, μM, average, n = 2) | |||
|---|---|---|---|
| Cell density (cells/well) | Hep3B | HepG2 | SK-Hep1 |
| 1000 | 0.37 | 11.4 | 8.5 |
| 2000 | 0.83 | 6.7 | 12.9 |
| 5000 | 1.01 | 14.1 | 29.3 |
Assay optimization
The assay was optimized to maximize the effect of the wnt pathway inhibitor on EpCAM(+) Hep3B cells while also maximizing the differential sensitivity between these cells and the relatively resistant EpCAM(−) SK-Hep1 cells. For this assay, XTT absorbance values of 1.5-2.0 (for untreated cells) are ideal as is a signal/background (S/B) ratio of > ~4 in order to obtain a Z’ of > 0.5 for the positive control compound. Variation in cell densities suggested that sufficient XTT signal and maximal differential responses were obtained with 1000-5000 cells/well (Table 1, Figure 1, and data not shown), although SK-Hep1 cells may have somewhat increased resistance at high density (5000 cells/well). A 2 d incubation provided maximal response and differential. After 3d, the resistant SK-Hep1 cells began to show sensitivity and the differential was diminished (Figure 1). IC50 values after 2 d were consistent across cell densities (1000-5000 cells/well) and averaged 0.92 ± 0.30 μM for Hep3B and 15.1 ± 1.6 μM for SK-Hep1. By 6 d, significant edge effects were observed (data not shown). For screening, therefore, 2000-3000 cells/well and a 2 d incubation time were identified as optimal. Under these conditions, S/B ranged from 15-20 and Z’ averaged 0.77 (for 10 μM AV606, Hep3B cells). To ensure that a 50% inhibition value was statistically significant (i.e., > 3 standard deviations from untreated control), CVs were required to be < 17%. The average CVs from screening plates were 8-9% across 1162 primary assay plates.
Figure 1.

Effect of treatment time and cell numbers on sensitivity of HCC cell lines to wnt pathway inhibition
Hep3B or SK-Hep1 cells were plated in 384-well tissue culture treated plates at 1000 or 2000 cells per well. After overnight attachment, the wnt pathway inhibitor, A606, was added (10 μM final concentration). Following 2d (panel A) or 3d (panel B) incubation, cell numbers were assessed using the XTT assay and normalized to untreated controls wells.
Open bars: 1000 cells/well, gray bars: 2000 cells/well. Error bars represent standard deviation (n = 3 plates, duplicate wells per plate).
Assay reproducibility
Identification of novel natural products as modulators of EpCAM-dependent cell growth is a future goal of this project. Past experience has shown that natural product extracts show a wide range of growth inhibitory activities against a variety of cancer cell lines. Therefore, reproducibility of the differential growth inhibition assay was assessed using a selection of 1408 extracts representing a wide range of taxonomic groups. The assay was repeated multiple times over multiple days at 1, 10, and 25 μg/ml. The results were assessed in terms of experiment-to-experiment reproducibility for each extract as well as for estimation of the rates of false conclusions regarding growth inhibition. Table 2 summarizes the results of multiple reassay of these samples with the two cell lines (Hep3B and SK-Hep1). The average variation for each cell line is in the 10-20% range (CVs). For analysis of identification of inhibitory extracts, the average effect across the multiple repeats was taken to represent the “true” value for the sample. Variation from the mean population response by > 3 standard deviations was taken to be a significant effect, i.e., growth inhibitory activity was considered to be significant if the treatment resulted in < 40% cell survival (compared to untreated control). A “false” reading was indicated if the result of an individual repeat resulted in significant inhibition while the average for multiple reads of the same sample indicated insignificant activity, and vice versa. Table 3 shows false positive and false negative rates estimated from repeated sampling of the 1408 extracts tested at 10 and 25 μg/ml. Data from cells treated with 1 μg/ml were insufficient for this analysis since no extracts significantly inhibited growth of either cell line at this concentration. Identification of false “hits” would combine false positive rates for Hep3B growth inhibition with false negative rates for SK-Hep1 inhibition and is further discussed below in the context of screening data. At 25 μg/ml, only 5 “true” hits were identified and at 10 μg/ml, only 1. Thus, meaningful analysis of overall false negative rates was not possible.
TABLE 2.
Reproducibility of cell growth inhibition by natural product extracts
| average CV (n = 3-4) a | |||
|---|---|---|---|
| Cell line | 1 μg/ml | 10 μg/ml | 25 μg/ml |
| Hep3B | 9.4% | 11.5% | 22.2% |
| SK-Hep1 | 13.3% | 13.8% | 23.8% |
CV (coefficient of variation, sd/ave x 100%) = average of CVs for each of 1408 individual samples.
TABLE 3.
Error rates predicted from multiple repeat assays
| growth inhibitory samples a | inactive samples b | |||||||
|---|---|---|---|---|---|---|---|---|
| Cell line | 10 μg/ml | 25 μg/ml | 10 μg/ml | 25 μg/ml | ||||
| true c | false d | true c | false d | true c | false d | true c | false d | |
| Hep3B | 0.62% | 0.21% | 1.79 | 0.43 | 99.0 | 0.17 | 97.7 | 0.05 |
| SK-Hep1 | 1.04 | 0.07 | 1.18 | 0.43 | 98.8 | 0.09 | 98.3 | 0.06 |
growth inhibitory samples are defined as those giving > 3 sd variation from overall average (across all plates, all samples at the same concentration).
inactive samples are defined as those giving < 3 sd variation from overall average.
True = % of individual assay reads correctly scored as “inhibitory” (i.e., the average of all repeats for that sample indicated significant inhibition) or “inactive.”
False = % of individual assay reads incorrectly scored as “inhibitory” (i.e., the average of all repeats for that sample was not inhibitory) or “inactive.”
Application to high-throughput screening
The differential growth inhibition assay was applied to libraries of pure compounds from a variety of sources and resulted in a total of 12 confirmed active compounds. The other hit compounds did not give a significantly differential growth inhibition effect in a dose-response format or were unavailable for resupply. Application of the assay to libraries of natural product extracts (totaling 107,741 samples) identified 67 confirmed, differentially-active samples.
False positive rates in screening
The goal of the screen is to identify samples that differentially affect growth of EpCAM(−) and EpCAM(+) HCCs. Thus, a “false positive” would result from identifying an inactive sample as growth inhibitory for Hep3B cells and/or identifying a growth inhibitory sample as inactive for SK-Hep1 cells. Based on repeatability experiments using extracts (Table 3) and expected statistical variation based on > 3 sd as a cutoff for a meaningful change, up to 0.4% of inactive sample might be expected to score as inhibitors of Hep3B cells and up to 0.06% of SK-Hep1 growth inhibitors might be expected to be inaccurately identified as inactive against SK-Hep1 cells. This was borne out for screening by analysis of hit confirmation/reassay data for pure compounds. Of 22,668 pure compounds screened, 92 (or 0.36%) were incorrectly identified as Hep3B inhibitors while 6 (or 0.02%) were inaccurately identified as inactive against SK-Hep1 cells. In general, the false positive samples were borderline hits (close to the 50% threshold) and hit confirmation criteria are quite stringent (95% confidence interval analysis upon reassay). “False negative” analysis (i.e., identification of inactive compounds that “should” have shown activity) is significantly more difficult and no analysis of screening results was attempted.
Characterization of selected active compounds
Three of the confirmed active compounds from the screening libraries were chosen for further analysis. These compounds have reported putative molecular targets (see Discussion section). Table 4 shows the effects of these compounds and the control compound on the four cell lines used in assay development. Use of an alternative cell quantitation assay (calcein) in these experiments corroborated XTT results. Previous results (8, 11) suggested that “stemness,” invasiveness, tumorigenicity, and proliferation of EpCAM(+) HCCs are dependent on EpCAM expression. Therefore, the confirmed active hit compounds were further assessed for their effects on EpCAM expression in Hep3B cells. Flow cytometry optimization experiments indicated that anti-EpCAM (clone EBA-1) and anti-human epithelial antigen (clone Ber-EP4) gave similar results with Hep3B cells and confirmed the EpCAM(−) status of SK-Hep1 cells (data not shown). Figure 2 shows histograms of Hep3B cell staining by anti-EpCAM (APC-labeled clone EBA-1) after 2 d treatment with the selected compounds. A clear shift in expression patterns can be observed for each compound after 1 d (not shown) or 2 d (Figure 2A-E). Quantitation of antibody binding by either % of cells that were EpCAM positive or by assessing the median cell signal is shown in Figure 2F (normalized to untreated control cells which were 79.7 ± 1.7% EBA-1-positive and 79.3 ± 3.6% Ber-EP4-positive). Although the proportion of cells expressing detectable EpCAM changed only modestly (average for all treatments: 67.8% EpCAM positive after 1 d, 59.6% after 2 d), a very large decrease in median expression was observed (i.e. accumulation of cells expressing lower levels of EpCAM). These experiments used the compounds at roughly their IC50 concentrations (1 μM for AV606, 10 μM for the others). Consistent with these concentrations and the time-course results obtained during assay development (Figure 1), when recovered cells were counted, little change was observed after 1 d (average 87.2 ± 7.5% of control), while cell numbers were reduced to 66.7 ± 11.4% of control after 2 d. Thus, reduction in median expression of EpCAM preceded detection of significant reduction in cell numbers by at least 1 d. Similarly, at 24 h, these compounds had insignificant effects on DNA fragmentation (using Roche Cell Death ELISA – data not shown). Finally, EpCAM-dependence of the differential cellular effects of hit compounds was further corroborated by use of a compound (doxorubicin) that showed non-specific growth inhibition/cytotoxicity in the screening assay. At 1 μM (~IC50), doxorubicin reduced Hep3B cell numbers to 52.5% of control after 2 d incubation. However, by contrast to differentially active compounds (Figure 2), after 2 d, median EpCAM expression of surviving cells was unaffected (106.3% of control) suggesting that initiation of cell death does not necessarily result in decrease in EpCAM expression.
TABLE 4.
Growth inhibition of HCC cells by selected hit compounds
| IC50 (μM) a | ||||
|---|---|---|---|---|
| Compound | Hep3B | SK-Hep1 | HepG2 | MHCC97 |
| AV606 | 0.92 ± 0.30 | 15.1 ± 1.6 | 0.89 ± 0.13b | NDc |
| Fiduxosin | 13.9 ± 5.3 | > 40 | 17.9 ± 2.8 | > 40 |
| NSC45291 | 6.8 ± 2.3 | > 40 | > 40 | > 40 |
| Pimozide | 5.0 ± 0.4 | 19.9 ± 4.3 | 5.6 ± 0.8 | 10.8 ±0.08 |
concentration giving half-maximal growth inhibition (average ± sd, n = 3 plates, duplicate wells per plate). Highest concentration tested was 40 μM
range (n = 2)
not determined
Figure 2.
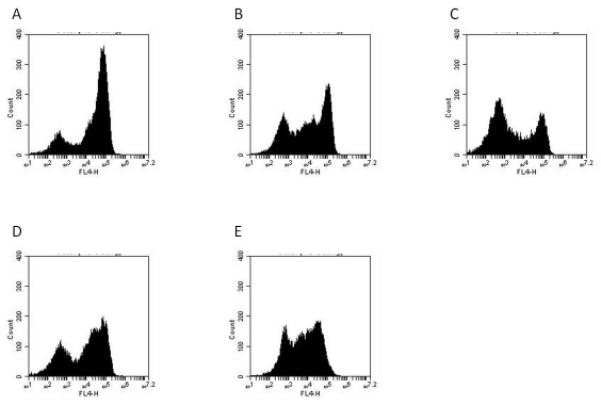

Effect of active compounds on EpCAM expression in Hep3B cells.
Cells were treated for 1-2 d with the indicated compound followed by harvesting as assessment of EpCAM expression by flow cytometry using APC-labeled anti-EpCAM (clone EBA-1).
A: untreated, B: AV606 (1 μM, 2 d), C: fiduxosin (10 μM, 2 d), D: NSC45291(10 μM, 2 d), E: pimozide (10 μM, 2 d).
“FL4-H” = fluorescence intensity (APC channel).
In panel F, data from flow cytometry experiments were analyzed for % of cells expressing any EpCAM and for median expression of EpCAM in the cell population. All results were normalized to the value for control (untreated) cells and presented at treated/control ratio (T/C).
Error bars represent sd or range (n = 2-4).
Open bars: total EpCAM-positive cells, 1 d incubation with compounds
Light gray bars: total EpCAM-positive cells, 2 d incubation with compounds
Dark gray bars: median EpCAM expression, 1 d incubation with compounds
Black bars: median EpCAM expression, 2 d incubation with compounds.
*p < 0.05, students t-test. For 2 d, data are insufficient for t-test analysis.
DISCUSSION
A differential growth inhibition assay has been developed for identification of substances able to target EpCAM/β-catenin signaling in hepatocarcinoma cell lines. In order to simplify and speed development and screening, two cell lines were chosen based on their similar growth characteristics, XTT color development, and maximal differential response to a model compound. As might be expected from such an assay, the overall confirmed hit rate was very low (< 0.1% for both pure compounds and natural product extracts). The vast majority (> 94%) of samples that inhibited the growth of the EpCAM(+) Hep3B cells also inhibited growth of the EpCAM(−) SK-Hep1 cells. Validity of use of the XTT assay was corroborated by inclusion of an alternative cell quantitation method and by cell counts in preparation for flow cytometry experiments.
In order to increase the probability that resultant “hits” are selective for EpCAM/β-catenin-expressing cells, after confirmation by multiple reassay, compounds were assessed against 4 cell lines (two EpCAM/β-catenin positive, two negative) in a dose-response format. The results of the screening/reassay/dose-response assay process for pure compound libraries provided several examples of potentially interesting modulators of EpCAM-dependent HCC cell growth. The numbers obtained also require attention to an aspect of screening that is not always discussed, that of apparent loss of activity upon resupply of compounds. For this assay, before dose-response assays were performed, compounds were either freshly prepared from archived stock aliquots or newly provided by the original suppliers. In a number of cases, the resulting material did not show differential activity in a dose-response format. Several possibilities could account for this observation. The cell lines themselves could be unstable. The entire screening campaign, including lead times for compound resupply, stretched over several months. It is possible that the cells underwent changes in culture that changed their relative sensitivity to the compounds. This is unlikely given that several other of the compounds continued to show significant and reproducible differential activities at multiple concentrations using fresh materials (Figure 2 for example). Although not included in every screening plate, the model wnt pathway inhibitor AV606 used in assay development was included as a control in the dose-response assays and continued to differentially affect EpCAM(+) and EpCAM(−) cells (data not shown). Because samples in screening plates are maintained in DMSO (at −20° C) and subjected to freeze-thaw cycles (albeit minimal based on storage aliquots, etc.), they could be breaking down in the freezers. Thus, the active material could be a degradation product. The purity of the compound and identity of possible contaminants in newly prepared samples could be different. When compounds are confirmed active by reassay, if they cannot be resynthesized or resupplied, their purity and identity are confirmed by HPLC, MS, and NMR analysis as appropriate. It should also be noted that in screening at only one or two concentrations, where there is a narrow window of differential activity, the possibility arises (and has been seen – data not shown) that a “fortuitous” concentration was chosen for the screen and that even minor variation from that concentration eliminates the differential activity. Any of these could easily affect the results and/or result in apparently active compounds not providing a significant differential activity in a dose-response format. Finally, the nature of the assay itself contributes to false positive rates in that relatively long incubation times (days) in cell-based assays often lead to lack of repeatability. This issues are compounded by the requirement in this assay that both cell lines give highly reproducible results.
In the case of extracts, preliminary dose-response analysis (using Hep3B and SK-Hep1 cells and the XTT assay only) suggested that 20-30 of the confirmed active extracts continued to be considered true actives in that they showed clear growth inhibitory activity (< 25% recovery of cells compared to control) against Hep3B cells and significant differential (>50%) compared to SK-Hep1 cells (data not shown). Analysis of the data obtained with natural product extracts revealed no patterns of activity with regard to taxonomy or type of extract (not shown). Similarly, confirmed active samples included extracts derived from both marine organisms and terrestrial plants (which together comprise a large majority of the samples tested). Within these broad categories, no patterns were observed.
At least three interesting compounds have been identified as putative modulators of EpCAM-specific cell growth (Table 4). These include pimozide, a dopamine antagonist recently identified as a STAT5 inhibitor (17), fiduxosin, an α adrenoceptor antagonist, and NSC45291 (N-phenyl-2-pyridinecarbothioamide), identified in the PubChem database as a possible inhibitor of the RORγ transcription factor (see http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=2551&preview=f). The structures of these three compounds are shown in Figure 3. Connections in the literature have been reported between wnt/β-catenin signaling components and/or EpCAM and STAT5 (18, 19) as well as RORγ (20). Connection to alpha adrenergic signaling is much more tenuous. Investigation of these possible connections as well as a more thorough analysis of molecular mechanisms of action for these compounds and any that are derived from natural product extracts will be an important future activity. For the purposes of screening, the connection to EpCAM expression (regardless of mechanism) has been established by the data presented in this report.
Figure 3.

Structures of selected confirmed active compounds.
Further assessment of these compounds and the control AV606 suggest that their differential activity is a result of effects on EpCAM expression. Flow cytometry results indicate that a reduction in EpCAM expression precedes cellular toxicity effects suggesting EpCAM dependence for cell growth and for the effects of the compounds. This is consistent with results previously reported (8, 11). It is particularly interesting to note that the changes in EpCAM expression observed (Figures 2) are not an all or none phenomenon. Instead, appearance of cells expressing intermediate levels of EpCAM suggest that there may be a threshold of EpCAM expression required for maintenance of cell viability and proliferative capacity in EpCAM-dependent cells. Further investigation of the effects of these compounds on EpCAM-dependent HCC lines should be useful for clarification of this issue and implications for maintenance of cancer stem cell function as well as mechanisms of cell death as a result of EpCAM down-regulation. Although, as noted, the three compounds further assessed appear to target widely divergent pathways, there are possible wnt/β-catenin connections.
An important consideration for characterization of active compounds from any cell-based HTS assay is the multidrug resistance (MDR) status of the cells being employed. Three of the cell lines used in this report, Hep3B, HepG2, and SK-Hep1, have been assessed for expression of p-glycoprotein (Pgp, the major contributor to most multidrug resistance) (21). The EpCAM(+) lines (Hep3B and HepG2) were also Pgp-positive whereas no Pgp expression was detected in SK-Hep1 cells. The expression was somewhat heterogeneous in cell populations. This would not account for relative resistance of EpCAM(−) cells to the compounds identified as hits.
Interestingly, NSC45291 did not affect growth of EpCAM(−) cells and inhibited growth of the EpCAM(+) Hep3B line, but did not affect the other EpCAM(+) line, HepG2. The target of this compound may be differentially expressed in these two lines, or HepG2 cells may be relatively more resistant based on higher Pgp expression (21). Multiple subsets of EpCAM(+) cells may exist and it may be possible to differentially target EpCAM(+) subpopulations.
In addition to these approaches, signaling by EpCAM itself was investigated using two compounds reported to inhibit EpCAM nuclear signaling (22). These compounds, a γ-secretase inhibitor (compound E) and a TACE inhibitor (TAPI) had no growth inhibitory or cytotoxic effect up to 40 μM (data not shown). Thus, signaling by EpCAM itself is probably not relevant in these assays.
CONCLUSIONS AND FUTURE DIRECTIONS
The identification of compounds that differentially affect EpCAM(+) HCCS and have multiple putative targets suggests that a range of molecular targets may be available for modulation of EpCAM-dependent cell growth. As other compounds, without known targets, are assessed, and as active components are identified from natural product extracts, the range of potential targets may well increase significantly providing multiple pathways to EpCAM modulation.
ACKNOWLEDGEMENTS
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
This research was supported in part by the Intramural Research Program of NIH, Frederick National Lab, the Center for Cancer Research, the National Cancer Institute (Z01 BC 010876 and ZIC BC 010469).
REFERENCES
- 1.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 2.Barker N, Clevers H. Mining the wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 3.Trosset JY, Calvit C, Knapp S, Fasolini M, Veronesi M, Mantegani S, Gianellini LM, Catana C, Sunstrom M, Stouten PF, Moll JK. Inhibition of protein-protein interactins: the discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins. 2006;64:60–67. doi: 10.1002/prot.20955. [DOI] [PubMed] [Google Scholar]
- 4.Kawamoto SA, Thompson AD, Coleska A, Nikolovska-Coleska Z, Yi H, Wang S. Analysis of the interaction of BCL9 with beta-catenin and development of fluorescence polarization and surface plasmon resonance binding assays for this interaction. Biochemistry. 2009;48:9534–9541. doi: 10.1021/bi900770z. [DOI] [PubMed] [Google Scholar]
- 5.Ewan K, Pajak B, Stubbs M, Todo H, Barbeau O, Quevedo C, Botfield H, Young R, Ruddle R, Samuel L, Battersby A, Raynaud F, Allen N, Wilson S, Latinkic B, Workman P, McDonald E, Blagg J, Aherne W, Dale T. A useful approach to identify novel small-molecule inhibitors of wnt-dependent transctiption. Cancer Res. 2010;70:5963–5973. doi: 10.1158/0008-5472.CAN-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Small-molecule antagonists of the oncogenic Tcr/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 7.Herlyn M, Steplewski Z, Herlyn D, Koprowski H. Colorectal carcinoma-specific antigen: detection by means of monoclonal antibodies. Proc Natl Acad Sci USA. 1979;76:1438–1442. doi: 10.1073/pnas.76.3.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang H-Y, Jia H, Ye Q, Qin L-X, Wauthier E, Reid LM, Minato H, Honda M, Kaneko S, Tang Z-Y, Wang XW. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69:5627–5629. doi: 10.1158/0008-5472.CAN-09-0654. [DOI] [PubMed] [Google Scholar]
- 10.Terris B, Carvard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatology. 2010;52:280–281. doi: 10.1016/j.jhep.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67:10831–10839. doi: 10.1158/0008-5472.CAN-07-0908. [DOI] [PubMed] [Google Scholar]
- 12.Oishi N, Wang XW. Novel therapeutic strategies for targeting liver cancer stem cells. Int J Bio Sci. 2011;7:517–535. doi: 10.7150/ijbs.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Gun BTF, Melchers LJ, Ruiters MHJ, de Leij LFMH, McLaughlin PMJ, Rots MG. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis. 2010;31:1913–1921. doi: 10.1093/carcin/bgq187. [DOI] [PubMed] [Google Scholar]
- 14.Patriarca C, Macchi RM, Marschner AK, Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treatment Reviews. 2012;38:68–75. doi: 10.1016/j.ctrv.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Kimura O, Takahashi T, Ishii N, Inoue Y, Ueno Y, Kogure T, Fukushima K, Shiina M, Yamagiwa Y, Kondo Y, Inoue J, Kakazu E, Iwasaki T, Kawagishi N, Shimosegawa T, Sugamura K. Characterization of epithelial cell adhesion molecule (EpCAM)+ cell population in hepatocellular carcinoma cell lines. Cancer Sci. 2010;101:2145–2155. doi: 10.1111/j.1349-7006.2010.01661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–4833. [PubMed] [Google Scholar]
- 17.Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgarrd JL, Santo L, Addorio MR, Ebert BL, Griffin JD, Frank DA. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117:3421–3429. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sahm C, Schonfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61:1451–1461. doi: 10.1007/s00262-012-1212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muraoka-Cook RS, Sandahl MA, Strunk KE, Miraglia LC, Husted C, Hunter DM, Elenius K, Chodosh LA, Earp HS., 3rd ErbB4 splice variants Cyt1 and Cyt2 differ by 16 amino acids and exert opposing effects on the mammary epithelium in vivo. Mol Cell Biol. 2009;29:4935–4948. doi: 10.1128/MCB.01705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R, Xie H, Huang Z, Ma J, Fang X, Ding Y, Sun Z. T cell factor 1 regulates thymocyte survival via a RORγt-dependent pathway. J Immunol. 2011;187:5964–5973. doi: 10.4049/jimmunol.1101205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong AW, Su D, Mues G, Tillery GW, Goldstein R, Klintmalm G, Stone MJ. Chemosensitization of human hepatocellular carcinoma cells with cyclosporine A in post-liver transplant patient plasma. Clin Cancer Res. 1996;2:531–539. [PubMed] [Google Scholar]
- 22.Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, Gires O. Nuclear signaling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11:162–171. doi: 10.1038/ncb1824. [DOI] [PubMed] [Google Scholar]