

Abstract
A serine-threonine protein kinase, WNK4, reduces Na+ reabsorption and K+ secretion in the distal convoluted tubule by reducing trafficking of the thiazide-sensitive Na-Cl cotransporter to and enhancing renal outer medullary potassium channel retrieval from the apical membrane. Epithelial sodium channels (ENaC) in the distal nephron also play a role in regulating Na+ reabsorption and are also regulated by WNK4, but the mechanism is unclear. In A6 distal nephron cells, transepithelial current measurement and single channel recording show that WNK4 inhibits ENaC activity. Analysis of the number of channel per patch shows that WNK4 reduces channel number but has no effect on channel open probability. Western blots of apical and total ENaC provide additional evidence that WNK4 reduces apical as well as total ENaC expression. WNK4 enhances ENaC internalization independent of Nedd4-2-mediated ENaC ubiquitination. WNK4 also reduced the amount of ENaC available for recycling but has no effect on the rate of transepithelial current increase to forskolin. In contrast, Nedd4-2 not only reduced ENaC in the recycling pool but also decreased the rate of increase of current after forskolin. WNK4 associates with wild-type as well as Liddle's mutated ENaC, and WNK4 reduces both wild-type and mutated ENaC expressed in HEK293 cells.
Keywords: WNK4, ENaC, Nedd4-2, ubiquitination
epithelial Na channels (ENaC) are channels located in the apical membrane that are found primarily in the polarized epithelia in distal nephron, lung, distal colon, and a few other organs. In the distal nephron, the activity of ENaC is rate limiting for Na+ reabsorption (19, 31); therefore, ENaC activity is critical in the physiological maintenance of systematic Na+ homeostasis and the control of long-term blood pressure. ENaC is composed of three subunits, α, β, and γ, and its activity is regulated by either changing channel surface expression or channel gating (4, 7, 37, 42, 43). ENaC surface expression is mainly regulated by the two natriferic hormones, vasopressin and aldosterone. Vasopressin-induced c-AMP/PKA signaling stimulates ENaC surface insertion from a recycling pool (4, 36). Aldosterone stimulates ENaC activity in several ways, one of which is through an increase in SGK1 expression, which prevents Nedd4-2-mediated ENaC internalization, thereby increasing the surface density of functional ENaC (13, 28, 59). Mutations of the C terminus of β- and γ-ENaC at their PPxY motif prevent Nedd4-2 binding and result in a decrease in ENaC internalization from the surface and lead to Liddle's syndrome, a disease characterized by hypertension and hypokalemia (22, 50). However, there are reports that aldosterone and SGK1 stimulate ENaC independently of Nedd4-2, i.e., ENaC in Liddle's syndrome mice retain the ability to respond, at least partially, to aldosterone (3, 12).
Recently, a unique family of serine-threonine protein kinases known as WNKs [with no lysine (K) kinases] have been implicated in controlling the ionic permeability of epithelial tissues. Two members of the family, WNK1 and WNK4, are genetically linked to a rare type of hypertension, pseudohypoaldosteronism type 2 (PHA2), a disease characterized by hypertension, hyperkalemia, and metabolic acidosis (29, 53). WNK kinases constitute a novel signaling pathway, but one that appears to be modulated by aldosterone in its overall role of regulating electrolyte homeostasis. Both WNK1 and WNK4 are associated with SGK1; WNK1 can phosphorylate and activate SGK1 and therefore stimulate ENaC activity (9, 54), while WNK4 and SGK1 are able to phosphorylate each other (24, 46). WNK kinases act at two sites in the nephron to regulate electrolyte balance: on Na-Cl cotransporter (NCC) and potassium channels in the apical membrane of the distal convoluted tubule (34, 40) and on ENaC and potassium channels in the connecting tubule and collecting duct (2, 17, 54). However, there have been contrary reports about the effects of WNK4 on ENaC activity. When WNK4 was coexpressed with ENaC in Xenopus oocytes, it strongly inhibits ENaC activity and this inhibition was eliminated when WNK4 is mutated to WNK4S1169D, which mimics the phosphorylation produced by SGK1 (45, 46). However, similar to other WNKs, WNK4 increases the amiloride-sensitive Na current when it was coexpressed with ENaC in HEK 293 cells (24). These contrary observations led us to explore the effect of WNK4 on endogenously expressed ENaC function as well as the mechanism by which WNK4 regulates ENaC. We have found that WNK4 decreases ENaC activity and surface expression in A6 cells. In addition, we show that WNK4 reduces ENaC expression independent of Nedd4-2-mediated ENaC ubiqutination.
EXPERIMENTAL PROCEDURES
Plasmids.
Human wild-type WNK4 in pCMV-Myc vector was previously generated (5, 60). For patch-clamp recordings, the myc-tagged WNK4 was subcloned into one cloning site of the pIRES-GFP vector (biscistronic vector containing GFP reporter gene at one locus; Clontech). Human wild-type as well as PY-motif-mutated ENaC were kind gifts from Dr. Peter M. Snyder at the University of Iowa, and both wild-type and mutated α-, β-, and γ-ENaC subunits were subcloned into the p3XFLAG-CMV vector (Sigma-Aldrich) with flag fused at the N terminus of these genes.
Cell cultural and transfection.
2F3 subclone of A6 cells (obtained from Dr. Dale Benos, University of Alabama) were maintained in plastic tissue culture flasks using standard tissue culture techniques and seeded on permeable supports (Transwell polyester membrane with a pore size of 0.4 μm; Nalge Nunc) as described previously (57). All experiments were performed on cells between passages 100 and 115. For permanent gene transfection, 2F3 cells were seeded in 100-mm tissue-culture petri-dishes, and at 70% confluency, 10 μg of pIRES-GFP (as control), pIRES-GFP-WNK4, or pIRES-RFP-Nedd4-2 were transfected by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Twenty-four hours after transfection, 1 mg/ml of G418 was added to select for Neomycin-resistant cells. For transient gene expression, A6 cells were seeded onto 12 mm × 12 or 24 mm × 6 permeable polyester inserts and at 90% confluency were transfected with 10 μg of plasmid DNA. To overcome the low level of gene expression in A6 cells, cells used for protein biochemistry assay or transepithelial current measurement were double transfected (transient expression after cells were subjected to selection pressure). For HEK293 cells, cell culture and ENaC gene expression were performed by the same method as Zhou et al. (59). To prevent overloading the cells with sodium, 6 h after ENaC gene expression, 4 μM amiloride were included in the cell cultural medium until cells were lysed.
Biotinylation.
Four days after cells reached confluency on permeable supports, the apical side of A6 cells was labeled with 0.5 mg/ml sulfo-NHS-biotin (Pierce Chemical) in borate buffer (85 mM NaCl, 4 mM KCl, and 15 mM Na2B4O7, pH 9.0) for 2 × 20 min on ice. Afterwards cells were quenched with 100 mM glycine in PBS for 10 min and lysed in RIPA buffer (PBS with 0.1% SDS, 1% Nonidet P-40, and 0.5% sodium deoxycholate) containing protease inhibitor cocktail (100 μM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 100 μM antipain, 100 μM 1-chloro-3-tosylamido-7-amino-2-heptanone, and 100 μM l-1-tosylamido-2-phenylethyl chloromethyl ketone). Biotinylated proteins were isolated by incubating cell lysate with immobilized NeutrAvidin beads (Pierce) overnight at 4°C with the amount of beads being adjusted to ensure complete recovery of all biotinylated proteins.
Coimmunoprecipitation.
Plasmids containing different relevant proteins were transfected into HEK 293 cells. Forty-eight hours after transfection, HEK cells were solubilized in buffer (150 mM NaCl and 50 mM Tris, pH 7.4) containing 1% Triton X-100 and protease inhibitor cocktail. When a monoclonal antibody was used for immunoprecipitation, protein G beads were used to precipitate the protein complex; when polyclonal antibody was used for immunoprecipitation, protein A beads were used. The amount of antibody and beads was adjusted for complete precipitation of the target proteins.
Real-time PCR.
Total RNA from A6 cells transfected with empty vector or WNK4 was extracted by Trizol reagent (GIBCO-BRL, Life Technologies), 5 μg of total RNA were converted to cDNA by QuantiTect reverse transcription kit (Qiagen), and real-time PCR was carried out with QuantiTect SYBR Green PCR kit (Qiagen) on 7500 fast system SDS fostware (Applied Biosystem). Primers of sense and anti-sense for α-ENaC were GGGGAACAACTGGAGAACTTCA and TGGAGGTCTCCATCCCGG; for β-ENaC were TGGAGCACATGGAGGCCC and CCTGATGGCAACCGAGTCTC; for γ-ENaC were LTATGGGCGGAGCAGAAT and GGCTTGGGTTGGTCATAATGAG; and GAPDH was used to normalize the signals. The results of two independent experiments each with three repeats were analyzed and expression levels calculated by ΔΔCt method.
Western blot and quantification.
Following standard methods for SDS-PAGE, proteins were transferred to PVDF membranes and probed with specific antibodies. Luminescence of Western blot signals was detected by the Kodak Gel logic 2000 Imaging system, and protein quantification was quantified by the Carestream MI program. Equal loading for total cellular ENaC subunit protein was confirmed by the amount of actin on the same Western blot, and the relative inhibition of WNK4 on ENaC expression was statistical tested by one-sample two-tailed z-test.
Antibody verification.
Polyclonal antibodies against Xenopus β-ENaC were kind gift from the laboratory of J. P Johnson (47), and α- and γ-ENaC were raised in rabbits using synthetic peptides derived from amino acid sequence 137CIPNNQRVKRDRA-GLPYLLELLPPGS161 and 600CVDNPICLGEEDPPTFNSALQLPQSQDSHVPRTPPPKYNTLRIQSAF647, respectively. The specificity of these antibodies were further verified by expressing ENaC genes in HEK 293 cell, using anti α- and γ-ENaC antigen peptide competition, and expressing Nedd4-2 in A6 to reduce ENaC expression (Fig. 1). Anti-Nedd4-2 polyclonal antibody was purchased from (Abcam). Anti-myc, anti-flag, and anti-HA monoclonal antibodies are from (Sigma-Aldrich). Anti-μ2 antibody was a kind gift from Dr. Guanping Chen at Emory University School of Medicine (25).
Fig. 1.

WNK4 expression inhibits amiloride-sensitive transepithelial current in 2F3 cells: 2F3 cells were transfected with a selectable, myc-tagged WNK4 construct when they were ∼70% confluent and then selected for WNK4 expression in the cell culture mediumuntil they were ∼90–95% confluent at which time they were transfected a second time. A: c-myc-tagged WNK4 was detected by Western blot at 180 kDa in these cells with an anti-c-myc antibody. B: transepithelial voltage and resistance were measured by EVOM (WPI) from control (Ctrl) and WNK4 doubly transfected cells at 4 or 5 days after confluency, and transepithelial current is calculated using Ohm's law. ***P < 0.01.
ENaC function detection and data analysis.
To examine the effect of WNK4 on ENaC trafficking, 1 μM brefeldin A (BFA) or 5 μM forskolin (FSK) was applied 4 days after gene transfection, and the drug removal was achieved in <30 s by washing cells three times with cell culture medium. Transepithelial current was measured by EVOM (World Precision Instruments) at different times after drug treatment, and at the end of measurement 10 μM of amiloride were added to determine amiloride sensitive Na current. The current/unit area was calculated as: I = [(V)/(R)]/filter area in centimeters squared, where I is current, V is voltage, and R is resistance. Transepithelial current measured at different time after BFA treatment was fitted as an exponential decrease, and the log of the current decline was plotted against the time of BFA exposure. Transepithelial current measured at different times after FSK treatment or wash out was fitted as an exponential growth or decay and the best-fit curve was superimposed on averaged data. A t-test was used to determine significant differences in the time constants in WNK4-expressing or control cells.
Patch-clamp single channel recording from A6 cells grown on permeable supports and data analysis were performed as previously described (57).
RESULTS
WNK4 inhibits endogenous ENaC activity in A6 cells.
We examined the effect of WNK4 on endogenous ENaC in 2F3 cells; a subclone of A6 cell originating from the distal nephron of Xenopus leavis. The effect of WNK4 on ENaC activity was first examined by measuring its effect on the amiloride-sensitive, transepithelial current, which composes 90–95% of the total transepithelial current in these cells. Since transient expression of transfected genes is very inefficient in epithelial cells (∼20% in A6 cell), we increased the efficiency by transfecting cells with a WNK4 construct containing a selectable marker. After selecting for WNK4-containing cells for several days, we transiently transfected cells a second time. The c-myc-tagged WNK4 expressed in A6 cells can be detected in Western blots with anti-myc antibody in the doubly transfected cells, and WNK4 reduced the amiloride-sensitive current by ∼30% compared with mock-transfected cells, (Fig. 1, A and B). Since the transepithelial current is calculated from transepithelial voltage and resistance, the value may be influenced by transepithelial resistance, tight junctional permeability, or the amiloride-sensitive Na/H transporter expressed at the basolateral side of epithelial cells (16, 21, 33). Therefore, to directly measure effect of WNK4 on ENaC activity in these cells, we used patch-clamp recordings to measure single channel activity in control and WNK4-expressing cells. Figure 2 shows single channel activity recorded from cells grown on permeable supports expressing control pIRES-GFP vector (bicistronic vector containing GFP reporter gene at one locus) or pIRES-GFP with WNK4 inserted at the second locus. Figure 2A is a representative single channel recording from control (top) and from WNK4-expressing cells (bottom). The average channel activity (NPo) recorded from many cells (cell number indicated as n) is summarized in Fig. 2B. Average channel NPo of WNK4-expressing cells is 0.44 ± 0.12 (n = 39), which is ∼50% of the NPo value of control cells expressing GFP alone: 0.92 ± 0.17 (n = 35; P = 0.022). Since the calculation of channel activity (NPo) combines both channel open probability (Po) and channel number (N) in a patch, as previously described by Marunaka and Eaton (38), NPo can be separated into N and Po with the degree of certainty dependent mostly on the total length of recording period and to a lesser extent on Po. In general, if the Po is between 0.1 and 0.9, and the recording period is near 10 min, N and Po can be determined with ∼95% confidence. We recorded each patch for 8–10 min, and the channel N within a patch was counted based on the maximum number of unitary current levels during the recording. Average channel N for control is 4.29 ± 0.42 (n = 35) and for WNK4-expressing cells is 2.55 ± 0.41 (n = 39; P < 0.01), which is ∼60% of the control (Fig. 2C). We calculated Po by dividing NPo by N in cells with active channels. Average Po for control and for WNK4-expressing cells is similar, 0.16 ± 0.02 (n = 33) vs. 0.15 ± 0.02 (n = 26), respectively, (Fig. 2D). These results demonstrated that WNK4 inhibits ENaC activity by reducing the active channel number at the apical membrane but has no effect on channel open probability.
Fig. 2.

WNK4 expression reduces epithelial sodium channels (ENaC) single channel activity. A: representative single channel recordings from 2F3 cells transfected with vector containing only GFP (top) or with WNK4 (bottom). Closed state of ENaC channels is indicated with a “c” and the channel open levels are marked with “o”. B: average ENaC activity (NPo) from cells expressing only GFP (Ctrl) and from cells expressing GFP and WNK4. Number over each bar indicates the number of patches from which recordings were made. C and D: average number (N) of channels within a patch and average channel open probability (Po) were calculated by first determining N and then dividing NPo by N in each patch to yield Po values. Number over each bar in D is the number of patches with active channels. **P < 0.02; ***P < 0.01.
WNK4 reduces the total amount of cellular ENaC as well as ENaC in the apical membrane of A6 cells.
Mature functional ENaC at the apical membrane undergoes several posttranslational modifications; these modifications convert ENaC from low activity forms into high activity forms (7, 14, 26, 27, 49). Our single channel recordings indicate that WNK4 reduces active channel number at the apical membrane, but it is not certain whether this reduction is due to WNK4-mediated impairment of ENaC posttranslational maturation or due to WNK4-mediated reduction in ENaC surface expression. Based on the mechanism of WNK4-mediated inhibition of NCC and renal outer medullary K+ channel (ROMK), it is likely that WNK4 reduces ENaC surface expression.
We first verified the specificity of our anti-xENaC antibodies. Figure 3A shows that, in A6 cells, three bands for α-subunit and two bands for γ-subunit were eliminated in the presence of competing antigenic peptides to which these antibodies were raised. Figure 3B shows that when all three ENaC subunits, α, β, and γ, are expressed together in HEK293 cells, individual subunits can be detected by our subunit specific antibodies. The ability of these antibodies to detect ENaC subunits was further demonstrated after Nedd4-2 was coexpressed with xENaC subunits. Nedd4-2 is usually thought to reduce amounts of ENaC in cells, and, in fact, Nedd4-2 expression reduces the expression of each ENaC subunit (Fig. 3C).
Fig. 3.

Anti-xENaC antibodies characterization. A: yotal protein lysate from 2F3 cells was probed with anti-α-ENaC or anti-γ-ENaC antibody. Molecular mass bands at 80, 52, and 48 kDa in the α-ENaC blot and the 80- and 70-kDa bands in the γ-ENaC blot are eliminated by competition with antigen peptides. B: Xenopus anti-ENaC antibodies specifically detect xENaC subunits expressed in HEK 293 cells. Lane 1: proteins of control cells expressing empty vector only; lane 2: proteins of mouse renal cells (mpK-CCD-I4) that endogenously express ENaC; lane 3: HEK 293 cells transfected with Xenopus ENaC subunits. (In the β-ENaC blot at bottom, the lane marked “sb” contains only sample buffer. Note that in this same blot, lanes 2 and 3 are reversed from the α- and γ-blots.) C: total proteins from 2F3 cells transfected with Nedd4-2 or sham transfected were probed with antibodies against xENaC. Nedd4-2 reduces the intensity of 3 bands for αENaC and 2 bands for γENaC, which are the same as those competed by their respective antigenic peptides. Nedd4-2 also reduced β-ENaC at 80 and 105 kDa, which are known to be the nonglycosylated and glycosylated forms of βENaC.
To investigate the effect of WNK4 on surface expression of ENaC, we compared ENaC surface expression in control vs. WNK4-expressing cells. A6 cells were apically biotinylated with membrane-impermeable EZ-link sulfo-NHS-SS-biotin 4 days after cells reached confluency, and the biotinylated proteins were recovered by neutravidin beads and detected in Western blot by antibodies specific to each subunit of Xenopus ENaC. Figure 4, A and B, shows Western blots and the quantification of biotinylated ENaC in control or WNK4-expressing A6 cells. The dashed line in this figure is the relative amount of ENaC in control cells expressing empty vector. The relative expression level of each ENaC subunit at molecular masses ∼80 kDa was quantified from 6 to 8 independent experiments by Carestream MI software (Kodak), and the inhibitory effect of WNK4 on each subunit was statistically tested by a one-sample two-tailed z-test. The z-values for all three subunits tested are larger than 3 with P < 0.01, indicating that WNK4 significantly inhibits each subunit expressed at the apical membrane of A6 cells. We also compared the total amount of ENaC in control and WNK4-expressing cells; densitometry quantification followed by z-tests shows that WNK4 significantly reduces the relative total amount of each subunit as well (with z > 4.0 and P < 0.01; Fig. 4, C and D). As we showed in Fig. 1, there are multiple bands for each ENaC subunit in A6 cells, three for α-ENaC, two for β-ENaC, and two doublets for γ-ENaC. The expression level of all these bands were reduced in WNK4-expressing cells.
Fig. 4.

WNK4 reduces total cellular and apical ENaC expression of 2F3 cells. A and C: apical biotin-labeled as well as total ENaC subunit protein was detected from empty vector (Ctrl) and WNK4-expressing (W4) 2F3 cells. B and D: densitometry quantification of Western blot from total protein (3–4 repeats) as well as biotin-labeled ENaC (6–8 repeats) of control and WNK4-expressing 2F3 cells. Relative expression levels of ENaC in control cells are indicated by the dashed lines; columns are relative expression levels of xENaC subunits in WNK4 doubly expressing cells relative to control.
Although WNK4 has been reported to reduce NCC and ROMK protein levels by regulating their trafficking, there is no specific information about whether WNK4 might also have an effect on gene transcription. To examine whether WNK4 affects ENaC gene expression, we performed real-time PCR to compare the mRNA level of each ENaC subunit in control vs. WNK4-expressing cells. ΔΔCt for α-, β-, and γ-subunit is 0.49 ± 0.06, 0.59 ± 0.12, and 0.8 ± 0.27, respectively. This result implies that WNK4 slightly inhibits ENaC gene expression.
WNK4 enhances ENaC internalization.
WNK4 inhibits both NCC and ROMK surface expression but by different mechanisms. WNK4 reduces NCC surface delivery by inhibiting its forward trafficking, i.e., insertion (51), but reduces ROMK surface expression by enhancing channel retrieval (23). Our patch-clamp recording and Western blot results show that WNK4 reduces endogenous ENaC surface expression in A6 cells. The net reduction in ENaC surface expression could be due to either WNK4 enhancing the rate of ENaC internalization or reducing its delivery to the apical membrane or a combination of both. BFA selectively and reversibly blocks plasma membrane protein delivery from the biosynthetic pathway to the cell surface (11, 15, 41); therefore, the rate of reduction in ENaC current after BFA is a measure of the rate of retrieval of ENaC from the surface membrane. We reasoned that if the sole effect of WNK4 is to reduce trafficking of ENaC to the membrane, then the BFA-induced reduction in current will be the same in the presence or absence of WNK4. On the other hand, if WNK4 increases the rate of ENaC retrieval, then the rate at which ENaC current decreases after BFA will be greater in WNK4-treated cells. We measured BFA-induced reduction in amiloride-sensitive, transepithelial current at 4 days after cell confluency in control and WNK4 transfected cells to compare whether WNK4 increases the rate of retrieval (Fig. 5A). After 3 h of BFA treatment (1 μM), the relative current of both control and WNK4-expressing cells decreased substantially, but this decrease was greater in WNK4-expressing cells. However, the relative current of both groups was restored to the initial level after BFA was washed off (indicated by the arrows in the figure). When we fit the decrease in current of the control and WNK4-expressing cells, a single decreasing exponential function gives time constants of 345 ± 47.6 and 145 ± 19.5 min, respectively (Fig. 5B).
Fig. 5.

Brefeldin A (BFA) and forskolin (FSK) effects on transepithelial current of WNK4 or Nedd4-2-expressing 2F3 cells. A: relative transepithelial current measured from control (■) or WNK4 (▲)-expressing 2F3 cells exposed to 500 nM BFA at time 0. Data in A were normalized to the initial current immediately before BFA treatment. Arrow indicates BFA wash off. B: when we fit the decrease in current of the control and WNK4-expressing cells, a single decreasing exponential function gives time constants of 345 ± 47.6 and 145 ± 19.5 min, respectively, implying that WNK4 more than doubles the rate of ENaC internalization. C: time course of the increase in transepithelial current (Isc) after application of FSK in control and Nedd4-2-expressing cells. D: same as B but recorded from control or WNK4-expressing cells rather than Nedd4-2 cells. E: decrease of transepithelial current recorded from control or WNK4-expressing cells after washing off of FSK.
Since ENaC internalization from the apical membrane is normally mediated via Nedd4-2-induced ubiquitination and subsequent retrieval, we compared the effect of expression of WNK4 or Nedd4-2 on ENaC retrieval in response to FSK. FSK is commonly used to raise the level of cellular cyclic AMP, which reversibly stimulates insertion of intracellular ENaC-containing vesicles into the apical membrane of distal nephron cells from a subapical recycling pool (4). Ordinarily, a significant fraction of ENaC that is retrieved from the surface membrane is returned to this pool while the remainder enters a degradative pathway. Overexpression of Nedd4-2 diverts more retrieved ENaC to degradation so that the amount of ENaC in the recycling pool is reduced; therefore, when FSK is applied, there is less ENaC to enter the apical membrane. We compared control A6 cells and A6 cells transfected for 2 days with either WNK4 or Nedd4-2 after which we treated the cells with 5 μM FSK. Figure 5C shows that overexpression of Nedd4-2 dramatically reduces the transepithelial current compared with control cells presumably by reducing the amount of ENaC available to recycle back to the apical membrane. FSK potentiates the transepithelial current of control and Nedd4-2-expressing A6 cells from 9.3 ± 1.5 to 52.7 ± 1.1 μA/cm2 (a 5.7 ± 0.92-fold increase) and 1.7 ± 0.3 to 8.4 ± 0.7 μA/cm2 (a 4.9 ± 0.96-fold increase), respectively. The best fit for the current increase in the control and Nedd4-2-expressing cells shows that the t1/2 of current in Nedd4-2-expressing cells is ∼9 min, which is much slower than that of control cells (5 min). The same experiment was performed with WNK4-expressing cells (Fig. 5D). FSK stimulates transepithelial current of WNK4-expressing A6 cells from 2.5 ± 0.24 to 13.5 ± 1.6 μA/cm2 (a 5.4 ± 0.82-fold increase). This fold change is similar to that produced by FSK stimulation of transepithelial current in matched control cells from 4.4 ± 0.4 to 32.1 ± 4.2 μA/cm2 (a 7.3 ± 1.2-fold increase). The steady-state current in WNK4-expressing A6 cells after FSK treatment is less than half of the control showing that ENaC available for FSK-stimulated insertion has been reduced. In spite of a reduction in current, the time courses (t1/2) of the increase in transepithelial current in WNK4-expressing cells vs. control are similar (∼4 min each), implying that WNK4 affects the pool of ENaC available for FSK-stimulated insertion but does not affect the rate at which FSK promotes insertion. Similarly, the rate of recovery (i.e., internalization) after FSK washout is similar in both groups of cells (t1/2 is ∼6 min), implying that the mechanism for ENaC internalization from the apical membrane has also not changed (Fig. 5E). The above results imply that Nedd4-2 and WNK4 both reduce the amount of ENaC at the surface membrane, but the mechanism by which each molecule reduces surface membrane ENaC appears to be different.
Inhibitory effects of WNK4 on ENaC activity is much smaller than the effect of Nedd4-2.
To determine whether WNK4 produces its inhibitory effect independent of the Nedd4-2-mediated increase in ENaC retrieval, we examined the combined effect of WNK4 and Nedd4-2 on ENaC activity using single channel recording. In these studies, WNK4 was expressed in a bicistronic vector, pIRES-GFP, as described above, while Nedd4-2 was expressed in a bicistronic vector pIRES-RED, which expresses dsRed monomer in the same cells as Nedd4-2. Patch-clamp recordings were obtained from cells expressing ds-Red (and Nedd4-2 expression) or expressing both ds-Red and GFP (both Nedd4-2 and WNK4 expression); meanwhile, a set of control cells expressing empty vectors was recorded from in parallel. Our single channel recordings show that NPo of control is 0.5 ± 0.18 (n = 37), and NPo of Nedd4-2-expressing cells is 0.16 ± 0.07 (n = 38). As expected, the channel activity is reduced significantly in Nedd4-2-expressing cells (P < 0.01); however, coexpression of WNK4 produces no additional inhibition, and NPo of cells expressing both genes is 0.2 ± 0.06 (n = 37; Fig. 6A). The effects of Nedd4-2 or Nedd4-2 and WNK4 on channel Po and N were further compared by determining N and Po of these cells. Po of Nedd4-2 or Nedd4-2 and WNK4 coexpressing cells is similar to that of control, both around 0.1 ± 0.02. As anticipated Nedd4-2 significantly reduced the average channel density, N, from a control value of 2.57 ± 0.4 to 1.13 ± 0.29 (P < 0.01) in each patch; again, the value for N when Nedd4-2 and WNK4 are coexpressed together is 1.4 ± 0.22 implying no further inhibition of channel N by WNK4 compared with expressing Nedd4-2 alone (Fig. 6B).
Fig. 6.

Effect of Nedd4-2 and WNK4 are not additive. A: average channel activity (Po) from control, Nedd4-2, and Nedd4-2 plus WNK4-expressing 2F3 cells; the number over each bar indicates the number of patches in each condition. Nedd4-2 reduces channel activity to very low levels and addition of WNK4 produces little further reduction. B: average channel number (N) within patches on these same cells. *P < 0.05; ***P < 0.01.
WNK4 inhibits ENaC independently of Nedd4-2.
The lack of an additive effect of WNK4 and Nedd4-2 on ENaC regulation does not provide a clear indication of whether WNK4 inhibition of ENaC is dependent on Nedd4-2. The results of coexpression could be due either to WNK4 and Nedd4-2 coordinately regulating ENaC, or Nedd4-2-dependent ENaC retrieval could be so large that WNK4 cannot produce a significant additional increase in retrieval, implying that the WNK4 and Nedd4-2 effects could still be independent. To further explore the relationship between WNK4 and Nedd4-2 in regulating ENaC, we next examined the physical association of these two proteins. We coexpressed myc-tagged WNK4 and Nedd4-2 in HEK 293 cells, and then both proteins were detected in Western blots by anti-myc or anti-Nedd4-2 antibodies, respectively (Fig. 7A, top). However, anti-myc antibody does not immunoprecipitate Nedd4-2, and Nedd4-2 antibody does not precipitate WNK4. In contrast, a known WNK4-interacting protein, the big potassium channel (BK or Maxi-K; Ref. 60), is precipitated with WNK4 (Fig. 7A, bottom). Thus Nedd4-2 and WNK4 do not appear to directly interact. Second, we examined the physical association of WNK4 and ENaC. Since all of our ENaC functional measurements were performed on A6 cells, we first examined the association between Xenopus ENaC and WNK4. All three xENaC subunits and myc-tagged WNK4 were coexpressed in HEK 293 cells. Two days after gene expression, anti-myc antibody was used to precipitate WNK4. In the Western blot of the immunoprecipitate, all three xENaC subunits were detected by specific antibodies (Fig. 7B) showing that WNK4 appears to directly interact with ENaC. Nedd4-2 promotes ENaC retrieval by promoting ENaC ubiquitination and endocytosis. WNK4 could promote ENaC retrieval by augmenting Nedd4-2 ubiquitination in a manner similar to that of ERK1/2 stimulation of Nedd4-2 ubiquitination (32). If this were the mechanism for WNK4 action, then in the absence of the Nedd4-2 ubiquitinat site WNK4 would no longer have an effect on ENaC retrieval. Therefore, to determine whether WNK4 inhibits ENaC independent of Nedd4-2-mediated ENaC ubiquitination, we examined the association of WNK4 with human wild-type and Liddle's mutated ENaC in which the PPxY motif of Nedd4-2 binding site was altered to αY644A, βY620A, and γY627A. Human ENaC gene constructs were kind gift from Dr. Peter M. Snyder at the University of Iowa (59). Taking advantage of 3Xflag-tagged vector for our protein biochemistry assay, we cloned wild-type and mutated α-, β-, and γ-ENaC subunits into this vector and used anti-flag antibody to precipitate ENaC followed by using anti-myc antibody to detect WNK4. Figure 7C shows that both wild-type and the PPxY-deficient ENaC associate with WNK4. This result shows that WNK4 and ENaC association is independent of Nedd4-2-mediated ENaC ubiquitination. We also examined whether WNK4 is still capable of inhibiting Liddle's mutated ENaC expression. We expressed wild-type or Liddle's mutated ENaC in which the β-ENaC subunit was tagged with 3Xflag in HEK293 cells with or without WNK4. Western blots of β-ENaC detected in total cellular protein are shown in Fig. 8A. With equal amounts of transfected DNA, the expression of Liddle's mutated ENaC is, as expected, much higher than wild-type ENaC, but in five repetitions, WNK4 reduced both wild-type and mutated ENaC in similar proportion. WNK4 inhibition of α- and γ-subunits of Liddle's mutated ENaC was also examined. Densitometric quantification of four independent experiments shows that expression of WNK4 reduces the expression of each ENaC subunit to <60% of the respective control without coexpression of WNK4 (indicated by a dashed line in Fig. 8B). Since WNK4 has been reported to stimulate amiloride-sensitive sodium current when it was coexpressed with ENaC in HEK 293 cells (24), we wondered whether WNK4 regulation of ENaC surface expression in HEK cells is different from the regulation in A6 cells. Therefore, we expressed WNK4 and Liddle's mutated ENaC in which the α-subunit was fused with a 3Xflag tag in HEK cells. We biotinylated the surface membranes of these cells and found that expression of WNK4 reduces the amount of α-ENaC at the plasma membrane. The extent of WNK4 inhibition of α-ENaC surface expression is shown in Fig. 8, C and D. This result shows that WNK4 inhibits ENaC in HEK cells as well as in A6 cells and that this inhibition is independent of Nedd4-2-mediated ENaC ubiquitination.
Fig. 7.
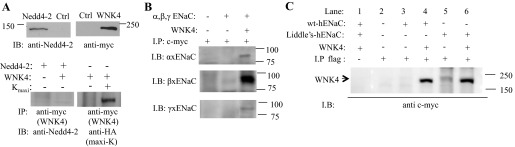
WNK4 associates with ENaC but not with Nedd4-2 in HEK cells. A: Nedd4-2 and myc-tagged WNK4 were detected by anti-Nedd4-2 and anti-c-myc antibodies, respectively (top) to demonstrate the efficacy of the antibodies. Anti-c-myc antibody does not coimmunoprecipitate Nedd4-2, but the same antibody can immunoprecipitate (IP) an HA-tagged Maxi-K, a known interacting partner of WNK4 (bottom); IB, immunoblot. B: xENaC and WNK4 were coexpressed in HEK 293 cells, all 3 ENaC subunits were coimmunoprecipitated with WNK4 by using anti-c-myc antibody to precipitate WNK4. Lanes from left to right: control empty vector-expressing cells; cells expressing xENaC; xENaC coexpressed with WNK4. C: WNK4 was coexpressed with human wild-type ENaC (α, β-3Xflag, and γ-3Xflag) or Liddle's mutated ENaC (αY644A, βY620A 3Xflag, and γY627A 3Xflag) in HEK 293 cells. Anti-flag antibody was used to precipitate the ENaC complex and c-myc tagged WNK4 was detected by anti-c-myc antibody in Western blots. Lane 1: wild-type ENaC + WNK4; lane 2: control empty vector-expressing cells; lane 3: wt ENaC only; lane 4: wt ENaC + WNK4; lane 5: mutated ENaC only; and lane 6: mutated ENaC + WNK4.
Fig. 8.

WNK4 reduces human wild-type as well Liddle's cell surface ENaC expressed in HEK 293 cells. A: Western blot of total wild-type ENaC or Liddle's mutated ENaC expressed in HEK 293 cells in which the β-subunit was tagged by 3Xflag. B: densitometric quantification shows that, relative to cells with no WNK4, WNK4 reduces the amount of the β-subunit of Wild-type and Liddle's mutated ENaC expressed in HEK 293 cells. C: Western blot of surface biotinylated Liddle's mutated ENaC, in which α-subunit was tagged by 3Xflag, expressed in HEK 293 cells. D: densitometric quantification of ENaC shows that WNK4 reduces plasma membrane α-subunit of Liddle's mutated ENaC expressed in HEK 293 cells.
DISCUSSION
Both WNK4 and ENaC play important roles in regulating Na+ reabsorption in the distal nephron, so it should not be surprising that WNK4 can alter ENaC activity. However, reports describing WNK4 regulation of ENaC are inconsistent with one report suggesting WNK4 inhibition of ENaC and another suggesting excitation (24, 45, 46). In this study, we show that endogenous ENaC single channel activity and amiloride-sensitive transepithelial current are reduced by WNK4. By using single channel recording we could unequivocally identify the channels in each patch as the highly selective, 5-pS ENaC channels consisting of the three α-, β-, and γ-subunits (6). By comparing average channel number within a patch from control and WNK4-expressing cells, we showed that WNK4 reduces the average number of functional ENaC in the apical membrane of A6 cells. We further determined from Western blots and cell surface biotinylation that the effect of WNK4 was to reduce both apical membrane ENaC expression and total cellular ENaC expression. Furthermore, when we coexpressed wild-type or Liddle's mutated ENaC (αY644A, βY620A, and γY627A) with WNK4 in HEK293 cells, our Western blot quantification shows again that WNK4 reduces both total and apically expressed wild-type and Liddle's mutated ENaC. Our results are consistent with the results of Gamba and colleagues (8) who showed that ENaC appears to be activated in WNK4 knockout animals. However, Heise et al. (24) reported a contrary effect of WNK4 on ENaC regulation: that WNK4 stimulates ENaC activity in HEK 293 cells. This opposite effect of WNK4 on ENaC might be due to several differences in the methods used in the experiments. First, Heise et al. measured amiloride-sensitive whole cells current from ENaC-expressing HEK cells. Amiloride-sensitive Na channels are formed from a combination of subunits from ENaC/degenerin super family, which includes in vertebrates ENaC, several stretch-sensitive cation channels, and acid-sensitive channels (ASICs; Ref. 31). The different members of the family have homologous sequences and strong domain similarity with all members containing two transmembrane α-helices and large extracellular domains with relatively small N- and C-terminal cytosolic domains (44). Heteromeric combination of subunits within the super family may form amiloride-sensitive cross-clade channels, which exhibit electrophysiological characteristics and regulation different from homomeric channels (39). Cross-clade channels may form in ENaC-expressing HEK 293 cells, because HEK 293 cells endogenously express ASIC subunits and, possibly, other family members (20, 35). In fact, in the many single channel recordings made by us from ENaC-expressing HEK293 cells, we only observe a 23-pS nonselective cation channel rather than the 4-to 5-pS conductance channel typical of heterotrimeric ENaC, implying that the cells express some other type of channel rather than traditional ENaC channels. In fact, as we have previously shown, the regulation of the highly selective, 5-pS channel and the higher conductance nonselective cation channel is quite different with some stimuli stimulating the nonselective channel while inhibiting the selective channel (10). Second, in the whole cell recordings of Heise et al. (24), cells are dialyzed with pipette solution, which may change the context of WNK4 signaling and therefore change the effects of WNK4. Three functionally distinct WNK4 variants have been identified including wild type, PHA2 mutants, and the SGK1 phosphorylated forms. Different forms of WNK4 exert different effects on Na and K transporters in the distal nephron, and therefore, WNK4 has been described as a molecular switch to regulate salt balance under different physiological demands (30, 52). For example, WNK4 was initially discovered based on the ability of WNK4 to inhibit NCC activity by reducing NCC surface expression (5, 55, 56). However, recent results demonstrate that in the presence of SGK1 and in the context of angiotensin II receptor (AT1R) activation, WNK4 phosphorylates a second kinase, SPAK, a Ste20-related upstream MAP kinase, which in turn phosphorylates and enhances NCC activity (48). Third, in our experiments, we included 4 μM of amiloride in HEK 293 cell culture medium after ENaC transfection to prevent excessive Na+ uptake in transfected cells unlike in the experiments of Heise et al (24). The volume-sensitive kinases OSR1/SPAK are regulated by WNK4 (1, 18), and different cell culture conditions may contribute to the differences in the observations. Regardless, our results are from native cells expressing endogenous ENaC. Therefore, we believe that our results probably may more accurately reflect renal regulation of ENaC by WNK4.
We have also investigated the mechanism by which WNK4 regulates ENaC. ENaC is a constitutive active channel whose activity is regulated by channel trafficking. The ubiquitin ligase Nedd4-2 is a major modulator that stimulates ENaC internalization. It has been suggested that WNK4 inhibition of ENaC is dependent on Nedd4-2-mediated ENaC ubiquitination because WNK4 no longer inhibits ENaC when the C-terminal tails of β- and γ-ENaC are truncated (45). However, truncation of the entire cytosolic domains of β- and γ-ENaC, besides removing the Nedd4-2 binding motif, also removes several kinase phosphorylation sites and clathrin adaptor binding motifs, and these motifs could be sites for WNK4-mediated ENaC regulation. Therefore, the question remains of whether WNK4 inhibition of ENaC is dependent on Nedd4-2. We attempted to answer this question in several ways. First, we compared the effect of Nedd4-2 and Nedd4-2 plus WNK4 on ENaC single channel activity, and we found that there was no additional WNK4 inhibition after initial Nedd4-2 inhibition of ENaC. This result might imply that WNK4-mediated inhibition of ENaC depends on Nedd4-2 function; however, it is also possible that the Nedd4-2 inhibition is so strong (see Fig. 6, A and B, and the 1st point in Fig. 5C) that it masks an independent effect of WNK4. On the other hand, WNK4 has no effect while Nedd4-2 significantly impairs the rate of accumulation (t1/2) of functional ENaC after FSK treatment. FSK is generally felt to promote movement of ENaC from a subapical recycling pool to the surface membrane. Nedd4-2 promotes movement, of retrieved ENaC into a degradative pathway rather than return of ENaC to a recycling pool, thus reducing the amount of ENaC available for insertion. Apparently WNK4 does not act in the same way. Third, Nedd4-2 and WNK4 are not associated with one another in the same protein complex, suggesting that WNK4 is unlikely to directly effect the ability of Nedd4-2 to ubiquitinate ENaC. Fourth, and the most convincing evidence, is that WNK4 not only associates with Liddle's ENaC with a single point mutation but also reduces the expression of mutated ENaC.
In this study, we have compared ENaC recycling in WNK4- and Nedd4-2-expressing cells. We found that WNK4 reduced the ENaC recycling pool but had no effect on the time course of ENaC recycling between early endosome and apical membrane. The t1/2 of ENaC exocytosis is ∼4 min in both control and WNK4-expressing cells after FSK treatment, which is consistent with the t1/2 (4.5 min) of ENaC exocytosis from early endosomes to the apical membrane of CCD cells (4). A similar value for the t1/2 of the decrease in current in WNK4 and control cells also implies that WNK4 has no effect on the rapid retrieval of ENaC from the apical membrane. In contrast to WNK4-expressing cells, the t1/2 of the decrease in current of Nedd4-2-expressing cells is much slower (∼9 min) implying that ENaC from deeper within cells in sorting vesicles or late endosomes is also sensitive to PKA regulation. Our results are consistent with observations that Nedd4-2 reduces ENaC surface expression by two distinct mechanisms, one which enhances ENaC retrieval from the cell surface and the other which enhances ENaC delivery to lysosomes for degradation. Both steps are sensitive to PKA or SGK1 phosphorylation (28, 58). Therefore, the slow exocytosis of ENaC in response to FSK in Nedd4-2-expressing A6 cells could be due to a FSK-induced reduction in the Nedd4-2-mediated ENaC from sorting endosomes or late endosomes.
We have also explored how WNK4 affects ENaC trafficking. Compared with untransfected cells, WNK4 transfection accelerates the BFA-induced decrease of transepithelial current, which suggests that WNK4 enhances ENaC internalization and retrograde trafficking (Fig. 5A). However, we cannot exclude the possibility that WNK4 promotes ENaC degradation from the interior of the cell. We found that only 2% of Liddle's ENaC is located at the surface of HEK cell (data not shown), while most ENaC is degraded without trafficking to the cell surface. Cotransfection of WNK4 into HEK cells reduces the level of Liddle's ENaC expression to ∼50% that of cells without WNK4 expression (Fig. 8, B and D), implying that it may also enhance ENaC degradation from intracellular pools.
GRANTS
This research was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants F32-DK-093255-01 (to A. A. Alli) and R37-DK-037963 (to D. C. Eaton), and Veteran's Health Administration Grant 1I01BX00099 (to H. Cai)
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.Y., H.C., A.A.A., O.A.-K., H.-F.B., and D.C.E. conception and design of research; L.Y., H.C., Q.Y., A.A.A., D.W., and O.A.-K. performed experiments; L.Y., Q.Y., D.W., and D.C.E. analyzed data; L.Y., H.C., A.A.A., O.A.-K., H.-F.B., and D.C.E. interpreted results of experiments; L.Y., Q.Y., H.-F.B., and D.C.E. prepared figures; L.Y. drafted manuscript; L.Y. and D.C.E. edited and revised manuscript; L.Y., H.C., Q.Y., A.A.A., D.W., O.A.-K., H.-F.B., and D.C.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank B. J. Duke for maintaining 2F3 cells in culture.
REFERENCES
- 1. Ahlstrom R, Yu AS. Characterization of the kinase activity of a WNK4 protein complex. Am J Physiol Renal Physiol 297: F685–F692, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beach CM, Wilson TA, Smart M, John Deutsch Institute for the Study of Economic Policy The 2006 Federal Budget: Rethinking Fiscal Priorities. Montreal, Canada: John Deutsch Institute for the Study of Economic Policy, 2007 [Google Scholar]
- 3. Bertog M, Cuffe JE, Pradervand S, Hummler E, Hartner A, Porst M, Hilgers KF, Rossier BC, Korbmacher C. Aldosterone responsiveness of the epithelial sodium channel (ENaC) in colon is increased in a mouse model for Liddle's syndrome. J Physiol 586: 459–475, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Butterworth MB, Edinger RS, Johnson JP, Frizzell RA. Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J Gen Physiol 125: 81–101, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cai H, Cebotaru V, Wang JH, Zhang XM, Cebotaru L, Guggino SE, Guggino WB. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int 69: 2162–2170, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of 3 homologous subunits. Nature 367: 463–467, 1994 [DOI] [PubMed] [Google Scholar]
- 7. Carattino MD, Hughey RP, Kleyman TR. Proteolytic processing of the epithelial sodium channel gamma subunit has a dominant role in channel activation. J Biol Chem 283: 25290–25295, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen W, Chen Y, Xu BE, Juang YC, Stippec S, Zhao YM, Cobb MH. Regulation of a third conserved phosphorylation site in SGK1. J Biol Chem 284: 3453–3460, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen XJ, Eaton DC, Jain L. Alveolar epithelial ion and fluid transport: β-adrenergic regulation of amiloride-sensitive lung sodium channels. Am J Physiol Lung Cell Mol Physiol 282: L609–L620, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Coupayegerard B, Kim HJ, Singh A, Blazeryost BL. Differential effects of brefeldin-A on hormonally regulated Na+ transport in a model renal epithelial cell line. Biochim Biophys Acta 1190: 449–456, 1994 [DOI] [PubMed] [Google Scholar]
- 12. Dahlmann A, Pradervand S, Hummler E, Rossier BC, Frindt G, Palmer LG. Mineralocorticoid regulation of epithelial Na+ channels is maintained in a mouse model of Liddle's syndrome. Am J Physiol Renal Physiol 285: F310–F318, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20: 7052–7059, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Firsov D, Robert-Nicoud M, Gruender S, Schild L, Rossier BC. Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC)–identification of cysteines essential for channel expression at the cell surface. J Biol Chem 274: 2743–2749, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Fisher RS, Grillo FG, SaribanSohraby S. Brefeldin A inhibition of apical Na+ channels in epithelia. Am J Physiol Cell Physiol 270: C138–C147, 1996 [DOI] [PubMed] [Google Scholar]
- 16. Frelin C, Barbry P, Vigne P, Chassande O, Cragoe EJ, Lazdunski M. Amiloride and its analogs as tools to inhibit Na+ transport via the Na+ channel, the Na+/H+ antiport and the Na+/Ca2+ exchanger. Biochimie 70: 1285–1290, 1988 [DOI] [PubMed] [Google Scholar]
- 17. Furgeson SB, Linas S. Mechanisms of type I and type II pseudohypoaldosteronism. J Am Soc Nephrol 21: 1842–1845, 2010 [DOI] [PubMed] [Google Scholar]
- 18. Gagnon KB, England R, Delpire E. Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am J Physiol Cell Physiol 290: C134–C142, 2006 [DOI] [PubMed] [Google Scholar]
- 19. Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997 [DOI] [PubMed] [Google Scholar]
- 20. Gunthorpe MJ, Davis JB, Randall AD. Characterisation of a human acid-sensing ion channel (hASIC1a) endogenously expressed in HEK293 cells. Pflügers Arch 442: 668–674, 2011 [DOI] [PubMed] [Google Scholar]
- 21. Haggerty JG, Cragoe EJ, Slayman CW, Adelberg EA. Na+/H+ exchanger activity in the pig-kidney epithelial-cell line, LLC-pk1: inhibition by amiloride and its derivatives. Biochem Biophys Res Commun 127: 759–767, 1985 [DOI] [PubMed] [Google Scholar]
- 22. Hansson JH, Nelsonwilliams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel gamma-subunit: genetic heterogeneity of Liddle syndrome. Nat Genet 11: 76–82, 1995 [DOI] [PubMed] [Google Scholar]
- 23. He GC, Wang HR, Huang SK, Huang CL. Intersectin links WNK kinases to endocytosis of ROMK1. J Clin Invest 117: 1078–1087, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heise CJ, Xu BE, Deaton SL, Cha SK, Cheng CJ, Earnest S, Sengupta S, Juang YC, Stippec S, Xu YD, Zhao YM, Huang CL, Cobb MH. Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem 285: 25161–25167, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang HD, Feng XY, Zhuang JQ, Frohlich O, Klein JD, Cai H, Sands JM, Chen GP. Internalization of UT-A1 urea transporter is dynamin dependent and mediated by both caveolae- and clathrin-coated pit pathways. Am J Physiol Renal Physiol 299: F1389–F1395, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong QS, Carattino MD, Johnson JP, Stockand JC, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem 279: 48491–48494, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Kabra R, Knight KK, Zhou R, Snyder PM. Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J Biol Chem 283: 6033–6039, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Kahle KT, Wilson FH, Lalioti M, Toka H, Qin H, Lifton RP. WNK kinases: molecular regulators of integrated epithelial ion transport. Curr Opin Nephrol Hypertens 13: 557–562, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Kahle KT, Wilson FH, Lifton RP. Regulation of diverse ion transport pathways by WNK4 kinase: a novel molecular switch. Trends Endocrinol Metab 16: 98–103, 2005 [DOI] [PubMed] [Google Scholar]
- 31. Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82: 735–767, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Ko B, Kamsteeg EJ, Cooke LL, Moddes LN, Deen PM, Hoover RS. RasGRP1 stimulation enhances ubiquitination and endocytosis of the sodium-chloride cotransporter. Am J Physiol Renal Physiol 299: F300–F309, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Labelle EF. Reconstituted amiloride-inhibited sodium transporter from rabbit kidney medulla is responsible for Na+-H+ exchange. Biochim Biophys Acta 770: 79–92, 1984 [DOI] [PubMed] [Google Scholar]
- 34. Lalioti MD, Zhang JH, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006 [DOI] [PubMed] [Google Scholar]
- 35. Lalo U, Pankratov Y, North RA, Verkhratsky A. Spontaneous autocrine release of protons activates ASIC-mediated currents in HEK293 cells. J Cell Physiol 212: 473–480, 2007 [DOI] [PubMed] [Google Scholar]
- 36. Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 8: 1246–1264, 2007 [DOI] [PubMed] [Google Scholar]
- 37. Ma HP, Chou CF, Wei SP, Eaton DC. Regulation of the epithelial sodium channel by phosphatidylinositides: experiments, implications, and speculations. Pflügers Arch 455: 169–180, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Marunaka Y, Eaton DC. Effects Of vasopressin and cAMP on single amiloride-blockable Na channels. Am J Physiol Cell Physiol 260: C1071–C1084, 1991 [DOI] [PubMed] [Google Scholar]
- 39. Meltzer RH, Kapoor N, Qadri YJ, Anderson SJ, Fuller CM, Benos DJ. Heteromeric assembly of acid-sensitive ion channel and epithelial sodium channel subunits. J Biol Chem 282: 25548–25559, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Ohta A, Rai T, Yui N, Chiga M, Yang SS, Lin SH, Sohara E, Sasaki S, Uchida S. Targeted disruption of the Wnk4 gene decreases phosphorylation of Na-Cl cotransporter, increases Na excretion and lowers blood pressure. Hum Mol Genet 18: 3978–3986, 2009 [DOI] [PubMed] [Google Scholar]
- 41. Pelham HR. Recycling of proteins between the endoplasmic reticulum and Golgi complex. Curr Opin Cell Biol 3: 585–591, 1991 [DOI] [PubMed] [Google Scholar]
- 42. Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens 17: 533–540, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na(+) inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y(2)-receptor tone. FASEB J 24: 2056–2065, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Renard S, Lingueglia E, Voilley N, Lazdunski M, Barbry P. Biochemical-analysis of the membrane topology of the amiloride-sensitive Na+ channel. J Biol Chem 269: 12981–12986, 1994 [PubMed] [Google Scholar]
- 45. Ring AM, Cheng SX, Leng Q, Kahle KT, Rinehart J, Lalioti MD, Volkman HM, Wilson FH, Hebert SC, Lifton RP. WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc Natl Acad Sci USA 104: 4020–4024, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA 104: 4025–4029, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rokaw MD, Wang JM, Edinger RS, Weisz OA, Hui D, Middleton P, Shlyonsky V, Berdiev BK, Ismailov I, Eaton DC, Benos DJ, Johnson JP. Carboxylmethylation of the beta subunit of xENaC regulates channel activity. J Biol Chem 273: 28746–28751, 1998 [DOI] [PubMed] [Google Scholar]
- 48. San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci USA 106: 4384–4389, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheng SH, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem 282: 20180–20190, 2007 [DOI] [PubMed] [Google Scholar]
- 50. Shimkets RA, Warnock DG, Bositis CM, Nelsonwilliams C, Hansson JH, Schambelan M, Gill JR, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP. Liddles syndrome: heritable human hypertension caused by mutations in the beta-subunit of the epithelial sodium-channel. Cell 79: 407–414, 1994 [DOI] [PubMed] [Google Scholar]
- 51. Subramanya AR, Liu J, Ellison DH, Wade JB, Welling PA. WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J Biol Chem 284: 18471–18480, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Welling PA, Chang YPC, Delpire E, Wade JB. Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int 77: 1063–1069, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Willams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001 [DOI] [PubMed] [Google Scholar]
- 54. Xu BE, Stippec S, Chu PY, Lazrak A, Li XJ, Lee BH, English JM, Ortega B, Huang CL, Cobb MH. WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc Natl Acad Sci USA 102: 10315–10320, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest 111: 1039–1045, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang SS, Yamauchi K, Rai T, Hiyama A, Sohara E, Suzuki T, Itoh T, Suda S, Sasaki S, Uchida S. Regulation of apical localization of the thiazide-sensitive NaCl cotransporter by WNK4 in polarized epithelial cells. Biochem Biophys Res Commun 330: 410–414, 2005 [DOI] [PubMed] [Google Scholar]
- 57. Yu L, Eaton DC, Helms MN. Effect of divalent heavy metals on epithelial Na+ channels in A6 cells. Am J Physiol Renal Physiol 293: F236–F244, 2007 [DOI] [PubMed] [Google Scholar]
- 58. Zhou RF, Kabra R, Olson DR, Piper RC, Snyder PM. Hrs controls sorting of the epithelial Na(+) channel between endosomal degradation and recycling pathways. J Biol Chem 285: 30523–30530, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou RF, Patel SV, Snyder PM. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J Biol Chem 282: 20207–20212, 2007 [DOI] [PubMed] [Google Scholar]
- 60. Zhuang JQ, Zhang XM, Wang DX, Li J, Zhou B, Shi Z, Gu DY, Denson DD, Eaton DC, Cai H. WNK4 kinase inhibits Maxi K channel activity by a kinase-dependent mechanism. Am J Physiol Renal Physiol 301: F410–F419, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]