

Abstract
Pancreatitis is classified into acute pancreatitis (AP) and chronic pancreatitis (CP). Apelin, a small regulatory peptide, is the endogenous ligand for the APJ receptor. Apelin and APJ are expressed in the pancreas. The aims of this study were to examine whether apelin influences the inflammatory and fibrosis responses to pancreatitis in mice and to identify mechanisms behind apelin's activities. Supramaximal cerulein induction of AP or CP caused significant (P < 0.05) elevations in pancreatic apelin and APJ expression. Levels declined during the recovery phases. In apelin gene-knockout mice with pancreatitis, pancreatic neutrophil invasion and myeloperoxidase activity were enhanced significantly, and apelin treatment suppressed both. Apelin exposure reduced CP-induced elevations of extracellular matrix-associated proteins. Apelin inhibited PDGF-simulated connective tissue growth factor production and proliferation of pancreatic stellate cells (PSCs). Serum granulocyte colony-stimulating factor and keratinocyte cytokine levels were higher in apelin gene-knockout than wild-type mice with pancreatitis. Apelin reduced AP- and CP-induced elevations in pancreatic NF-κB activation. Together, these findings imply that the pancreatic apelin-APJ system functions to curb the inflammatory and fibrosis responses during pancreatitis. Furthermore, findings suggest that apelin reduces inflammation and fibrosis by reducing neutrophil recruitment and PSC activity. Inhibition of neutrophil invasion may be mediated by reduced keratinocyte cytokine and granulocyte colony-stimulating factor secretion. Apelin-induced reductions in PSC proliferation and connective tissue growth factor production are putative mechanisms underlying apelin's inhibition of extracellular matrix production. The apelin-associated changes in NF-κB binding may be linked to apelin's regulation of pancreatic inflammatory and fibrosis responses during pancreatitis.
Keywords: collagen, inflammation, neutrophils, chemoattractants
acute pancreatitis (AP) and chronic pancreatitis (CP) are debilitating diseases associated with significant morbidities and mortalities (2, 26). Inflammatory cell infiltration and fibrosis are key components of pancreatitis. Pancreatic recruitment of neutrophils and macrophages results in parenchymal inflammation, acinar damage, and fibrosis (2, 49, 57). Pancreatic fibrosis is characterized by induction of profibrotic growth factors and activation of quiescent stellate cells, which have major roles in the remodeling process.
In pancreatitis patients, activated pancreatic stellate cells (PSCs) alter their phenotype to cells characterized by production of α-smooth muscle actin and extracellular matrix (ECM) proteins. CP-associated structural and functional changes in the pancreas result in deterioration of pancreatic exocrine and endocrine function. Additionally, activated PSCs and pancreatic acinar cells amplify the inflammatory process by attracting neutrophils and macrophages. Similar to CP, repetitive episodes of AP can result in fibrotic remodeling of pancreatic parenchyma (1, 2, 21).
Apelin (APLN), a small peptide, is the endogenous ligand for the APJ receptor [also known as the apelin receptor (APLNR) or ANG II receptor-like 1 (Agtrl1)], a receptor structurally related to the ANG II receptor AT1 (30, 48, 55). The apelin-APJ axis is expressed in the gastrointestinal tract and pancreas (33, 37, 46, 55). Apelin has been reported to influence pancreatic exocrine secretion (18). Additionally, apelin exerts a broad anti-inflammatory activity in the body. Apelin has an inhibitory activity against human immunodeficiency virus infection (3, 58), reduces proinflammatory cytokine production in spleen cells (13), and, in the mouse cardiovascular system, lowers abdominal aortic aneurysm formation by reducing disease-associated vascular inflammation (23). Our laboratory (16) reported that intestinal apelin is involved in the inflammatory response of the intestine. Emerging data indicate a role for apelin in regulation of fibrosis in the kidney, heart, and liver (24, 29, 32, 45). Whether the apelin-APJ signaling system is involved in regulation of pancreatic fibrosis during pancreatitis has not been investigated.
Objectives for the present study are as follows: 1) to define the influence of experimental AP and CP on expression levels of apelin and APJ in the mouse pancreas, 2) to examine the influence of apelin exposure on pancreatic neutrophil recruitment, serum chemoattractant levels, and pancreatic production of ECM-associated proteins during pancreatitis, 3) to examine the effects of apelin on PDGF-simulated proliferation and connective tissue growth factor (CTGF) production in cultured PSCs, and 4) because NF-κB is an important transcription factor thought to play a major role in regulation of pancreatic inflammation and fibrosis during pancreatitis (11, 12, 17, 31, 47), to examine the influence of apelin exposure on pancreatic NF-κB binding in mice with AP and CP.
Our findings demonstrate that pancreatic apelin expression and APJ expression are increased robustly during experimental AP and CP in mice. Further data imply that the enhanced apelin-APJ signaling functions to regulate the pancreatic inflammatory and fibrosis responses during pancreatitis. In apelin-deficient mice with pancreatitis, pancreatic neutrophil recruitment and ECM-associated protein production are enhanced significantly. Apelin treatment will reduce neutrophil infiltration and ECM protein production. Further data suggest that inhibition of neutrophil invasion is mediated by an apelin-induced reduction of keratinocyte chemokine (KC) and granulocyte colony-stimulating factor (G-CSF) secretion. In apelin-deficient mice with AP or CP, pancreatic NF-κB activation is enhanced, and apelin administration in CP suppresses NF-κB activation. Together, our findings imply that the pancreatic apelin-APJ axis exerts an inhibitory influence on pancreatic inflammation, fibrosis, and acinar degeneration during pancreatitis. Apelin's salutary effects may be mediated partly by inhibition of pancreatic NF-κB activity.
MATERIALS AND METHODS
Animals
All animal experiments were done in accordance with mandated standards of humane care and were approved by the University of Texas Medical Branch Institutional Animal Care and Use Committee. Global apelin-deficient [apelin−/−, or apelin-knockout (APKO)] mice were generated by targeted homologous recombination (4, 43). APKO mice are on a mixed 129SvJ-C57Bl/6 background. Wild-type (WT), age-matched littermates were used as control mice. All mice were fed a standard diet and water ad libitum. AP was induced in adult male mice by administration of supramaximal doses of cerulein (50 μg/kg ip, 7 injections hourly, including 0 h, for 6 h) (8). CP was induced by five hourly injections of cerulein (50 μg/kg ip) on Monday, Wednesday, and Friday mornings for three consecutive weeks (28, 41, 50). Mice were euthanized at selected intervals after the start of cerulein injections, and the pancreas was removed immediately for tissue fixation and preparation of total cellular RNA and total protein or nuclear protein extracts. In one experiment, mice were subjected to two AP episodes 24 h apart. For measurement of serum amylase levels, trunk blood samples were collected upon euthanization, and serum was prepared by centrifugation. Serum amylase levels were measured using the Phadebas amylase test (Pharmacia and Upjohn Diagnostics, Uppsala, Sweden) (8). Serum levels of G-CSF and KC were measured using the Bio-Plex Pro mouse cytokine 23-plex panel (Bio-Rad) according to the manufacturer's instructions (10). Pancreatic myeloperoxidase (MPO) activity was measured as described elsewhere (54).
Pancreatic Acinar Cells and Islets
Mouse and human acinar cells are isolated by the enzymatic dissociation method (1, 22, 56). Briefly, the pancreas was perfused with saline, minced with fine scissors, and digested for 15 min in 3 ml of warm isolation buffer [PBS with Ca2+ and Mg2+, 0.01% soybean trypsin inhibitor, 0.1% BSA, and collagenase type IV (0.3 mg/ml)]. Digestion was facilitated mechanically by continuous pipetting for 15 min. Digested tissues were washed with 6 ml of cold isolation buffer and collected by centrifugation at 1,000 rpm for 2 min. The washing step was repeated twice to remove small debris and blood cells. Acini were further purified by filtration through a sterile 860-μm mesh to remove large debris. Acini were collected by centrifugation at 1,000 rpm for 2 min.
Mouse pancreatic islets were isolated using the manual technique (22). Briefly, mice were anesthetized, a midline incision was made, the liver was flipped, and the pancreatic duct was clamped at the duodenal ampulla. After isolation, the pancreatic duct was cannulated, and cold (4°C) enzyme solution (collagenase, ∼1.2 mg/ml) was injected to completely distend the pancreas, which was then extirpated. The pancreas was digested, and cells were separated by density gradients.
Human pancreatic islets were isolated in a US Food and Drug Administration-registered isolation facility. The isolation was performed following a semiautomated method (35, 36). Briefly, pancreatic islets were isolated under sterile conditions from the pancreas of human cadaveric multiorgan donors. All experiments generating human islets and PSCs were done with permission of the University of Texas Medical Branch Internal Review Board for Use of Human Materials. The pancreas was divided at the neck-head junction, and the pancreatic duct was cannulated on both specimens. Human Liberase solution at 4°C was injected at programmed pressure using a perfusion apparatus to completely distend the glands. The pancreas was then minced into seven to nine pieces and placed in an acrylic digestion chamber with stainless beads and a screen. Fluid was recirculated through the digestion system, increasing the temperature to 37–38°C, while the chamber was shaken using a mechanical shaker. The pancreas was digested, and samples were observed through an inverted microscope following dithizone staining. When free islets were observed, digestion was interrupted and tissue was collected. Purification was performed by density gradients using a COBE 2991 cell separator.
PSC Cultures
Mouse and human PSC cultures were prepared by the outgrowth method (1, 6, 22) from normal 129Sv mice or from freshly resected, nonpathological human pancreas. Human PSCs were cultured in high-glucose DMEM containing 10% FBS, insulin-transferrin-selenium supplement X (1%), antibiotic-antimycotic (1%), gentamicin (50 μg/ml), and nonessential amino acids. Mouse PSCs were cultured in high-glucose DMEM containing 10% FBS. Histological examination of stellate cell cultures showed a typical stellate morphology with reduced levels of vitamin A and cells immunopositive for fibronectin and α-smooth muscle actin (data not shown) (6).
Chemicals
All chemicals were obtained from Sigma (St. Louis, MO), Fisher Scientific (Pittsburgh, PA), Invitrogen (Carlsbad, CA), and MP Biomedicals (Santa Ana, CA), unless noted otherwise. Cell culture media were purchased from Mediatech (Herndon, VA) and Life Technologies (Carlsbad, CA). RNAqueous kits (Ambion, Foster City, CA) were used to prepare total cellular RNA from cultured cells. Cerulein was purchased from Bachem California (Torrance, CA); human Liberase from Roche Applied Science (Pleasanton, CA); collagen-4, p50, p65, CTGF, and fibronectin antibodies from Santa Cruz Biotechnology; and IκBα and IκBβ antibodies from Cell Signaling Technology (Danvers, MA).
Real-Time RT-PCR Analysis of Apelin, APJ, and 18S rRNA Expression Levels
For measurement of pancreatic apelin and APJ mRNA and 18S rRNA expression levels, total cellular RNA was extracted and purified from pancreatic homogenates according to published procedures (8, 16, 55). For measurement of apelin expression levels in cell lines, total cellular RNA was extracted and purified using RNAqueous kits.
The real-time RT-PCR analysis was run in two steps under the conditions specified. The ΔΔCT analysis method was used. RNA samples for real-time analysis were quantified using a spectrophotometer (Nanodrop Technologies) and quantified by analysis on an RNA Nano or Pico chip using the Agilent 2100 Bioanalyzer (Agilent Technologies). Synthesis of cDNA was performed with 1 μg of total RNA in a 20-μl reaction using the reagents in the TaqMan reverse transcription reagents kit (catalog no. N8080234, Applied Biosystems). The reaction conditions were as follows: 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min. Quantitative PCR (qPCR) amplifications (in triplicate) were done using 1 μl of cDNA in a total volume of 25 μl using TaqMan MGB probes with the TaqMan Universal PCR Master Mix (catalog no. 4304437, Applied Biosystems), as specified by the manufacturer. The final concentrations of the probe and primers were 250 and 900 nM, respectively. Relative RT-qPCR assays were performed with 18S rRNA as a housekeeping gene. Absolute analysis was performed using known amounts of a synthetic transcript of the gene of interest. All PCR assays were run in the Prism 7500 sequence detection system (Applied Biosystems) under the following conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles at 95°C for 15 s and then at 60°C for 1 min. For mouse apelin and APJ (Agtrl1), TaqMan gene expression assay ID no. Mm00443562_m1, TaqMan gene expression assay ID no. Mm00442191_s1, and TaqMan rRNA control reagents (catalog no. 4308329) were used.
RNA Slot-Blot and Western Blot Analyses
Pancreatic collagen-1α1 and collagen-4 mRNA levels and 18S rRNA levels were measured using previously described methods (8, 9, 16, 55). Pancreatic collagen-4, IκBα, IκBβ, and CTGF protein levels were measured by Western blot analysis (51, 52). An enhanced chemiluminescence detection kit was used to develop blots for visualization.
Preparation of Nuclear Protein Extracts and EMSA
Pancreatic nuclear protein extracts were prepared according to a previously published protocol described in detail elsewhere (14, 15). Protein concentrations in nuclear extracts were determined by protein assay (Bio-Rad Laboratories, Hercules, CA). EMSAs were done using a double-stranded oligonucleotide containing the consensus sequences for NF-κB (5′-AGT TGA GGG GAC TTT CCC AGG C-3′; Promega). Approximately 50,000 cpm of [32P]DNA were added to the nuclear extract, and the preparations were incubated with 50 ng/μl poly(dI-dC)μ-poly (dI-dC) and 5 ng/μl poly l-lysine in binding buffer [20 mM HEPES (pH 7.6), 1 mM DTT, 30 mM KCl, 10 mM (NH4)2SO4, 0.2% Tween 20, and 1 mM EDTA] at room temperature for 20 min. DNA-protein complexes were resolved on a 8% polyacrylamide gel in 0.5 μM TBE (Tris base, boric acid, and EDTA) buffer. Gels were dried and autoradiographed with intensifying screens at −70°C. For competition experiments, nuclear extracts were incubated with a 100-fold molar excess of WT or mutated nonradiolabeled oligonucleotides (Santa Cruz Biotechnology) at room temperature for 10 min before addition of radiolabeled probe. For supershift experiments, ∼6 μg of specific antibodies against NF-κB protein p65 or p50 were incubated in the binding reaction mixture for 60 min at room temperature before addition of radiolabeled probe.
EMSAs were used to assess the influence of apelin exposure on NF-κB activation in mice subjected to AP or CP. In mice not subjected to intraperitoneal injections or subjected to intraperitoneal saline injections, EMSA studies showed nominal pancreatic NF-κB activation (data not shown). In this study, nuclear NF-κB binding was measured in pancreata harvested before (controls), 1 h, and 1 and 3 wk after single/repetitive intraperitoneal saline injection(s) (n = 5 mice per time point).
Histological Analyses
For histological assessment, pancreatic tissue was fixed in 10% buffered formaldehyde, embedded in paraffin, and sectioned. For neutrophil quantitation in pancreatic sections, neutrophils were immunolabeled with a Gr-1 monoclonal antibody (BD Biosciences, San Jose, CA). The Gr-1 antibody will identify monocytes as well as neutrophils. Monocytes are involved in AP pathogenesis (39, 44). Gr-1-immunopositive cells were counted in at least five high-power (×40) fields. For assessment of pancreatic fibrosis, entire pancreatic tissue sections immunostained for fibronectin were scored on a scale of 0–5. Pancreatic acinar degeneration was assessed by scoring acinar vacuole (0–4) and pancreatic tubular complex (0–5) densities. For each mouse, the pancreas was assessed blindly at three levels. Images were captured with a Nikon Eclipse E600 microscope equipped with a digital camera using SPOT imaging software (Diagnostic Instruments, Sterling Heights, MI). Localization of pancreatic NF-κB was determined by immunostaining for p65.
Experiments
Experiment 1.
The aims of this experiment were 1) to measure apelin and APJ expression levels in islets, acinar cells, and stellate cells of the mouse and human pancreas and 2) to define the influence of AP or CP on pancreatic apelin and APJ expression (mRNA) levels.
Experiment 2.
The aim of this experiment was to examine the influence of apelin exposure on pancreatitis-induced elevations in pancreatic neutrophil recruitment and MPO activity. Two AP episodes were induced 24 h apart by supramaximal cerulein administration. Synthetic pyro-apelin-13 (50 μg per ip injection) or saline was given daily (at 0800 and 1800) starting at the time of cerulein administration. In some mice, serum was harvested for measurement of serum G-CSF and KC levels.
Experiment 3.
The aim of this experiment was to examine the influence of apelin exposure on production of pancreatic ECM-associated proteins in mice with CP.
Experiment 4.
The aim of this experiment was to determine whether apelin exposure affects PSC density and CTGF production.
Experiment 5.
By means of EMSAs, the aim of this experiment was to analyze the influence of apelin exposure on pancreatic nuclear NF-κB accumulation during induction of AP or CP. In addition, in mice subjected to CP, cytosol levels of IκBα and IκBβ were measured by Western blot analysis. Cellular localization of NF-κB was examined in the pancreas by immunohistochemistry in mice subjected to CP.
Statistical Analyses
Values are means ± SE. Data were analyzed by t-test or one-way ANOVA and subsequently with Newman-Keuls test when appropriate. In the neutrophil experiment (experiment 2), data were analyzed using the Kruskal-Wallis test. For all comparisons, P < 0.05 was considered significant.
RESULTS
Apelin and APJ Are Expressed in Pancreatic Cells: AP and CP Trigger Upregulation of Pancreatic Apelin and APJ Expression
Mouse and human pancreatic islets and acinar and stellate cells were isolated for measurements of apelin and APJ expression levels. Table 1 shows that apelin and APJ are expressed in mouse and human pancreatic islets and acinar and stellate cells.
Table 1.
Relative apelin and APJ expression in normal pancreas
| Acini | Islets | PSCs* | |
|---|---|---|---|
| Mouse | |||
| Apelin | 1.4 | 0.6 | 12.5 |
| APJ | 1.2 | 0.3 | 1.3 |
| Human | |||
| Apelin | 0.03 | 1.0 | 19.0 |
| APJ | 5.1 | 10.0 | 0.1 |
Apelin and APJ mRNA and 18S rRNA levels were measured by real-time RT-PCR; apelin and APJ mRNA levels are normalized to 18S rRNA levels. PSCs, pancreatic stellate cells.
Isolated by outgrowth method.
In AP and CP experiments, cerulein-induced AP or CP was confirmed by acute elevations of serum amylase levels and histological evaluation of the pancreas (data not shown). In mice with cerulein-induced CP, serum amylase levels increased during each cerulein-induced cycle of AP and then declined to control or near-control levels following each pancreatitis episode.
In mice with AP, pancreatic apelin mRNA levels decreased marginally, but significantly, 7 h after the start of AP induction compared with control apelin mRNA levels (0 h = 1st cerulein injection; Fig. 1A). Pancreatic apelin expression levels increased approximately two- and fivefold 24 and 48 h, respectively, after the start of AP induction. Pancreatic apelin expression levels continued to increase at 72 h (9-fold) and 96 h (12-fold) after the start of AP induction. At 20 days after the start of AP induction, pancreatic apelin mRNA levels declined significantly but remained significantly elevated compared with pancreatic apelin expression levels before induction of AP.
Fig. 1.

Relative pancreatic apelin and APJ expression levels at selected intervals in mice with cerulein-induced acute pancreatitis (AP) or chronic pancreatitis (CP). Apelin and APJ mRNA and 18S rRNA levels were measured by real-time RT-PCR. Apelin and APJ are normalized to 18S rRNA levels. Values are means ± SE; n = 6–8 mice per group. *P < 0.05 vs. control (Con). †P < 0.05 vs. preceding value.
In mice with AP, pancreatic APJ mRNA levels increased significantly (∼1.6-fold) 7 h after the start of AP induction (Fig. 1B). Pancreatic APJ mRNA levels continued to increase 5.9- and 7.3-fold at 24 and 48 h, respectively, after the start of AP induction. Pancreatic APJ mRNA levels remained elevated (∼7-fold) at 72 and 96 h after the start of AP induction. At 20 days after the start of AP induction, pancreatic APJ mRNA levels declined significantly but remained significantly elevated compared with pancreatic APJ expression levels before induction of AP.
CP was induced by three series of cerulein injections administered weekly for three consecutive weeks (see materials and methods). Pancreata were harvested at selected intervals during CP induction. Pancreatic apelin expression levels increased marginally, but significantly, 24 and 48 h following a single series of cerulein injections (Fig. 1C). Pancreatic apelin expression levels continued to increase progressively during weeks 2 and 3 of cerulein injections. Apelin expression levels were maximal at the 3-wk sampling time. Pancreatic apelin expression levels increased ∼46-fold compared with pre-cerulein levels. Pancreatic APJ expression levels increased marginally, but significantly, 24 and 48 h following a single series of cerulein injections (Fig. 1D). Pancreatic APJ expression levels continued to increase progressively during weeks 2 and 3 of cerulein injections. APJ expression levels were maximal at the 3-wk sampling time. Pancreatic APJ expression levels increased ∼80-fold compared with pre-cerulein levels. In mice subjected to CP and then allowed to recover for 1 wk (i.e., no injections during recovery), pancreatic apelin and APJ mRNA levels decreased significantly compared with 3-wk expression levels but remained elevated significantly compared with pre-cerulein (control) levels. In mice subjected to CP and allowed to recover for 5 wk, pancreatic apelin and APJ expression levels did not differ significantly from control levels.
A potential influence of saline injections on pancreatic apelin and APJ expression was assessed in mice euthanized before (controls, no injections) and at selected intervals (20 min; 1, 4, 8, and 24 h; and 1 and 3 wk) after intraperitoneal saline injection(s) (n = 5 mice per time point). Relative apelin expression levels fluctuated between 0.5 ± 0.1 and 1.2 ± 0.14. Relative APJ expression levels fluctuated between 0.5 ± 0.1 and 1.0 ± 0.17.
Enhanced Neutrophil Recruitment and MPO Activity in APKO Mice With AP
In this experiment, two AP episodes 24 h apart were induced by supramaximal cerulein administration. In pancreata of WT and APKO mice before induction of pancreatitis, few neutrophils were observed (data not shown).
Mice subjected to two AP episodes were treated with vehicle or apelin during cerulein injections. Neutrophil invasion, 48 and 72 h after induction of the first AP episode, was significantly higher in APKO than WT mice (Table 2, Fig. 2). At 48 h after the start of cerulein injections, apelin treatment of WT or APKO mice reduced neutrophil invasion. Neutrophil invasion, however, remained higher in apelin-treated APKO than WT mice. At 72 h, apelin treatment of APKO mice reduced neutrophil density. In agreement with neutrophil data (Table 2), apelin treatment resulted in reduced pancreatic MPO levels. In apelin-treated WT and APKO mice, MPO levels were 64 ± 13 and 71 ± 8% of respective MPO levels in vehicle-treated groups at 48 h. In apelin-treated WT and APKO mice, MPO levels were 91 ± 13 and 76 ± 10% of respective MPO levels in vehicle-treated groups at 72 h.
Table 2.
Pancreatic neutrophil densities 48 and 72 h after first cerulein injection
| 48 h | 72 h | |
|---|---|---|
| WT + vehicle | 5.0 ± 0.7 | 0.5 ± 0.2 |
| WT + apelin | 2.2 ± 0.3* | 0.4 ± 0.1 |
| APKO + vehicle | 14.9 ± 1.0† | 4.6 ± 1.4† |
| APKO + apelin | 8.6 ± 1.3*† | 1.7 ± 0.2*† |
Values are means ± SE. Neutrophil density, number of neutrophils per unit area; WT, wild-type; APKO, apelin-knockout. Mice were subjected to 2 sequential acute pancreatitis (AP) episodes. Vehicle or apelin (50 μg per ip injection, twice per day (at 0800 and 1800)] was administered daily. Neutrophils were identified by GR1 immunostaining.
P < 0.05 vs. vehicle.
P < 0.05 vs. WT.
Fig. 2.

Photomicrographs showing that apelin treatment reduces neutrophil recruitment in wild-type (WT) and apelin-knockout (APKO) mice subjected to 2 sequential AP episodes induced 24 h apart. Vehicle or apelin [50 μg per ip injection, twice per day (at 0800 and 1800)] was administered daily. Neutrophils are identified by Gr-1 immunostaining. Histological samples were harvested 48 h after start of AP induction. Brown-stained cells are neutrophils (see inset).
Increased Systemic G-CSF and KC Levels in APKO Mice With AP
Serum levels of G-CSF and KC were significantly higher in APKO than WT mice with AP (Table 3). At 24 h after the start of AP induction, serum G-CSF and KC levels were approximately one- to threefold greater in APKO than WT mice.
Table 3.
Elevated serum G-CSF and KC levels in APKO mice with pancreatitis
Values are means ± SE; n =5–6 mice/group. G-CSF, granulocyte colony-stimulating factor; KC, keratinocyte chemokine. Serum was harvested 24 h after AP induction.
P < 0.05 vs. WT.
Apelin Exposure Reduces ECM-Associated Protein Levels in CP
Collagen-4 mRNA expression and protein levels were significantly higher in APKO than WT mice subjected to CP (Fig. 3, A and B). Control (pre-CP induction) collagen-4 protein levels did not differ in WT and APKO mice. In WT and APKO mice subjected to CP and allowed to recover for 1 wk, pancreatic collagen-1α1 expression levels were elevated significantly compared with control mice (pre-CP; Fig. 3C). Collagen-1α1 mRNA levels were significantly higher in vehicle-treated APKO than WT mice. Apelin treatment during the recovery period reduced collagen-1α1 expression levels significantly in WT and APKO mice; however, collagen-1α1 expression levels remained higher in APKO mice.
Fig. 3.

A and B: pancreatic collagen-4 mRNA and protein levels are significantly higher in APKO than WT mice with CP. C: apelin treatment (25 μg per ip injection, twice per day) during a 1-wk recovery period following CP induction suppresses CP-induced elevations of pancreatic collagen-1α1 expression in WT and APKO mice. C: pancreatic collagen-1α1 expression levels are significantly higher in vehicle- or apelin-treated APKO than corresponding WT mice. Values are means ± SE; n = 6 mice per group. *P < 0.05 vs. WT. †P < 0.05 vs. control/pre-CP. ΔP < 0.05 vs. CP + 1-wk recovery (vehicle-treated).
Apelin Inhibits PSC Proliferation and CTGF Protein Levels
Acute PDGF-BB treatment increased density of cultured mouse PSCs by ∼27% (P < 0.05) compared vehicle-treated PSCs (Table 4). Apelin treatment during PDGF-BB treatment reduced PSC density significantly by ∼24%. PDGF-BB treatment also stimulated increased CTGF protein levels in cultured human PSCs (Fig. 4). Apelin treatment significantly inhibited PDGF-BB-stimulated CTGF protein production.
Table 4.
Apelin reduces PDGF-stimulated PSC density
| PDGF + Vehicle | PDGF + Apelin |
|---|---|
| 127 ± 12 | 103 ± 7* |
Values (means ± SE) are %increase in PSC density compared with PSCs not treated with PDGF; n = 7 wells per group. Cultures of mouse PSCs were exposed to PDGF-BB (10−8 M) + vehicle or apelin-13 (10−6 M) for 2 days.
P < 0.05 vs. PDGF + vehicle.
Fig. 4.

Apelin (10−6 M) treatment reduces PDGF-BB (10 nM)-stimulated connective tissue growth factor (CTGF) production in cultured human pancreatic stellate cells (PSCs). Top: Western blot of CTGF protein levels. Western blots represent results for 2 wells per treatment. Bottom: densitometric analysis of Western blots. Values are means ± SE; n = 6 wells per treatment. *P < 0.05 vs. control. †P < 0.05 vs. PDGF + vehicle.
Inhibitory Effect of Apelin Treatment on NF-κB Activation in Mice With AP or CP
Pancreatic NF-κB activation was significantly higher in APKO than WT mice with AP (Fig. 5). The specificity of pancreatic NF-κB binding activity in EMSAs was shown by cold competition and supershift experiments. As depicted in Fig. 6, inclusion of excess, nonradioactive WT, but not mutated, κB oligonucleotide in the EMSA incubation mixture reduced NF-κB binding intensity. Inclusion of p65 or p50 antibodies in the EMSA incubation supershifted the complex. Together, these findings confirm the specificity of NF-κB binding and indicate that p50 and p65 proteins are present in the NF-κB complexes (Figs. 6 and 7).
Fig. 5.

Increased pancreatic NF-κB binding in APKO mice with AP. Top: EMSA showing enhanced pancreatic NF-κB binding in APKO mice with AP compared with WT littermates. Bottom: NF-κB band intensities. Values are means ± SE; n = 6 mice per group. *P < 0.05 vs. control (control/pre-AP values not shown). †P < 0.05 vs. WT.
Fig. 6.

Increased pancreatic NF-κB binding in APKO mice with CP. Right: EMSA showing enhanced pancreatic NF-κB binding in APKO mice with CP compared with WT littermates. Specificity of pancreatic NF-κB binding detected by EMSAs was shown by nonradioactive competition and supershift experiments. In competition experiments, 100× excess of nonradioactive WT, but not mutated, κB oligonucleotide inhibited NF-κB binding (lanes 5 and 6). Inclusion of a p65 or p50 antibody, but not an antibody to an unrelated protein (IgG, lanes 7–9), in the EMSA incubation supershifted the complex (∗). White space between WT and APKO lanes indicates that the 2 lanes for each group are extracted from the same gel, but they are not immediately adjacent to each other. Left: NF-κB band intensities. Values are means ± SE; n = 6 mice per group. *P < 0.05 vs. control (control/pre-CP values not shown). †P < 0.05 vs. WT.
Fig. 7.

Apelin treatment reduces pancreatic NF-κB binding in WT or APKO mice with CP. Vehicle or apelin (50 μg per ip injection, twice per day) treatment was given during a 1-wk recovery period following CP induction. Top: EMSA showing significant reduction of pancreatic NF-κB binding in apelin-treated WT and APKO mice. Bottom: NF-κB band intensities. Values are means ± SE; n = 6 mice per group. *P < 0.05 vs. vehicle-treated group. †P < 0.05 vs. WT.
In mice subjected to CP with and without 1 wk of recovery, pancreatic NF-κB activation was increased significantly in APKO mice compared with WT mice (Figs. 6 and 7). In the mice subjected to CP and allowed to recover for 1 wk, apelin administration to WT or APKO mice reduced nuclear NF-κB activation significantly compared with vehicle-treated mice (Fig. 7).
Cellular localization of activated NF-κB was examined by immunostaining for p65 in mice subjected to CP. Immunostained nuclei were observed primarily in acinar cells (Fig. 8).
Fig. 8.
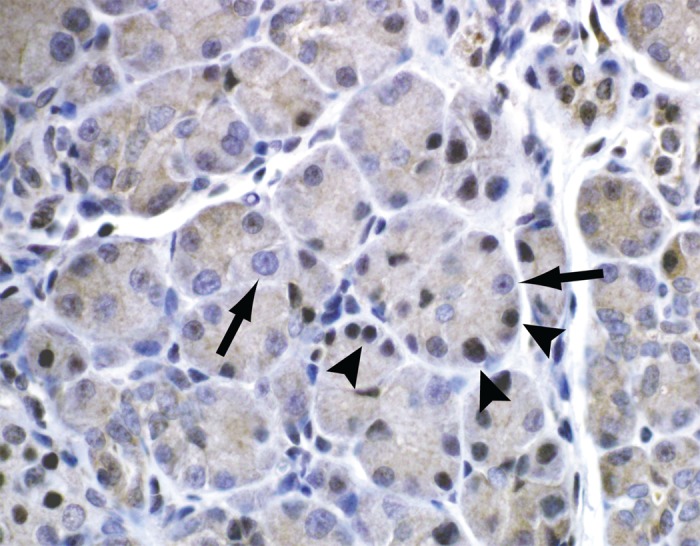
Immunohistochemistry of immunoreactive p65 (NF-κB) in pancreas of mice with CP. Arrowheads identify cells with p65-positive nuclei; arrows identify cells without p65-positive nuclei. p65-positive nuclei occur primarily in acinar cells.
IκBα and IκBβ protein levels were measured in cytosolic protein extracts of pancreata harvested from mice with CP (Fig. 9). Pancreata were harvested prior to (CON) and following the final series (CP) of cerulein injections. CON levels of nonphosphorylated IκBα (IκBα) were significantly lower in APKO than WT mice. In CP mice, IκBα levels decreased significantly compared with CON levels. Phosphorylated IκBα was not detected (data not shown). CON levels of nonphosphorylated IκBβ (IκBβ) and phosphorylated IκBβ (p-IκBβ) did not differ in WT and APKO mice. In WT mice, CP levels of IκBβ, but not p-IκBβ, decreased significantly compared with CON levels. In APKO mice, CP levels of IκBβ and p-IκBβ decreased significantly compared with CON levels in APKO mice.
Fig. 9.

Top: Western blots of pancreatic IκBα and IκBβ protein levels in WT and APKO mice with CP. Pancreata were harvested prior to (CON) and following (CP) the final series of cerulein injections for Western blot analyses. Cerulein injections were given for 3 wk. Bottom: IκBα and IκBβ band intensities. Densitometric values are means ± SE; n = 6 mice per group. p-IκBβ, phosphorylated IκBβ. *P < 0.05 vs. WT. †P < 0.05 vs. corresponding CON value.
Apelin Treatment Improves CP Markers
The salutary influence of apelin exposure on CP markers (inflammation, fibrosis, vacuole, and tubular complex densities) is shown in Table 5. In WT and APKO mice with CP, apelin treatment during a subsequent 1-wk recovery period (50 μg per ip injection, twice per day) significantly reduced pancreatic MPO activity, extent of fibrosis, and acinar degeneration.
Table 5.
Apelin treatment improves CP markers
| Acinar Degeneration |
||||
|---|---|---|---|---|
| Inflammation, MPO activity | Fibrosis | Vacuole density | Tubular complexes | |
| WT + vehicle | 100 ± 20 | 2.7 ± 0.3 | 2.5 ± 0.2 | 4.8 ± 0.1 |
| WT + apelin | 41 ± 5* | 1.7 ± 0.3* | 1.7 ± 0.2* | 3.1 ± 0.3* |
| APKO + vehicle | 100 ± 12 | 3.8 ± 0.2† | 2.6 ± 0.3 | 4.2 ± 0.2† |
| APKO + apelin | 68 ± 13*† | 2.4 ± 0.2*† | 1.3 ± 0.2* | 2.9 ± 0.2* |
Values are means ± SE. Chronic pancreatitis (CP) was induced by repetitive cerulein administration for 3 wk. Vehicle or apelin was given daily during a 1-wk recovery period after CP induction (50 μg ip, twice per day). Myeloperoxidase (MPO) activity (ng/mg protein, transformed to percentage of vehicle-treated) was measured in pancreatic extracts harvested after treatment. Fibrosis (fibronectin-stained slides; scale of 0–5) and acinar vacuole (scale of 0–4) and tubular complex (scale of 0–5) densities were scored for each mouse at 3 different levels.
P < 0.05 vs. vehicle.
P < 0.05 vs. WT.
DISCUSSION
Prior studies indicate that apelin exerts a broad anti-inflammatory activity. Apelin has inhibitory activity against human immunodeficiency virus infection (3, 58), reduces proinflammatory cytokine production in spleen cells (13) and in the mouse cardiovascular system, and lowers abdominal aortic aneurysm formation by reducing disease-associated vascular inflammation (23). Recent reports demonstrate an emerging role for the apelin-APJ axis in regulation of liver, kidney, and heart fibrosis (24, 29, 32, 45).
The present findings show robust elevations in pancreatic apelin and APJ expression during induction of experimental AP or CP in mice. For example, during cerulein-induced AP, pancreatic apelin and APJ expression levels increase ∼7-fold; in mice with CP, pancreatic apelin and APJ expression levels increase ∼46- and 80-fold, respectively. Maximal elevations in pancreatic apelin and APJ expression levels are much higher (∼7- and 11-fold, respectively) in mice with CP than in mice with a single episode of AP. The higher elevations in pancreatic apelin and APJ expression in mice with CP may be linked to the recurrent nature of pancreatic insults during experimental CP induction. Three AP episodes were induced weekly for three consecutive weeks. In both models of pancreatitis, pancreatic apelin and APJ expression levels declined to prepancreatitis or approximate prepancreatitis levels during the postpancreatitis recovery periods.
The large elevations in pancreatic apelin expression during AP or CP were not unexpected. AP and CP are inflammatory diseases, and findings from earlier studies predict increased pancreatic apelin and APJ expression during pancreatitis. In humans with inflammatory bowel disease and in rodents with experimental colitis, intestinal apelin production is increased (16).
The elevations in pancreatic apelin and APJ expression during AP or CP provoke several key questions. 1) In what cell types do apelin and APJ expression levels increase? 2) What mechanism triggers the large elevations in apelin and APJ expression during pancreatitis? 3) What are the physiological functions of these pancreatic apelin and APJ elevations.
Although the present study does not identify in which specific pancreatic cells apelin and APJ expression increase during pancreatitis, our findings confirm and extend earlier reports (18, 37, 46) that the apelin-APJ axis is expressed in islets and acinar cells. This is the first description of the apelin-APJ signaling axis in PSCs. Earlier work by other laboratories indicates that islet and acinar APJ are functional (18, 37, 46). Interestingly, on the basis of the relative magnitudes of apelin expression (Table 1), PSC-derived apelin may be a key source of apelin in autocrine/paracrine apelin-APJ pathways during pancreatitis. Furthermore, our findings demonstrate that PSC APJ is functional, since apelin exposure inhibits PSC proliferation and CTGF production in vitro. Autocrine and/or paracrine apelin-APJ signaling axes are also postulated for the cardiovascular and pulmonary vascular systems (4, 19). Elevations in pancreatic apelin and APJ expression during pancreatitis may partly reflect invasion of immune cells and elevated PSC proliferation. APJ expression occurs in macrophages (G. H. Greeley, Jr., unpublished observations).
Although we did not elucidate mechanisms that trigger the pancreatitis-associated elevations in apelin and APJ expression, results from earlier studies imply that pancreatic hypoxia and elevated proinflammatory cytokine levels drive pancreatic apelin expression during pancreatitis. In several tissues, cellular hypoxia is a potent activator of apelin expression (7, 15, 38). During pancreatitis, pancreatic ischemia is prevalent and O2 tension is expected to decline (5). Furthermore, our laboratory demonstrated that hypoxia activates apelin transcription via a putative hypoxia-inducible factor-binding element in the apelin promoter (15). Hypoxia inducible factor is a transcription factor overexpressed in response to tissue O2 deprivation. Hypoxia can also ramp up APJ transcription (15, 43), and a greatly increased elevation in pancreatic APJ expression is expected to enhance apelin signaling during pancreatitis. Apelin expression is most likely amplified by another mechanism. Pancreatic production of proinflammatory cytokines increases during pancreatitis (21, 27, 34, 42), and proinflammatory cytokines will stimulate apelin transcription via a putative Stat3 binding site in the apelin promoter (14).
A characteristic feature of CP is increased pancreatic synthesis and deposition of ECM-associated proteins, including several collagen types, fibronectin, and laminin (2, 42). Based on our results, we propose that the pancreatic apelin-APJ axis functions to regulate negatively the pancreatic fibrosis response during pancreatitis. This hypothesis is supported by results indicating enhanced collagen-1α1 expression, as well as enhanced collagen-4 expression and protein levels, in APKO mice with CP. Furthermore, apelin treatment following CP induction suppressed the elevation in collagen-1α1 expression. In addition, apelin treatment during a 1-wk recovery period following CP induction reduced the extent of pancreatic fibrosis (Table 5). Pancreatic fibrosis was scored by assessing the amount of fibronectin immunostaining.
In contrast to the inhibitory effect of apelin on pancreatic fibrosis, hepatic fibrosis is stimulated by apelin exposure (24). In human liver disease, apelin seems to mediate profibrogenic gene induction promoted by ANG II and endothelin-1 in hepatic stellate cells. In agreement with the findings in the pancreas, apelin exerts antifibrosis activities in the kidney and heart (29, 32, 45).
The present study shows that apelin exerts a potent inhibitory effect on the inflammatory process during pancreatitis. Neutrophil and macrophage infiltration is a major characteristic of human and experimental pancreatitis, resulting in parenchymal damage and a dysfunctional pancreas (2, 31, 42, 50). In APKO mice subjected to two sequential episodes of AP, neutrophil recruitment was enhanced greatly compared with WT mice. The implied inhibitory role of endogenous apelin on neutrophil invasion is bolstered by the reduced pancreatic neutrophil invasion and MPO levels in apelin-treated WT and APKO mice. In these experiments, the amplified neutrophil recruitment in the pancreas agrees with the enhanced elevations in systemic levels of G-CSF and KC in APKO mice. Serum G-CSF and KC levels were 2.4- to 3.6- and 1-fold higher, respectively, in APKO than WT mice with pancreatitis. G-CSF and KC are important regulators of neutrophil homeostasis. G-CSF exerts a variety of trophic actions on neutrophils to increase their density, and KC is a key chemoattractant for neutrophils (2, 31). The present study also shows that apelin administration during a 1-wk recovery period following CP induction reduced pancreatic MPO levels.
This apelin-induced inhibition of neutrophil recruitment may be one mechanism behind the reduction in pancreatic fibrosis with apelin exposure. During pancreatitis, invading immune cells stimulate PSCs to produce ECM-associated proteins. More importantly, the present studies show, for the first time, inhibition of PSC proliferation and of PDGF-induced CTGF protein production by apelin exposure. These may be additional mechanisms underlying the inhibitory activity of apelin on PSC activity and on ECM protein levels in mice with CP.
The present study demonstrates that apelin exposure exerts an inhibitory influence on pancreatic NF-κB activation during AP and CP. In APKO mice, pancreatic NF-κB activation was enhanced, and apelin treatment in APKO and WT mice lowered NF-κB activation. Previous studies show that nuclear accumulation of NF-κB during pancreatitis involves p50/p65 activation and is mediated by degradation of cytosolic IκBα and IκBβ proteins (11). Our results show decrements in pancreatic IκBs with NF-κB activation. NF-κB activation occurs in all experimental pancreatitis models (11, 12, 31, 47); however, the functional role of NF-κB activation during CP is less studied (50). A recent report shows that prolonged NF-κB activation in a transgenic mouse model results in CP (17). In the present study, apelin-induced reductions of pancreatic ECM protein levels were associated temporally with apelin-induced reductions in pancreatic NF-κB activation, implying that apelin's induced reductions in pancreatic ECM levels are linked to its inhibition of NF-κB activation. Interestingly, evidence of NF-κB involvement in human AP has been reported (40).
Activation of the NF-κB pathway is dependent on phosphorylation of cytosolic IκB proteins with subsequent degradation by the proteasome. In the present studies, higher pancreatic IκB levels agree with a lower nuclear NF-κB accumulation in WT than APKO mice with CP. An important consideration is identity of pancreatic cells wherein apelin exerts its inhibitory effects on NF-κB activation. The present study did not document the locus of apelin's activity; however, prior findings indicate that the bulk of NF-κB activation during pancreatitis occurs in acinar cells (53). Invading inflammatory cells (53) also contribute, implying that acinar and immune cells are primary targets of apelin activity. Results of the present study suggest that apelin's induced reductions in immune cell invasion and PSC proliferation will contribute to reduced NF-κB activation in CP.
Together, our findings indicate that apelin exposure can exert a protective effect on the pancreas during CP. Apelin exposure lowers pancreatic inflammation (MPO levels and immune cell invasion), extent of fibrosis, and features of acinar degeneration (acinar vacuoles and tubular complexes).
It should be mentioned that the apelin-APJ signaling system is expressed outside the gastrointestinal tract and pancreas (20, 33). Therefore, pancreatic changes in APKO mice might reflect a deficit in systemic apelin levels. However, a recent report shows that bioactive apelin does not exist in the general circulation (25). Another consideration is that experimental apelin administration acts at extrapancreatic APJ sites to release substances into the systemic circulation that influence pancreatic function. This possibility will be investigated in future studies by comparing the effects of global and selective pancreatic APJ deletion on pancreatic function during pancreatitis.
GRANTS
This work is supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK-035608.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.H., E.W.E., G.A.G., J.F.A., T.Q., and G.H.G.J. are responsible for conception and design of the research; S.H., J.F.A., C.R., D.K., and R.K.K. performed the experiments; S.H., E.W.E., G.A.G., and G.H.G.J. analyzed the data; S.H., E.W.E., G.A.G., and J.F.A. interpreted the results of the experiments; S.H., E.W.E., and G.H.G.J. prepared the figures; S.H., E.W.E., and G.H.G.J. drafted the manuscript; S.H., E.W.E., G.A.G., and G.H.G.J. edited and revised the manuscript; S.H., E.W.E., G.A.G., J.F.A., C.R., R.P.G., D.K., T.Q., R.K.K., and G.H.G.J. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Eileen Figueroa, Karen Martin, and Steve Schuenke (Department of Surgery, University of Texas Medical Branch) for manuscript and figure preparation and submission.
REFERENCES
- 1. Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A, Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115: 421–432, 1998 [DOI] [PubMed] [Google Scholar]
- 2. Braganza JM, Lee SH, McCloy RF, McMahon MJ. Chronic pancreatitis. Lancet 377: 1184–1197, 2011 [DOI] [PubMed] [Google Scholar]
- 3. Cayabyab M, Hinuma S, Farzan M, Choe H, Fukusumi S, Kitada C, Nishizawa N, Hosoya M, Nishimura O, Messele T, Pollakis G, Goudsmit J, Fujino M, Sodroski J. Apelin, the natural ligand of the orphan seven-transmembrane receptor APJ, inhibits human immunodeficiency virus type 1 entry. J Virol 74: 11972–11976, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY, Finsterbach TP, Leeper NJ, Ernst KV, Chen MM, Ho YD, Chun HJ, Bernstein D, Ashley EA, Quertermous T. Endogenous regulation of cardiovascular function by apelin-APJ. Am J Physiol Heart Circ Physiol 297: H1904–H1913, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dembinski A, Warzecha Z, Ceranowicz P, Tomaszewska R, Dembinski M, Pabianczyk M, Stachura J, Konturek SJ. Ischemic preconditioning reduces the severity of ischemia/reperfusion-induced pancreatitis. Eur J Pharmacol 473: 207–216, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Gao X, Cao Y, Yang W, Duan C, Aronson JF, Rastellini C, Chao C, Hellmich MR, Ko TC. BMP2 inhibits TGF-β-induced pancreatic stellate cell activation and extracellular matrix formation. Am J Physiol Gastrointest Liver Physiol 304: G804–G814, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glassford AJ, Yue P, Sheikh AY, Chun HJ, Zarafshar S, Chan DA, Reaven GM, Quertermous T, Tsao PS. HIF-1 regulates hypoxia- and insulin-induced expression of apelin in adipocytes. Am J Physiol Endocrinol Metab 293: E1590–E1596, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomez G, Englander EW, Wang G, Greeley GH., Jr Increased expression of hypoxia-inducible factor-1α, p48, and the Notch signaling cascade during acute pancreatitis in mice. Pancreas 28: 58–64, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Gomez G, Lambertz I, Udupi V, Qi X, Thompson JC, Greeley GH., Jr Influence of nicotine on gastrin and peptide YY in the rat. Regul Pept 67: 55–61, 1996 [DOI] [PubMed] [Google Scholar]
- 10. Guerrero-Plata A, Kolli D, Hong C, Casola A, Garofalo RP. Subversion of pulmonary dendritic cell function by paramyxovirus infections. J Immunol 182: 3072–3083, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gukovsky I, Gukovskaya A. Nuclear factor-κB in pancreatitis: jack-of-all-trades, but which one is more important? Gastroenterology 144: 26–29, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-κB activation is associated with hormone-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 275: G1402–G1414, 1998 [DOI] [PubMed] [Google Scholar]
- 13. Habata Y, Fujii R, Hosoya M, Fukusumi S, Kawamata Y, Hinuma S, Kitada C, Nishizawa N, Murosaki S, Kurokawa T, Onda H, Tatemoto K, Fujino M. Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim Biophys Acta 1452: 25–35, 1999 [DOI] [PubMed] [Google Scholar]
- 14. Han S, Wang G, Qi X, Englander EW, Greeley GH., Jr Involvement of a Stat3 binding site in inflammation-induced enteric apelin expression. Am J Physiol Gastrointest Liver Physiol 295: G1068–G1078, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han S, Wang G, Qi X, Lee HM, Englander EW, Greeley GH., Jr A possible role for hypoxia-induced apelin expression in enteric cell proliferation. Am J Physiol Regul Integr Comp Physiol 294: R1832–R1839, 2008 [DOI] [PubMed] [Google Scholar]
- 16. Han S, Wang G, Qiu S, de la Motte C, Wang HQ, Gomez G, Englander EW, Greeley GH., Jr Increased colonic apelin production in rodents with experimental colitis and in humans with IBD. Regul Pept 142: 131–137, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Huang H, Liu Y, Daniluk J, Gaiser S, Chu J, Wang H, Li ZS, Logsdon CD, Ji B. Activation of nuclear factor-κB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 144: 202–210, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kapica M, Jankowska A, Antushevich H, Pietrzak P, Bierla JB, Dembinski A, Zabielski R. The effect of exogenous apelin on the secretion of pancreatic juice in anaesthetized rats. J Physiol Pharmacol 63: 53–60, 2012 [PubMed] [Google Scholar]
- 19. Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, Erzurum SC, Chun HJ. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 19: 74–82, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleinz MJ, Skepper JN, Davenport AP. Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept 126: 233–240, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Kloppel G, Detlefsen S, Feyerabend B. Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch 445: 1–8, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Kruse ML, Hildebrand PB, Timke C, Folsch UR, Schafer H, Schmidt WE. Isolation, long-term culture, and characterization of rat pancreatic fibroblastoid/stellate cells. Pancreas 23: 49–54, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Leeper NJ, Tedesco MM, Kojima Y, Schultz GM, Kundu RK, Ashley EA, Tsao PS, Dalman RL, Quertermous T. Apelin prevents aortic aneurysm formation by inhibiting macrophage inflammation. Am J Physiol Heart Circ Physiol 296: H1329–H1335, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Melgar-Lesmes P, Casals G, Pauta M, Ros J, Reichenbach V, Bataller R, Morales-Ruiz M, Jimenez W. Apelin mediates the induction of profibrogenic genes in human hepatic stellate cells. Endocrinology 151: 5306–5314, 2010 [DOI] [PubMed] [Google Scholar]
- 25. Mesmin C, Dubois M, Becher F, Fenaille F, Ezan E. Liquid chromatography/tandem mass spectrometry assay for the absolute quantification of the expected circulating apelin peptides in human plasma. Rapid Commun Mass Spectrom 24: 2875–2884, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Mullady DK, Yadav D, Amann ST, O'Connell MR, Barmada MM, Elta GH, Scheiman JM, Wamsteker EJ, Chey WD, Korneffel ML, Weinman BM, Slivka A, Sherman S, Hawes RH, Brand RE, Burton FR, Lewis MD, Gardner TB, Gelrud A, DiSario J, Baillie J, Banks PA, Whitcomb DC, Anderson MA. Type of pain, pain-associated complications, quality of life, disability and resource utilisation in chronic pancreatitis: a prospective cohort study. Gut 60: 77–84, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muniraj T, Gajendran M, Thiruvengadam S, Raghuram K, Rao S, Devaraj P. Acute pancreatitis. Dis Mon 58: 98–144, 2012 [DOI] [PubMed] [Google Scholar]
- 28. Neuschwander-Tetri BA, Bridle KR, Wells LD, Marcu M, Ramm GA. Repetitive acute pancreatic injury in the mouse induces procollagen α1I expression colocalized to pancreatic stellate cells. Lab Invest 80: 143–150, 2000 [DOI] [PubMed] [Google Scholar]
- 29. Nishida M, Okumura Y, Oka T, Toiyama K, Ozawa S, Itoi T, Hamaoka K. The role of apelin on the alleviative effect of angiotensin receptor blocker in unilateral ureteral obstruction-induced renal fibrosis. Nephron Extra 2: 39–47, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 136: 355–360, 1993 [DOI] [PubMed] [Google Scholar]
- 31. Orlichenko LS, Behari J, Yeh TH, Liu S, Stolz DB, Saluja AK, Singh VP. Transcriptional regulation of CXC-ELR chemokines KC and MIP-2 in mouse pancreatic acini. Am J Physiol Gastrointest Liver Physiol 299: G867–G876, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau-Frugier C, Schaak S, Seguelas MH, Wanecq E, Valet P, Parini A, Kunduzova O. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J 33: 2360–2369, 2012 [DOI] [PubMed] [Google Scholar]
- 33. Pope GR, Roberts EM, Lolait SJ, O'Carroll AM. Central and peripheral apelin receptor distribution in the mouse: species differences with rat. Peptides 33: 139–148, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rakonczay Z, Jr, Hegyi P, Takacs T, McCarroll J, Saluja AK. The role of NF-κB activation in the pathogenesis of acute pancreatitis. Gut 57: 259–267, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW. Automated method for isolation of human pancreatic islets. Diabetes 37: 413–420, 1988 [DOI] [PubMed] [Google Scholar]
- 36. Ricordi C, Rastellini C. Automated method for pancreatic islets separation. In: Methods in Cell Transplantation, edited by Ricordi C. Austin, TX: Landes, 1995, p. 433–438 [Google Scholar]
- 37. Ringstrom C, Nitert MD, Bennet H, Fex M, Valet P, Rehfeld JF, Friis-Hansen L, Wierup N. Apelin is a novel islet peptide. Regul Pept 162: 44–51, 2010 [DOI] [PubMed] [Google Scholar]
- 38. Ronkainen VP, Ronkainen JJ, Hanninen SL, Leskinen H, Ruas JL, Pereira T, Poellinger L, Vuolteenaho O, Tavi P. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. FASEB J 21: 1821–1830, 2007 [DOI] [PubMed] [Google Scholar]
- 39. Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol 69: 249–269, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Satoh A, Masamune A, Kimura K, Kaneko K, Sakai Y, Yamagiwa T, Satoh M, Kikuta K, Asakura T, Shimosegawa T. Nuclear factor-κB expression in peripheral blood mononuclear cells of patients with acute pancreatitis. Pancreas 26: 350–356, 2003 [DOI] [PubMed] [Google Scholar]
- 41. Schmitz JC, Protiva P, Gattu AK, Utsumi T, Iwakiri Y, Neto AG, Quinn M, Cornwell ML, Fitchev P, Lugea A, Crawford SE, Chung C. Pigment epithelium-derived factor regulates early pancreatic fibrotic responses and suppresses the profibrotic cytokine thrombospondin-1. Am J Pathol 179: 2990–2999, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider E, Schmid-Kotsas A, Zhao J, Weidenbach H, Schmid RM, Menke A, Adler G, Waltenberger J, Grunert A, Bachem MG. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am J Physiol Cell Physiol 281: C532–C543, 2001 [DOI] [PubMed] [Google Scholar]
- 43. Sheikh AY, Chun HJ, Glassford AJ, Kundu RK, Kutschka I, Ardigo D, Hendry SL, Wagner RA, Chen MM, Ali ZA, Yue P, Huynh DT, Connolly AJ, Pelletier MP, Tsao PS, Robbins RC, Quertermous T. In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol Heart Circ Physiol 294: H88–H98, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shrivastava P, Bhatia M. Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J Gastroenterol 16: 3995–4002, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Siddiquee K, Hampton J, Khan S, Zadory D, Gleaves L, Vaughan DE, Smith LH. Apelin protects against angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J Hypertens 29: 724–731, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sorhede Winzell M, Magnusson C, Ahren B. The APJ receptor is expressed in pancreatic islets and its ligand, apelin, inhibits insulin secretion in mice. Regul Pept 131: 12–17, 2005 [DOI] [PubMed] [Google Scholar]
- 47. Steinle AU, Weidenbach H, Wagner M, Adler G, Schmid RM. NF-κB/Rel activation in cerulein pancreatitis. Gastroenterology 116: 420–430, 1999 [DOI] [PubMed] [Google Scholar]
- 48. Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 251: 471–476, 1998 [DOI] [PubMed] [Google Scholar]
- 49. Thrower EC, Gorelick FS, Husain SZ. Molecular and cellular mechanisms of pancreatic injury. Curr Opin Gastroenterol 26: 484–489, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Treiber M, Neuhofer P, Anetsberger E, Einwachter H, Lesina M, Rickmann M, Liang S, Kehl T, Nakhai H, Schmid RM, Algul H. Myeloid, but not pancreatic, RelA/p65 is required for fibrosis in a mouse model of chronic pancreatitis. Gastroenterology 141: 1473–1485, 2011 [DOI] [PubMed] [Google Scholar]
- 51. Udupi V, Lee HM, Kurosky A, Greeley GH., Jr Prohormone convertase-1 is essential for conversion of chromogranin A to pancreastatin. Regul Pept 83: 123–127, 1999 [DOI] [PubMed] [Google Scholar]
- 52. Udupi V, Townsend CM, Jr, Greeley GH., Jr Stimulation of prohormone convertase-1 mRNA expression by second messenger signaling systems. Biochem Biophys Res Commun 246: 463–465, 1998 [DOI] [PubMed] [Google Scholar]
- 53. Vaquero E, Gukovsky I, Zaninovic V, Gukovskaya AS, Pandol SJ. Localized pancreatic NF-κB activation and inflammatory response in taurocholate-induced pancreatitis. Am J Physiol Gastrointest Liver Physiol 280: G1197–G1208, 2001 [DOI] [PubMed] [Google Scholar]
- 54. Vigna SR, Shahid RA, Nathan JD, McVey DC, Liddle RA. Leukotriene B4 mediates inflammation via TRPV1 in duct obstruction-induced pancreatitis in rats. Pancreas 40: 708–714, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang G, Anini Y, Wei W, Qi X, O'Carroll AM, Mochizuki T, Wang HQ, Hellmich MR, Englander EW, Greeley GH., Jr Apelin, a new enteric peptide: localization in the gastrointestinal tract, ontogeny, and stimulation of gastric cell proliferation and of cholecystokinin secretion. Endocrinology 145: 1342–1348, 2004 [DOI] [PubMed] [Google Scholar]
- 56. Watanabe H, Saito H, Rychahou PG, Uchida T, Evers BM. Aging is associated with decreased pancreatic acinar cell regeneration and phosphatidylinositol 3-kinase/Akt activation. Gastroenterology 128: 1391–1404, 2005 [DOI] [PubMed] [Google Scholar]
- 57. Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 132: 1557–1573, 2007 [DOI] [PubMed] [Google Scholar]
- 58. Zhou N, Zhang X, Fan X, Argyris E, Fang J, Acheampong E, DuBois GC, Pomerantz RJ. The N-terminal domain of APJ, a CNS-based coreceptor for HIV-1, is essential for its receptor function and coreceptor activity. Virology 317: 84–94, 2003 [DOI] [PubMed] [Google Scholar]