

Abstract
A disintegrin and metalloproteinase 17 (ADAM17), or tumor necrosis factor (TNF)-α-converting enzyme, is a key metalloproteinase and physiological convertase for a number of putative targets that play critical roles in cytokine and growth factor signaling. These interdependent pathways are essential components of the signaling network that links liver function with the compensatory growth that occurs during liver regeneration following 2/3 partial hepatectomy (PH) or chemically induced hepatotoxicity. Despite identification of many soluble factors needed for efficient liver regeneration, very little is known about how such ligands are regulated in the liver. To directly study the role of ADAM17 in the liver, we employed two cell-specific ADAM17 knockout (KO) mouse models. Using lipopolysaccharide (LPS) as a robust stimulus for TNF release, we found attenuated levels of circulating TNF in myeloid-specific ADAM17 KO mice (ADAM17 m-KO) and, unexpectedly, in mice with hepatocyte-specific ADAM17 deletion (ADAM17 h-KO), indicating that ADAM17 expression in both cell types plays a role in TNF shedding. After 2/3 PH, induction of TNF, TNFR1, and amphiregulin (AR) was significantly attenuated in ADAM17 h-KO mice, implicating ADAM17 as the primary sheddase for these factors in the liver. Surprisingly, the extent and timing of hepatocyte proliferation were not affected after PH or carbon tetrachloride injection in ADAM17 h-KO or ADAM17 m-KO mice. We conclude that ADAM17 regulates TNF, TNFR1, and AR in the liver, and its expression in both hepatocytes and myeloid cells is important for TNF regulation after LPS injury or 2/3 PH, but is not required for liver regeneration.
Keywords: partial hepatectomy, tumor necrosis factor-α converting enzyme, lipopolysaccharide, tumor necrosis factor receptor, amphiregulin
following 2/3 partial hepatectomy (PH), the murine liver restores its preoperative mass by compensatory hyperplasia of the remaining parenchyma. Fully differentiated hepatocytes enter the cell cycle, undergo one to two rounds of replication, and return to quiescence. Numerous studies have shown that disruption of several signaling pathways can delay liver regeneration, indicating the requirement for multiple interdependent cytokine and growth factor pathways for efficient regeneration. These studies have established the importance of cytokines during the hepatocyte transition from G0 to G1 of the cell cycle and the central role of growth factors to promote progression through S phase for autonomous replication.
Initiation of the cytokine network in liver regeneration is associated with increased levels of circulating tumor necrosis factor (TNF) and requires TNF receptor 1 (TNFR1) (41). The primary enzyme that cleaves membrane-bound TNF is ADAM17, which belongs to the a disintegrin and metalloproteinase (ADAM) family and is also called TNF-α-converting enzyme (6). ADAM17 activity is posttranslationally regulated by tissue inhibitor of metalloproteinases 3 (TIMP3), its only known physiological inhibitor (1), and TIMP3 knockout (KO) mice have high levels of ADAM17 activity (25). After PH in TIMP3 KO mice, elevated TNF levels initially accelerate the hepatocyte cell cycle, but persistent TNF elevation inhibits complete restoration of liver mass and results in high morbidity (25). In addition to TNF's proliferative effects, this pleiotropic cytokine can stimulate apoptosis during tissue repair or exacerbate liver damage by inducing excessive inflammation (21). Murthy et al. demonstrated that Fas-induced hepatotoxicity is delayed in TIMP3 KO mice and is abrogated in those mice when combined with TNF or TNFR1 deficiency (27). We have previously shown that mice lacking TNFR1 have deficient hepatocyte proliferation after inflammatory injury induced by carbon tetrachloride (CCl4) (40). These studies collectively suggest that the physiological effects of TNF release vary greatly depending on the stage and context of liver injury and demonstrate that ADAM17 is likely to be involved in regulation of the hepatocyte response to resection or injury.
In addition to TNF, ADAM17 has been shown in vitro to shed other proteins from their latent precursor forms, including the epidermal growth factor receptor (EGFR) ligands heparin-binding epidermal growth factor-like growth factor (HB-EGF), amphiregulin (AR), and transforming growth factor-α (TGF-α) (36). Following the priming phase of regeneration, these growth factors, among others, drive cell cycle progression through S phase (37). Binding of these ligands to EGFR activates well-characterized mitogenic pathways, such as those of ERK1/2 and protein kinase B (Akt), but the mechanisms that generate ligand availability are not well understood and may involve ADAM17.
We have previously shown that TNF exerts its proliferative influence in cultured hepatocytes through EGFR transactivation, during which ADAM17 liberates pro-TGF-α to a soluble form that binds EGFR (2). These in vitro data suggest that TNF may trigger EGFR transactivation by stimulating ADAM17 activity during liver regeneration. Furthermore, ADAM17 protein was recently shown to be upregulated in the livers of rats following PH (20). Taken together, these studies suggest that ADAM17 is essential for normal liver regeneration. Despite the evidence demonstrating the importance of ADAM17 substrates in liver regeneration, there are no published studies in which ADAM17's role has been directly examined and no data regarding the relative contribution to ADAM17 activity by the different cell types of the liver.
Although hepatocytes account for the majority of liver mass and volume, nonparenchymal cells (NPCs), including Kupffer cells (KCs), which are the resident liver macrophages, play key roles in the liver's response to injury. Activation of KCs is essential for optimal regeneration after PH, possibly via production of TNF and IL-6 (5). Given the intimate relationship between NPCs and hepatocytes, we aimed to define the in vivo role of ADAM17 in both cell types during liver injury and regeneration. To determine the requirement for ADAM17 expression in hepatocytes, we evaluated systemic lipopolysaccharide (LPS) injury, 2/3 PH, and acute CCl4 injury in mice with hepatocyte-specific ADAM17 inactivation or control mice. To complement those experiments and to determine the role of ADAM17 in KCs, we also assessed liver injury and regeneration in mice lacking ADAM17 in the myeloid cell lineage.
MATERIALS AND METHODS
Animals.
The targeting construct in which exon 5 of the Adam17 gene is flanked by loxP sites (Adam17FlNeo/FlNeo mice) has been previously described and demonstrated to enable efficient ablation of ADAM17 expression using a ubiquitous or cell-specific Cre recombinase driver (38). Mouse embryos (E17.5) that are homozygous for ubiquitous exon 5 deletion exhibit the same open-eye phenotype as embryos harboring the Adam17ΔZn/ΔZn construct originally developed by Peschon et al. (30). Adam17FlNeo/FlNeo mice were generated on a C57BL/6J background. Hepatocyte-specific inactivation of ADAM17 was achieved by breeding Adam17FlNeo/FlNeo mice with mice expressing Cre recombinase under control of the albumin promoter (Alb-Cre+), purchased from The Jackson Lab (Bar Harbor, ME), yielding ADAM17 h-KO mice. Mice expressing Cre under control of the M lysozyme promoter (LysM-Cre+) were purchased from The Jackson Lab and backcrossed four additional generations on the C57BL/6J background to achieve a total of 10 backcrosses onto C57BL/6J. Myeloid-specific inactivation of ADAM17 was achieved by breeding Adam17FlNeo/FlNeo mice with LysM-Cre+ mice (9), yielding ADAM17 m-KO mice. ADAM17 h-KO and ADAM17 m-KO mice were born with normal Mendelian ratios and had normal fecundity, health, and lifespans. Furthermore, no discernable differences were seen in liver morphology in Cre-negative;Adam17FlNeo/FlNeo, Alb-Cre;Adam17FlNeo/FlNeo, or LysM-Cre;Adam17FlNeo/FlNeo mice when compared with C57BL/6J mice, as assessed by hematoxylin and eosin (H&E) staining. Mx1- Cre+ mice were also purchased from The Jackson Lab (no. 003556), and Cre-negative;Adam17FlNeo/FlNeo mice were used as controls for all experiments. All mice were housed in a specific pathogen-free facility with 12:12-h light-dark cycles and free access to standard chow and water. All experiments were approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at the University of Washington.
Surgical procedures and administration of LPS and CCl4.
Male mice (8–12 wk old) were fasted overnight before operations performed between 8:00 A.M. and 12:00 P.M. Mice were anesthetized via isoflurane inhalation (4–5% for induction and 1–2% for maintenance) and underwent 2/3 PH or sham laparotomy performed by the same surgeon (R. McMahan) using a modification of the Higgins and Anderson method as previously described (7, 14). For LPS experiments, mice were fasted overnight and then injected intraperitoneally with 1 μg/g body wt Escherichia Coli 055:B9 LPS (L-2880; Sigma-Aldrich, St. Louis, MO) diluted in saline. Another cohort of mice (8–12 wk old) was injected intraperitoneally with a single dose of CCl4 (0.3 ml/kg) diluted 1:10 in olive oil. At the indicated times, mice were killed by CO2 inhalation. Blood was collected via cardiac puncture or retro-orbital eye bleed. Liver tissue was snap-frozen for protein or RNA extraction or fixed in 10% formalin/PBS and embedded in paraffin for histological analyses. For determining liver-to-body weight ratios after PH, liver remnants were weighed after removal of necrotic stumps and sutures and compared with postoperative body weight.
Histology.
For all mice killed 24 h or later after PH or 48 h or later after CCl4 injection, bromodeoxyuridine (BrdU) (50 μg/g body wt) was injected intraperitoneally 2 h before death. Liver lobes were fixed and stained with a monoclonal anti-BrdU antibody (Dako, Carpentaria, CA) as previously described (7). The Vectastain mouse on mouse kit (Vector Labs, Burlingame, CA) was used to detect BrdU labeling in both NPCs and hepatocytes. Mitotic indexes were determined by counting the number of hepatocyte mitotic figures (nuclei in mitosis) per ∼3,000 hepatocytes, using H&E-stained tissue. The area of necrosis on H&E-stained tissue was determined semiquantitatively using NIH Image J software.
RNA isolation and real-time RT-PCR analysis.
Total RNA was isolated from whole liver, primary hepatocytes, or NPCs and extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). For cDNA synthesis, 1–2 μg RNA were reverse transcribed using the Retroscript kit (Invitrogen), and real-time RT-PCR was performed using FAM-labeled Taqman probes for Adam17 (Mm01231075_m1 or Mm00456428_m1), Tnf (Mm00443258_m1), Adam10 (Mm00545742_m1), and 18s (Hs99999901_s1) from Applied Biosystems (Carlsbad, CA) and a RG 3000 thermocycler (Corbett Research, San Francisco, CA). Relative gene expression was calculated using 18s rRNA expression as an internal control, followed by normalization of all values to those of male, age-matched, nonoperated Cre- controls using the ΔΔCt method.
ELISA.
Serum or tissue levels of proteins were determined using ELISA kits from BD Biosciences (Franklin Lakes, NJ) for TNF (no. 555268) and IL-6 (no. 555240), and from R&D Systems (Minneapolis, MN) for AR (DY989) and TNFR1 (DY425), according to the manufacturer's instructions.
Liver transaminase and caspase-3 activity assays.
Serum levels of the hepatocellular enzymes alanine transaminase and aspartate transaminase were measured by kinetic assays at 37°C on a microplate reader (680XR; Bio-Rad, Hercules, CA) as previously described (18). For caspase-3 activity assays, whole liver lysates were prepared in Nonidet P-40 buffer and quantified as described previously (7). Caspase-3 activity in whole liver lysates (50 μg) was determined using a fluorogenic substrate (Ac-DEVD-AMC; Enzo Life Sciences, Farmingdale, NY) as previously described (8).
Isolation of primary hepatocytes and NPCs.
Primary hepatocytes were isolated from ADAM17 h-KO and control mice by collagenase perfusion and Percoll gradient enrichment as previously described (16). For isolation of NPCs, hepatocytes were removed from collagenase perfusates by centrifugation for 3 min at 50 g. NPCs were then pelleted from the supernatant by centrifugation for 15 min at 300 g. The pellets were resuspended in 17.5% Opti-Prep Density Gradient Medium (Sigma-Aldrich) in DMEM + F-12 (Invitrogen) and then topped off with 2 ml DMEM + F-12. The gradients were centrifuged for 15 min at 1,400 g, yielding a small layer of sinusoidal cells, collected by pipette. The cell suspensions were washed with 10 ml DMEM + F-12, centrifuged for 10 min at 1,400 g, and resuspended in DMEM + F-12 (35).
Liver macrophage enrichment.
For collection of liver macrophages, CD11b+ cells were isolated from liver sinusoidal cell suspensions by magnetic separation. Briefly, cells were blocked with anti-mouse CD16/31 Fc blocking antibody (eBioscience, San Diego, CA) at 0.5 μg per 1 × 106 cells. Cells were then incubated with Alexafluor 647-conjugated anti-CD11b antibody (eBioscience), washed, and incubated with anti-Alexafluor 647 microbeads (Miltenyi Biotec, Auburn, CA). After additional washes, cell suspensions were applied to MACS MS separation columns (Miltenyi Biotec) in a magnetic field. The columns were rinsed with buffer to collect CD11b-negative cells and then removed from the magnetic field to collect CD11b+ cells.
Statistical analyses.
Statistical analysis was done by two-way ANOVA with Bonferroni posttest or nonparametric analysis, using either the Kruskal-Wallis test followed by Dunn's test or an unpaired t-test with Welch's correction. Data are presented as averages ± SE, with P < 0.05 considered statistically significant. Prism 4 software (GraphPad Software, La Jolla, CA) was used for all statistical analyses.
RESULTS
Generation of liver cell type-specific ADAM17 KO mouse models.
Because constitutive deletion of ADAM17 causes perinatal lethality (30), we developed two conditional ADAM17 KO mouse models in which ADAM17 is specifically inactivated in hepatocytes (ADAM17 h-KO) or myeloid cells (ADAM17 m-KO). The hepatocyte-specific albumin promoter is established as an effective driver of Cre expression (31), but we sought to determine its efficiency with a floxed Adam17 target. We isolated RNA from whole liver extracts and performed real-time RT-PCR using primers that detect endogenous Adam17 transcripts, but not those from which the loxP-flanked exon 5 has been excised. We found a 65–70% reduction in endogenous Adam17 in ADAM17 h-KO whole livers compared with controls (Fig. 1A), consistent with excision from hepatocytes, but not NPCs. To subtract residual expression in NPCs, we performed collagenase perfusions and cell separations and found that, in Percoll-purified, hepatocyte-enriched perfusates, endogenous Adam17 levels were reduced >95% in ADAM17 h-KO mice compared with controls (Fig. 1B). As expected, Adam17 expression is similar in CD11b+ liver NPCs from control and ADAM17 h-KO mice (Fig. 1C), as well as in CD11b-negative NPCs (data not shown). These data indicate that ADAM17 inactivation in hepatocytes is efficient and specific, and the residual expression observed in whole liver extracts from ADAM17 h-KO can be primarily attributed to NPCs.
Fig. 1.
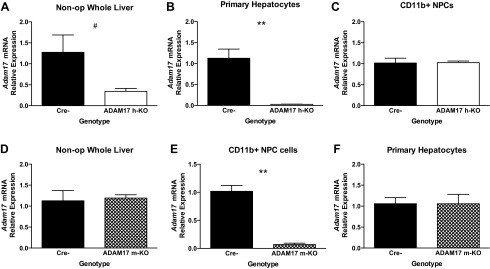
Tissue-specific inactivation of a disintegrin and metalloproteinase 17 (ADAM17). Data presented are the results of real-time RT-PCR analyses using 18S expression for normalization. A: reduced expression of Adam17 in whole liver RNA from unoperated mice with hepatocyte-specific ADAM17 deletion (ADAM17 h-KO) (open bars) (n = 4–5). B: confirmed knockdown of Adam17 mRNA expression in primary hepatocytes from ADAM17 h-KO mice (n = 4–6). C: Adam17 mRNA levels in CD11b+ nonparenchymal cells (NPC) are similar in Cre- (black bars) and ADAM17 h-KO mice (n = 2–4). D: similar expression of Adam17 mRNA in whole liver RNA from Cre- and myeloid-specific ADAM17 KO (ADAM17 m-KO) mice (hatched bars) (n = 5). E: abrogated Adam17 expression in CD11b+ NPC cells from ADAM17 m-KO mice (n = 3–4) compared with controls. F: primary hepatocytes isolated from Cre- and ADAM17 m-KO mice express similar levels of Adam17 mRNA (n = 5). Data are presented as fold change using the values from Cre- controls as the denominator and are presented as means ± SE. #P = 0.09 and **P < 0.01. Non-op, unoperated mice.
As the resident liver macrophages, KCs are the major source of TNF after PH (5). We therefore developed a second, complementary mouse model to enable studies of the relative contribution of KCs to ADAM17 activity in the liver after PH. We crossed Adam17FlNeo/FlNeo mice with mice expressing LysM-Cre, which specifically targets cells of the myeloid lineage, including KCs (9), to generate ADAM17 m-KO mice. As expected, given that the majority of liver cells are hepatocytes, we detected similar basal levels of Adam17 transcript in livers from control and ADAM17 m-KO mice (Fig. 1D). To determine whether ADAM17 was successfully inactivated in KCs, we isolated primary NPCs after collagenase perfusion and further selected for CD11b+ cells by magnetic sorting. We found that endogenous Adam17 mRNA levels in the CD11b+ fraction were significantly reduced (by ∼93%) in ADAM17 m-KO mice compared with controls (Fig. 1E), whereas levels in the CD11b-negative fractions were not significantly different between the two groups (data not shown). Adam17 expression in primary hepatocytes isolated from ADAM17 m-KO mice was nearly identical to that of controls, as expected (Fig. 1F). These data indicate that ADAM17 expression is efficiently inactivated in the CD11b+ cell population in the liver, which includes a large subset of KCs.
Both hepatocyte and myeloid ADAM17 play a role in LPS-stimulated TNF release.
To test the effect of ADAM17 deficiency on TNF shedding, we injected ADAM17 m-KO, ADAM17 h-KO, and control mice with LPS and measured serum TNF levels by ELISA 90 min later. We observed a significant elevation of circulating TNF after LPS injection in control mice that was abrogated in mice with myeloid ADAM17 deficiency, as previously reported (15) (Fig. 2A). Unexpectedly, we found that release of TNF in the serum was also reduced in ADAM17 h-KO mice (Fig. 2A). This was surprising since Kupffer cells are thought to be the major source of TNF during hepatic inflammatory processes (13). To exclude the possibility that low levels of serum TNF in ADAM17-deficient mice were due to diminished Tnf expression, we performed real-time RT-PCR to measure Tnf transcription in the liver. We found that, 90 min after LPS injection, Tnf mRNA was markedly induced in all groups, suggesting that the effect of ADAM17 deficiency on serum TNF was the result of posttranscriptional processes such as shedding, and not inhibition of Tnf expression (Fig. 2B).
Fig. 2.

Inhibition of tumor necrosis factor (TNF) and tumor necrosis factor receptor 1 (TNFR1) release after lipopolysaccharide (LPS) injection in ADAM17 m-KO and ADAM17 h-KO mice. Data presented are from samples collected 90 min after injection of saline or LPS (1 μg/g body wt). A: serum levels of TNF, as determined by ELISA, are significantly lower in ADAM17 h-KO and ADAM17 m-KO mice after injection of LPS compared with controls (n = 4–5). B: LPS induction of Tnf mRNA in the liver is similar between Cre-, ADAM17 m-KO, and ADAM17 h-KO mice (black, hatched, and open bars, respectively) (n = 2–5). C: serum levels of TNFR1, as determined by ELISA, are lower in ADAM17 h-KO and ADAM17 m-KO mice after injection of LPS compared with controls (n = 3–4). D: serum levels of IL-6 are significantly lower in ADAM17 h-KO mice, but not ADAM17 m-KO mice, compared with control mice after LPS injection (n = 5). ELISA data are presented as means ± SE. Real-time RT-PCR data are presented as fold change using the values from untreated Cre- controls as the denominator and are presented as means ± SE. *P < 0.05 and **P < 0.01 compared with Cre- group and normalized to 18S expression. ND, not detectable. Cre- represents control Adam17FlNeo/FlNeo mice.
We next examined levels of TNFR1, another putative ADAM17 substrate, whose soluble form is elevated in the sera of mice after LPS injection (3, 34). We found that soluble TNFR1 increased significantly in control mice after LPS injection, but this increase was diminished in both ADAM17 m-KO and ADAM17 h-KO mice (Fig. 2C). These results suggest that myeloid and hepatocyte-expressed ADAM17 are each partially responsible for shedding TNFR1 after LPS injection. To determine the net effect of ADAM17 deficiency on TNFR1 signaling, we measured levels of circulating IL-6 and found that release of IL-6 was slightly reduced in ADAM17 m-KO mice and significantly attenuated in ADAM17 h-KO mice (Fig. 2D). However, NF-κB activation, as assessed by upregulation of target genes Il-6, Tnfiap3, Birc2, and Nfkbia was not affected by ADAM17 deficiency 90 min after LPS injection (data not shown). Collectively, these data demonstrate effective reduction of ADAM17 activity in ADAM17 h-KO and ADAM17 m-KO mice and for the first time demonstrate a role for hepatocyte-expressed ADAM17 in LPS-stimulated TNF shedding and IL-6 release.
Liver expression of cytokine and growth factor substrates of ADAM17 is reduced in ADAM17 h-KO mice after PH.
Given that TNF and IL-6 are important in the early phases of liver regeneration (11), and our previous demonstration that ADAM17 links TNF to growth factor signaling in vitro (2), we hypothesized that ADAM17 is critical to normal liver regeneration. To determine whether hepatocyte-expressed ADAM17 is important during liver regeneration, we examined levels of ADAM17 substrates in ADAM17 h-KO mice after PH using specific ELISAs. We found significantly less TNF in the livers of ADAM17 h-KO mice at baseline and an even greater deficit 6 h after PH compared with controls (Fig. 3A). Serum levels of TNFR1 transiently increase after PH in rats, but little is known about how it is shed (12). We therefore examined hepatic levels of TNFR1 and observed a transient increase in the livers of control mice 6 h after PH that did not occur in ADAM17 h-KO mice (Fig. 3B), similar to the pattern observed for TNF. Interestingly, IL-6 release was unaffected in ADAM17 h-KO mice, suggesting that the combined effect of deficient TNF and TNFR1 shedding did not alter signaling downstream of TNFR1 after PH (Fig. 3C), as it did after LPS injection.
Fig. 3.

Suppression of cytokine and growth factor induction in the liver after partial hepatectomy (PH) in ADAM17 h-KO mice, determined by specific ELISAs. A: levels of TNF in liver tissue after PH are significantly lower in ADAM17 h-KO mice (open bars) compared with controls (black bars) (n = 3–5). B: levels of TNFR1 are lower in liver tissue from ADAM17 h-KO mice compared with controls after PH (n = 3–5). C: levels of serum IL-6 are similar in ADAM17 h-KO mice and control mice after PH (n = 2–6). D: upregulation of amphiregulin protein is abrogated after PH in ADAM17 h-KO mice compared with controls (n = 3–6). Data are presented as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with Cre- controls; n = 3–5 mice.
Given that in vitro studies have implicated ADAM17 as a sheddase for AR (36), and that AR-deficient mice have impaired hepatocyte proliferation after PH (4), we measured levels of AR protein in the liver after PH. We found that liver expression of AR was significantly lower in ADAM17 h-KO mice at baseline, and that the induction of AR observed in control mice after PH was abrogated in ADAM17 h-KO mice, which had ∼85% less AR than controls at 6 h (Fig. 3D). Collectively, these data indicate that ADAM17 expressed in hepatocytes is an important regulator of key cytokine and growth factor signaling pathways that are activated after PH.
DNA replication after PH does not require ADAM17 in hepatocytes.
Given the profound effects of hepatocyte ADAM17 deficiency on levels of substrates known to be involved in liver regeneration, we asked whether Adam17 expression is upregulated after 2/3 PH. We found that expression of Adam17 mRNA is indeed induced, beginning 6 h after PH and reaching statistical significance at 36–40 h (Fig. 4A). We next examined hepatocyte proliferation in ADAM17 h-KO mice after PH. DNA synthesis, as determined by BrdU incorporation, peaked 36–40 h after PH as expected in Cre- control mice, but was not significantly affected by ADAM17 deficiency (Fig. 4B). Interestingly, ADAM17 h-KO mice had fewer mitotic figures 48 h after PH but no deficits at other time points (Fig. 4C). Restoration of liver mass, as assessed by liver-to-body weight ratio, was also unperturbed in ADAM17 h-KO mice (Fig. 4D). PH also induces proliferation of macrophages and other liver NPCs following hepatocyte proliferation (22). We found similar levels of DNA synthesis in NPCs of ADAM17 h-KO and control mice at 72 h after PH (data not shown). These data indicate that expression of ADAM17 in hepatocytes is not required for liver regeneration after PH. We were surprised that the AR deficit in ADAM17 h-KO mice did not lead to impaired liver regeneration and thus hypothesized that other EGFR ligands compensated for the lack of AR. We measured activation of the EGFR after PH by phosphoimmunoblotting (Tyr1068), and found no difference in the activation of this receptor in ADAM17 h-KO mice compared with controls (data not shown). Activation of Akt and ERK, two pathways downstream of the EGFR and assessed by phosphoimmunoblotting, was also similar after PH in ADAM17 h-KO mice and controls. Thus, there are likely ADAM17-independent proliferative pathways that compensate for the deficits in AR in ADAM17 h-KO mice.
Fig. 4.

Liver regeneration after PH is largely unaffected in ADAM17 h-KO mice. A: liver Adam17 expression in Cre- controls is upregulated after PH compared with basal levels (n = 3–5). B: staining for bromodeoxyuridine (BrdU) indicates no effect of hepatic ADAM17 deficiency on DNA synthesis after PH (n = 3–7). C: the overall extent of mitosis after PH is similar in control and ADAM17 h-KO mice, except at the 48-h time point (n = 3–6). D: restoration of liver mass, assessed by liver-to-body weight (BW) ratio, after PH is unaffected by ADAM17 deficiency in hepatocytes (n = 3–6). Data are presented as means ± SE. *P < 0.05 compared with Cre- mice.
Myeloid ADAM17 expression regulates TNF induction after PH.
The minimal effect of hepatocyte ADAM17 deficiency on liver regeneration was unexpected, considering the impaired regeneration reported in TNFR1 and AR KO mice and the abrogated induction of these substrates in ADAM17 h-KO mice. We reasoned that liver NPCs might represent another important source of ADAM17 activity after PH, since our analysis of ADAM17 expression indicated that ∼30% of liver Adam17 mRNA is in NPCs (Fig. 1A). To determine the relative importance of ADAM17 activity in KCs, we performed PH on ADAM17 m-KO mice and Cre- controls. In ADAM17 m-KO mice, induction of TNF in the liver was completely blocked, as it was in ADAM17 h-KO mice, suggesting that ADAM17 activity in both cell types regulates TNF levels after PH (Fig. 5A). Levels of TNFR1 in the liver were similar in controls and ADAM17 m-KO mice before and after PH, however (Fig. 5B). Hepatocyte DNA synthesis at 40 and 48 h after PH was not affected by ADAM17 deficiency in myeloid cells (Fig. 5C). Consistent with these data, the number of mitotic hepatocytes at 48 h was similar in ADAM17 m-KO mice and Cre- mice (Fig. 5D), and liver:-to-body weight ratios of ADAM17 m-KO and control mice were nearly identical during the first 5 days after PH (Fig. 5E). Collectively, these results suggest that myeloid ADAM17 regulates TNF induction but is unnecessary for hepatocyte proliferation after PH.
Fig. 5.

Induction of liver TNF after PH is abrogated in ADAM17 m-KO mice, but liver regeneration is not affected. A: levels of TNF protein in the liver after PH are significantly lower in ADAM17 h-KO mice compared with controls (n = 3–5). B: liver levels of TNFR1 protein in ADAM17 m-KO mice are not significantly different from controls at baseline or 6 h after PH (n = 3). C and D: DNA synthesis, as determined by BrdU incorporation (n = 4) (C) and mitosis (n = 4–5) (D) are unaffected in ADAM17 h-KO mice compared with Cre- littermates. E: restoration of liver mass, determined by liver-to-body weight ratio, is similar in ADAM17 h-KO mice and Cre- controls (n = 3–6). Data are presented as means ± SE. ***P < 0.001.
Liver regeneration after CCl4-induced hepatotoxicity does not require ADAM17 expression in hepatocytes or myeloid cells.
Administration of CCl4 induces significant hepatocyte proliferation in mice but differs from PH in that the response to CCl4 occurs in an environment of significant inflammation and centrilobular necrosis (19). We have previously demonstrated that mice lacking TNFR1 have deficient liver regeneration induced by CCl4 (40), so we next asked whether ADAM17 is required for liver regeneration in this context. We first assessed liver injury by serum levels of alanine transaminase and aspartate transaminase and found similar levels in ADAM17 h-KO and control mice after injection of CCl4 (data not shown). We examined hepatocyte proliferation by BrdU incorporation and mitotic indexes and found no significant differences in ADAM17 h-KO mice compared with controls (data not shown), indicating that ADAM17 expression in hepatocytes is not required for CCl4-induced proliferation.
Release of TNF from KCs is thought to exacerbate liver damage in mice after CCl4 treatment. We thus asked whether ADAM17 inactivation in myeloid cells has a protective effect against CCl4-induced liver damage. Circulating levels of liver transaminases at 48 h after injection were significantly attenuated in ADAM17 m-KO mice compared with controls, suggesting that ADAM17 activity in KCs may increase hepatocyte death (Fig. 6, A and B). However, direct assessment of liver injury by measuring necrotic area on H&E sections revealed no differences between ADAM17 m-KO mice and controls (Fig. 6, C–E). To assess the extent of apoptosis after CCl4, we measured caspase-3 activity and found that it is only modestly induced, and its induction is independent of ADAM17 (Fig. 6F). Consistent with this finding, hepatocyte proliferation was unaffected in ADAM17 m-KO mice after CCl4 injection (Fig. 6, G and H). Taken together, these data indicate that expression of ADAM17 in hepatocytes or myeloid cells does not affect the extent of liver injury after CCl4 injection and is not required for hepatocyte proliferation in this model.
Fig. 6.

Liver regeneration after carbon tetrachloride (CCl4)-induced hepatotoxicity in ADAM17 m-KO mice. A and B: CCl4-induced release of liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) is significantly reduced in ADAM17 m-KO mice compared with controls at 48 h (n = 3–4). TACE: tumor necrosis factor-α-converting enzyme. C and D: significant necrosis (traced) is evident in representative images of liver sections from Cre- and ADAM17 h-KO mice 48 h after CCl4 injection. E: necrotic liver area, as determined using NIH Image J on hematoxylin and eosin (H&E)-stained sections, is similar in ADAM17 m-KO mice and controls (n = 3–6). F: caspase-3 activity in liver tissue is unaffected by ADAM17 inactivation in myeloid cells (n = 3–5). G and H: DNA synthesis and mitosis after CCl4 injection are unaffected by ADAM17 deficiency in myeloid cells (n = 3–6). Data are presented as means ± SE. ***P < 0.001 compared with Cre- mice. Scale bars = 500 μm.
DISCUSSION
Liver regeneration after 2/3 PH or chemically induced injury requires the interplay of cytokine and growth factor signaling between multiple cell types. Although the essential cytokines and growth factors have been defined, the enzymes responsible for shedding and activating these molecules during liver regeneration are largely unknown. The work presented here marks the first direct examination of the sheddase ADAM17's function in liver regeneration. In the present study, we used cell-specific KO mice to inactivate ADAM17 in hepatocytes or myeloid cells and investigated the role of ADAM17 in the response to LPS injection, 2/3 PH, and acute CCl4-induced hepatotoxicity. The main findings of this study are: 1) both myeloid and hepatocyte expression of ADAM17 play a role in LPS-induced TNF shedding; 2) ADAM17 is a major regulator of TNF, TNFR1, and AR after PH; and 3) ADAM17 expressed by hepatocytes or myeloid cells is not essential for hepatocyte proliferation after PH or acute CCl4 injury.
Our laboratory has previously established the importance of TNFR1 activation after PH (41), and experiments in mice deficient for TIMP3, the only known endogenous ADAM17 inhibitor, indirectly implicate ADAM17's role in regulation of TNF signaling in the liver (25, 27). Because TIMP3 inhibits multiple proteases, it was unclear from these latter studies whether ADAM17 is a primary regulator of TNF during liver regeneration. We show here that ADAM17 regulates hepatic TNF and TNFR1 induction after PH, since lack of ADAM17 correlates with loss of both ligand and receptor protein. It was therefore expected that the deletion of ADAM17 in either hepatocytes or myeloid cells would impair liver regeneration. However, our examination of multiple time points after PH shows unequivocally that reduced levels of TNF and TNFR1 after surgery do not result in significantly altered hepatoctye proliferation.
Although Kupffer cells are believed to be the major source of TNF during liver regeneration (5, 23), very little is known about TNF shedding during regeneration. We show that roughly 1/3 of baseline Adam17 expression in the liver is expressed in NPCs, consistent with the observation that myeloid-expressed ADAM17 contributes to TNF induction after PH. It was surprising, however, to see a lack of induction of TNF when ADAM17 was deleted from hepatocytes. These results suggest that both myeloid and hepatocyte ADAM17 play a role in TNF induction after PH and that there is cross talk between these cell types with regard to TNF regulation in the liver. Because this potential interaction may be more broadly applicable to TNF regulation, it will be important to confirm the mechanisms involved.
After PH, priming by cytokines such as TNF and IL-6 is followed by stimulation of hepatocyte proliferation by growth factors. Transcription of Ar, an EGFR ligand, is rapidly induced after PH, and hepatocyte proliferation is impaired in AR KO mice (4). We show that the induction of AR protein is abrogated in ADAM17 h-KO mice, implicating ADAM17 as a major regulator of AR in the liver. EGFR activation is key to hepatocyte cell cycle progression, and rodents in which liver EGFR is conditionally inactivated have impaired liver regeneration after PH (28, 29). TGF-α and HB-EGF, which are other EGFR ligands and hepatocyte mitogens, are also cleaved by ADAM17 in cell culture (32). Unfortunately, currently available commercial antibodies are unable to detect cleaved TGF-α or HB-EGF in whole liver lysates from mice, precluding our assessment of the effects of ADAM17 deficiency on their shedding in vivo. Regardless, we could not detect any differences in activation of the EGFR or downstream signaling pathways in ADAM17 h-KO mice (data not shown). Furthermore, although the frequency of mitotic figures observed in ADAM17 h-KO mice at 48 h after PH was statistically lower than controls, the deficit was short-lived and did not affect recovery of liver mass (Fig. 4, C and D). The absence of a phenotype similar to that reported in AR KO mice could be attributed to a requirement for only low levels of AR protein to maintain EGFR activation, signaling mediated by membrane-bound AR, or compensation by other EGFR ligands in ADAM17 h-KO mice that does not occur in AR KO mice.
Given the decreased hepatic levels of TNF, TNFR1, and AR after PH in ADAM17 h-KO mice, we predicted that liver regeneration would be delayed. However, we observed only a transient decrease in mitosis in these mice, rather than a robust delay in hepatocyte proliferation. A number of explanations are possible for our findings. First, it is possible that ADAM17 has different substrates depending on the nature of the regenerative stimuli. Thus, to complement our PH experiments, we also examined liver regeneration after acute CCl4 injury, wherein hepatocyte proliferation occurs in the context of necrosis and inflammation. However, we observed only minimal effects of ADAM17 deficiency on liver regeneration in this injury model. Second, it is possible that sufficient amounts of TNFR1 may remain membrane-bound to facilitate downstream TNF signaling events even in the presence of reduced levels of TNF ligand. This notion is supported by our observation of unaffected IL-6 serum levels after PH in ADAM17 h-KO mice, which is consistent with intact TNFR1 signaling. Thus the net effect of decreased levels of both TNF and TNFR1 was a nearly normal regenerative response. Third, other ADAMs may play critical regulatory roles in liver regeneration, in addition to ADAM17. We found that Adam10 expression increases significantly after PH in both control and ADAM17 h-KO mice (data not shown). If enzymatic redundancy exists, however, it is not sufficient to prevent the marked attenuation of substrate induction observed in ADAM17 h-KO mice. Finally, it is possible that cytokines or growth factors that are regulated in an ADAM17-independent manner may compensate or act redundantly when ADAM17-dependent signaling is lost. Indeed, hepatocyte growth factor (HGF) has not been shown to be an ADAM17 substrate but is a major regulator of liver regeneration (24).
Given that ADAM17 is expressed on multiple liver cell types, it is plausible that ADAM17 expressed on the nontargeted cell types may be sufficient to allow normal liver regeneration. To address the possibility of cellular redundance, we performed PH in Adam17FlNeo/FlNeo mice crossed with mice expressing an inducible Mx1-Cre, enabling total liver knockout and reducing the window for adaptive mechanisms to arise (17). Unfortunately, we found that the Mx1-Cre inducer, polyinosinic:polycytidylic acid, independently delayed hepatocyte proliferation after PH (data not shown), making it impossible to dissociate the effects of ADAM17 inactivation from those of the inducing agent.
To assess TNF shedding in our models, we employed LPS injection, which is a robust stimulator of TNF shedding and is commonly used to model endotoxemia in mice (13, 26). Knockout of TIMP3 leads to increased LPS-induced release of TNF and TNFR1, a net increase in TNFR signaling, and increased LPS-induced mortality in mice (34). Additionally, mice with myeloid ADAM17 deficiency have been previously demonstrated to have attenuated TNF shedding after LPS injection, suggesting that myeloid cells are the primary source of circulating TNF (15). The significant reduction of LPS-induced serum TNF that we observed in ADAM17 h-KO mice, which have intact myeloid ADAM17, was thus surprising, although it was corroborated by our similar findings after PH.
TNFR1 shedding is thought to regulate TNF signaling, balancing resistance to bacterial infection and ensuing inflammation (39). In another model of TNF-mediated liver injury, Fas-induced hepatitis, excessive TNFR1 shedding protected TIMP3 KO mice, indirectly implicating ADAM17 activity, which should be elevated in these mice (27). It is possible that decreased circulating TNF in ADAM17 h-KO mice could be attributed to prolonged presence of hepatocyte-bound TNFR1, to which soluble TNF could immediately bind. However, our data do not support this mechanism, since it would lead to increased signaling downstream of TNFR1, and increased IL-6 release. Instead, ADAM17 m-KO and ADAM17 h-KO mice actually have lower levels of serum IL-6 after LPS injection than controls. We examined the expression of multiple NF-kB-dependent genes and did not see a difference in activation in ADAM17-deficient mice at 90 min after LPS injection (data not shown). Interestingly, coculture of primary NPCs with hepatocytes augments release of TNF from LPS-treated NPCs, and this effect requires close cell interactions (33). We found that, in primary cultures, ADAM17 KO hepatocytes treated with LPS released slightly less TNF into the media, but a large dose (100 μg/ml) was required to stimulate shedding above baseline levels (data not shown). These data suggest that relatively little TNF is expressed in hepatocytes and are consistent with a requirement for myeloid-hepatocyte interactions for TNF shedding. We hypothesize that ADAM17-dependent cross talk or trans activity may occur to enable TNF release, a mechanism that warrants investigation in future studies.
This study is the first to implicate ADAM17 as the primary regulator of TNF, TNFR1, and AR after PH. Intriguingly, we show that ADAM17 expression in both hepatocytes and myeloid cells regulates TNF in the liver. Efficient hepatocyte proliferation without normal induction of these key ligands raises the possibility that ADAM17-independent compensatory signaling pathways are involved, perhaps through other promitogenic pathways such as HGF/met as mentioned above. This notion is supported by reports of interactions between EGFR and Met pathways during liver regeneration (10, 29). Elucidation of the mechanisms by which hepatocyte and myeloid cell interactions regulate signaling by TNF and EGFR ligands will have important clinical ramifications in both liver disease and other TNF-mediated inflammatory conditions.
GRANTS
This work was funded by National Cancer Institute Grants CA-23226, CA-174131, and CA-127228 (to N. Fausto and J. S. Campbell), the Herbert Coe Foundation and the American College of Surgeons (to K. J. Riehle), and an National Institute of Environmental Health Sciences training program T32-ES-007032 predoctoral fellowship (to R. S. McMahan).
DISCLOSURES
The authors have no conflicts of interest to report.
AUTHOR CONTRIBUTIONS
Author contributions: R.S.M., N.F., and J.S.C. conception and design of research; R.S.M. performed experiments; R.S.M. analyzed data; R.S.M., K.J.R., N.F., and J.S.C. interpreted results of experiments; R.S.M. prepared figures; R.S.M. drafted manuscript; R.S.M., K.J.R., N.F., and J.S.C. edited and revised manuscript; R.S.M., K.J.R., N.F., and J.S.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Carole Wilson and Elaine Raines for providing the Adam17FlNeo/FlNeo mice. We thank Sebastian Yuen, Renay Bauer, and Vicki Hoagland for technical assistance and Brian Hayes and Jocelyn Wright for critiquing this manuscript.
REFERENCES
- 1.Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knäuper V, Docherty AJ, Murphy G. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett 435: 39–44, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Argast GM, Campbell JS, Brooling JT, Fausto N. Epidermal growth factor receptor transactivation mediates tumor necrosis factor-induced hepatocyte replication. J Biol Chem 279: 34530–34536, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Bemelmans MH, Gouma DJ, Buurman WA. LPS-induced sTNF-receptor release in vivo in a murine model. Investigation of the role of tumor necrosis factor, IL-1, leukemia inhibiting factor, and IFN-gamma. J Immunol 151: 5554–5562, 1993 [PubMed] [Google Scholar]
- 4.Berasain C, García-Trevijano ER, Castillo J, Erroba E, Lee DC, Prieto J, Avila MA. Amphiregulin: an early trigger of liver regeneration in mice. Gastroenterology 128: 424–432, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int 26: 1175–1186, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385: 729–733, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Campbell JS, Riehle KJ, Brooling JT, Bauer RL, Mitchell C, Fausto N. Proinflammatory cytokine production in liver regeneration is Myd88-dependent, but independent of Cd14, Tlr2, and Tlr4. J Immunol 176: 2522–2528, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Chaisson ML, Brooling JT, Ladiges W, Tsai S, Fausto N. Hepatocyte-specific inhibition of NF-kappaB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J Clin Invest 110: 193–202, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277, 1999 [DOI] [PubMed] [Google Scholar]
- 10.Factor VM, Seo D, Ishikawa T, Kaposi-Novak P, Marquardt JU, Andersen JB, Conner EA, Thorgeirsson SS. Loss of c-Met disrupts gene expression program required for G2/M progression during liver regeneration in mice. PLoS One 16: 5, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology 43: S45–S53, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Fulop AK, Pocsik E, Brozik M, Karabelyos C, Kiss A, Novak I, Szalai C, Dobozy O, Falus A. Hepatic regeneration induces transient acute phase reaction: systemic elevation of acute phase reactants and soluble cytokine receptors. Cell Biol Int 25: 585–592, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, Murakami T, Drutskaya LN, Förster I, Clausen BE, Tessarollo L, Ryffel B, Kuprash DV, Nedospasov SA. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity 22: 93–104, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol 186–202, 1931 [Google Scholar]
- 15.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol 179: 2686–2689, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Kovalovich K, Li W, DeAngelis R, Greenbaum LE, Ciliberto G, Taub R. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-xL. J Biol Chem 276: 26605–26613, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science 269: 1427–1429, 1995 [DOI] [PubMed] [Google Scholar]
- 18.Langdale LA, Hoagland V, Benz W, Riehle KJ, Campbell JS, Liggitt DH, Fausto N. Suppressor of cytokine signaling expression with increasing severity of murine hepatic ischemia-reperfusion injury. J Hepatol 49: 198–206, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leevy CM, Hollister RM, Schmi DR, MacDonald RA, Davidson CS. Liver regeneration in experimental carbon tetrachloride intoxication. Proc Soc Exp Biol Med 102: 672–675, 1959 [DOI] [PubMed] [Google Scholar]
- 20.Lin XM, Liu YB, Zhou F, Wu YL, Chen L, Fang HQ. Expression of tumor necrosis factor-alpha converting enzyme in liver regeneration after partial hepatectomy. World J Gastroenterol 14: 1353–1357, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luster MI, Simeonova PP, Gallucci RM, Bruccoleri A, Blazka ME, Yucesoy B. Role of inflammation in chemical-induced hepatotoxicity. Toxicol Lett 120: 317–321, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Malik R, Selden C, Hodgson H. The role of non-parenchymal cells in liver growth. Semin Cell Dev Biol 13: 425–431, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Meijer C, Wiezer MJ, Diehl AM, Schouten HJ, Meijer S, van Rooijen N, van Lambalgen AA, Dijkstra CD, van Leeuwen PA. Kupffer cell depletion by CI2MDP-liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver 20: 66–77, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Michalopoulos GK. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Am J Pathol 176: 2–13, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B, Yeh WC, Khokha R. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet 36: 969–977, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Mohler KM, Sleath PR, Fitzner JN, Cerretti DP, Alderson M, Kerwar SS, Torrance DS, Otten-Evans C, Greenstreet T, Weerawarna K. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 370: 218–220, 1994 [DOI] [PubMed] [Google Scholar]
- 27.Murthy A, Defamie V, Smookler DS, Di Grappa MA, Horiuchi K, Federici M, Sibilia M, Blobel CP, Khokha R. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Invest 120: 2731–2744, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci USA 104: 17081–17086, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paranjpe S, Bowen WC, Tseng GC, Luo JH, Orr A, Michalopoulos GK. RNA interference against hepatic epidermal growth factor receptor has suppressive effects on liver regeneration in rats. Am J Pathol 176: 2669–2681, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26: 149–150, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164: 769–779, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott MJ, Liu S, Su GL, Vodovotz Y, Billiar TR. Hepatocytes enhance effects of lipopolysaccharide on liver nonparenchymal cells through close cell interactions. Shock 23: 453–458, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol 176: 721–725, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Steffan AM, Gendrault JL, McCuskey RS, McCuskey PA, Kirn A. Phagocytosis, an unrecognized property of murine endothelial liver cells. Hepatology 6: 830–836, 1986 [DOI] [PubMed] [Google Scholar]
- 36.Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, Lee DC. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem 277: 12838–12845, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Webber EM, Bruix J, Pierce RH, Fausto N. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology 28: 1226–1234, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Wilson CL, Gough PJ, Chang CA, Chan CK, Frey JM, Liu Y, Braun KR, Chin MT, Wight TN, Raines EW. Endothelial deletion of ADAM17 in mice results in defective remodeling of the semilunar valves and cardiac dysfunction in adults. Mech Dev 130: 272–289, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xanthoulea S, Pasparakis M, Kousteni S, Brakebusch C, Wallach D, Bauer J, Lassmann H, Kollias G. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J Exp Med 200: 367–376, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada Y, Fausto N. Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am J Pathol 152: 1577–1589, 1998 [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA 94: 1441–1446, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]