

Abstract
SOX9 regulates cell lineage specification by directly regulating target genes in a discrete number of tissues, and previous reports have shown cell proliferative and suppressive roles for SOX9. Although SOX9 is expressed in colorectal cancer, only a few direct targets have been identified in intestinal epithelial cells. We previously demonstrated increased proliferation in Sox9-deficient crypts through loss-of-function studies, indicating that SOX9 suppresses cell proliferation. In this study, crypt epithelial cells isolated from Sox9-deficient mice were used to identify potential target genes of SOX9. Insulin-like growth factor (IGF)-binding protein 4 (IGFBP-4), an inhibitor of the IGF/IGF receptor pathway, was significantly downregulated in Sox9-deficient intestinal epithelial cells and adenoma cells of Sox9-deficient ApcMin/+ mice. Immunolocalization experiments revealed that IGFBP-4 colocalized with SOX9 in mouse and human intestinal epithelial cells and in specimens from patients with primary colorectal cancer. Reporter assays and chromatin immunoprecipitation demonstrated direct binding of SOX9 to the IGFBP-4 promoter. Overexpression of SOX9 attenuated cell proliferation, which was restored following treatment with a neutralizing antibody against IGFBP-4. These results suggest that SOX9 regulates cell proliferation, at least in part via IGFBP-4. Furthermore, the antiproliferative effect of SOX9 was confirmed in vivo using Sox9-deficient mice, which showed increased tumor burden when bred with ApcMin/+ mice. Our results demonstrate, for the first time, that SOX9 is a transcriptional regulator of IGFBP-4 and that SOX9-induced activation of IGFBP-4 may be one of the mechanisms by which SOX9 suppresses cell proliferation and progression of colon cancer.
Keywords: SOX9, IGFBP-4, colorectal cancer, ApcMin/+ mice, intestinal epithelial cell proliferation
the sex-determining region y-box 9 (SOX9) gene belongs to a superfamily that is characterized by the presence of a homologous high-mobility group (HMG) sequence. The SOX HMG domain binds to specific DNA sequences and activates transcription of target genes. SOX9 regulates cell lineage specification by directly regulating target genes in a discrete number of tissues (2, 3, 6, 9, 24, 30). In normal intestinal epithelium, SOX9 is localized to the nuclei of crypt epithelial cells, which include terminally differentiated Paneth cells, stem cells, and a subset of transit-amplifying (TA) cells (25). Recent reports have shown that SOX9 is also expressed in tuft cells, an additional type of secretory cells that comprise 0.4% of epithelial cells in the villi and crypts, although SOX9 is not critical for their differentiation (17). We and others previously reported that inactivation of the Sox9 gene in mouse intestinal epithelium resulted in increased proliferation, indicating that SOX9 suppresses proliferation in vivo (5, 25). Furthermore, it has been reported that ectopic expression of SOX9 at high levels suppresses proliferation of the intestinal epithelial cell line IEC-18 (16). However, the mechanism by which SOX9 suppresses proliferation or the target for SOX9 activity has not been determined.
Both oncogenic and suppressive roles of SOX9 in various cancers, including breast, prostate, and ovarian cancers and melanomas, have been reported. In intestinal epithelium, some reports have suggested an antioncogenic role for SOX9 (1, 12, 20, 34), while others have demonstrated an oncogenic role (22, 23). A recent genome-scale analysis of human colorectal cancer (CRC) identified SOX9 as one of the frequently mutated genes, and mutations in SOX9 were frame-shift or nonsense mutations (8). However, the role of SOX9 in tumorigenesis has not been determined, and only a few direct targets of SOX9 in intestinal epithelial cells have been identified. These targets include the tumor suppressor carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM 1) (34), PKCα (14), and Bmi1 (23).
IGFBP-4, an insulin-like growth factor binding protein (IGFBP), is expressed in a variety of tissues, and its expression is regulated by different mechanisms in a cell type-specific manner. IGFBPs have rapidly gained attention as key players in a wide range of cellular events, such as proliferation, differentiation, apoptosis, and transformation (21). IGFBPs are important secreted proteins that exhibit high-affinity binding to growth factors (IGF-I and IGF-II) and modulate their bioavailability (11, 13). IGFBP-4 is expressed in a variety of tumor cells, including human CRC cells, where it inhibits the function of IGFs and, thus, reduces cell proliferation and DNA synthesis (31). Epidemiological studies have also shown that high circulating levels of IGF-I and IGF-II are associated with an increased risk for several types of cancers, including colorectal, breast, prostate, lung, and pancreatic cancers (10, 18, 27, 32, 33).
In this study, we identified IGFBP-4 as a SOX9 target gene that contributes to the suppression of proliferation in normal mouse intestinal epithelium and ApcMin/+ adenomas. The antiproliferative activity of SOX9 was further tested in vivo in Sox9-deficient ApcMin/+ mice. We demonstrate for the first time that SOX9 directly regulates IGFBP-4 gene expression in the intestinal epithelium and in CRC and that the resulting expression of IGFBP-4 suppresses cell proliferation in normal intestinal epithelium and CRC cells.
MATERIALS AND METHODS
Mice.
Sox9 was inactivated specifically in the intestinal epithelium (Sox9flox/flox;VilCre+/−), and the phenotypes of these mice are described elsewhere (25). Mice were backcrossed to a C57BL/6 background for >10 generations prior to the experiments. C57BL/6 ApcMin/+ heterozygous mice were purchased from Jackson Laboratory (Bar Harbor, ME) and crossed with Sox9-deficient mice to generate double-mutant (Sox9flox/flox;VilCre+/−;ApcMin/+) mice. All mice were bred and maintained in specific pathogen-free conditions at Baylor College of Medicine. All experiments were performed with the approval of the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Collection of crypt cells.
Intestinal epithelial cells were collected from crypts of jejunum of 3-mo-old Sox9-deficient mice and their littermate controls and subjected to RNA extraction for microarray, as well as quantitative RT-PCR (qRT-PCR). Briefly, inverted small intestinal samples on glass rods were incubated in 1 mM EDTA-Ca2+- and Mg2+-free Hanks' balanced salt solution at 37°C for 5 min and subjected to controlled vibration in Hanks' solution. Cells were collected in ∼12 successive fractions with 5-min incubations in 1 mM EDTA solution between each fraction collection. The final crypt-rich fractions (>98% crypts, on the basis of microscopic examination) were used as crypt-rich samples for the experiments.
RNA extraction and qRT-PCR.
Total RNA was extracted from intestinal crypt epithelial cells of Sox9-deficient mice and their littermate controls or cultured cells using the TRIzol reagent (Life Technologies, Carlsbad, CA) and treated with RNase-free DNase (Roche, Indianapolis, IN). Total RNA from laser capture microdissection samples was isolated using the RecoverAll Total Nucleic Acid Isolation Kit (Life Technologies).
The cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit (Roche). Real-time qRT-PCR analysis was performed using FastStart Universal SYBR Green Master Mix (Roche) and amplified (model 7900HT, Applied Biosystems). Primer sequences are as follows: 5′-CATCAAGACGGAGCAGCTGAG-3′ (forward) and 5′-ATGGTCAGCGTAGTCGTATTG-3′ (reverse) for mouse Sox9, 5′-TCGTGTGTGTGTGTTTATAG-3′ (forward) and 5′-ATTCCTATTGCTACACTCAG-3′ (reverse) for mouse Igfbp-4, 5′-TGCAGGAGGAGAAGAGAAGG-3′ (forward) and 5′-GTGGCCAGTTCACAGCTGC-3′ (reverse) for human SOX9, 5′-CGCAACGGCAACTTCCACC-3′ (forward) and 5′-CAGGCCTCACTCTCGAAAGC-3′ (reverse) for human IGFBP-4, 5′-CGCCCCCAGCAGACTTCAC (forward) and 5′-CTCCTCTTTTGCACCCCTCCCATTT (reverse) for human SOX2, 5′-GGCCTCGAGCTGGGAATCGC (forward) and 5′-GCCCACTCGGGGTCTTGCAC (reverse) for human SOX4, 5′-AGGATCTCGCTGGAAATCAA (forward) and 5′-CTGCCTCATCTCCTGTCTCC (reverse) for human SOX6, and 5′-GAGGCTGAAGAGGCTGACAG (forward) and 5′-ATCTTTCAGTGTGGGTGC (reverse) for human SOX10. The relative changes in gene expression were analyzed using the 2−ΔΔCt method.
Immunohistochemistry and immunofluorescence.
Formalin-fixed and paraffin-embedded (FFPE) sections were used for immunohistochemistry and immunofluorescence. Human primary CRC specimens were obtained from the Dan L. Duncan Cancer Center Tissue Bank at Baylor College of Medicine. The following antibodies were used for immunohistochemistry: rabbit anti-SOX9 (1:1,000 dilution; Millipore, Billerica, MA), rabbit anti-IGFBP-4 (1:200 dilution; Abcam, Cambridge, MA), and ImmPRESS anti-rabbit Ig (peroxidase) polymer detection kit (Vector Laboratories, Burlingame, CA). Bound antibodies were visualized by diaminobenzidine, and sections were counterstained with hematoxylin-eosin. For immunofluorescence, the following antibodies were used: mouse anti-SOX9 (1:1,000 dilution; Abcam), rabbit anti-IGFBP-4 (1:500 dilution; Abcam), rabbit anti-doublecortin-like kinase 1 (DCLK-1, 1:500 dilution; Abcam), and Alexa 488 anti-mouse IgG, Alexa 555 anti-rabbit IgG, and Alexa 488 anti-rabbit IgG (Life Technologies). The slides were mounted in SlowFade Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (Life Technologies).
Counting of IGFBP-4-positive villus cells.
Immunofluorescence of IGFBP-4 in wild-type mouse jejunum sections was used to count IGFBP-4-positive cells in the villus. The number of IGFBP-4-positive cells per 100 villus cells was counted from 20 randomly selected microscopic views at ×400 magnification.
Plasmid construction.
SOX9-pcDNA and HMG-mutated SOX9 plasmids were generously provided by Dr. Benoit deCrombrugghe and Dr. Hideyo Yasuda (University of Texas, M. D. Anderson Cancer Center). The fragment of 2-kb human and mouse IGFBP-4 promoter was cloned into a pGL3 luciferase vector (Promega, Madison, WI). The QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used to make 2-bp mutations in the putative SOX9 binding sites.
Cell culture and transfection.
Caco-2 cells were cultured in Eagle's MEM containing 10% FBS. DLD1, HT-29, HCT116, LS-174T, and Lim6 cells were cultured in DMEM containing 10% FBS. After overnight culture, the basal levels of SOX9 and IGFBP-4 in these cells were evaluated using Western blotting. To overexpress SOX9, SOX9-pcDNA was transfected using FuGENE 6 transfection reagent (Roche). For SOX9 knockdown, human SOX9 small interfering RNA (siRNA; ON-TARGETplus SMARTpool, Thermo Scientific, Lafayette, CO) was transfected using DharmaFECT 1 siRNA Transfection Reagent (Thermo Scientific). For Western blot and quantitative PCR analysis, cells were collected 48 h after transfection. For cell proliferation assays, cells were analyzed 72 h after transfection. Western blot experiments were performed in triplicate using independent samples from treated cells. Rabbit anti-SOX9 (Millipore) and rabbit anti-IGFBP-4 (Santa Cruz Biotechnology, Dallas, TX) were used. Bands were quantified for statistical analysis, and P values were obtained.
Cell proliferation assays.
For the bromodeoxyuridine (BrdU) incorporation assay and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, Caco-2, DLD1, HCT116, and HT-29 cells were seeded at a density of 5,000 cells per well in 96-well plates. After overnight incubation, cells were transiently transfected with SOX9-expressing plasmids or SOX9 siRNA. Each transfection was performed in nine replicate wells and repeated twice. For the experiments in which cultured cells were treated with a neutralizing antibody against IGFBP-4 (Santa Cruz Biotechnology), the antibody was added to each well (final concentration 1 μg/ml) 2 days after transfection. At 3 days after transfection, BrdU incorporation was measured using the Cell Proliferation ELISA-BrdU Kit (Roche) and MTT assays were performed using the MTT Cell Proliferation Assay Kit (American Type Culture Collection, Manassas, VA) according to the manufacturers' instructions. Absorbance from the experimental wells was corrected by basal absorbance from control wells containing culture medium alone, without cells.
Reporter assays.
Cells were transfected with 200 ng of pGL3-reporter plasmids or the pGL3 empty vector, 100 or 400 ng of the expression plasmid, or the empty control vector and 2 ng of the TK standardization vector used to normalize the transfection efficiency. At 48 h after the transfection, luciferase activity was measured using the Dual Luciferase Reporter Assay system (Promega). Renilla luciferase activity was used as the transfection control. Assays were performed in triplicate.
Chromatin immunoprecipitation analysis.
Chromatin immunoprecipitation (ChIP) experiments were performed using the ChIP-IT Express Kit (Active Motif, Carlsbad, CA) with minor modifications. Cells were cross-linked with 1% formaldehyde, and the chromatin was isolated by addition of lysis buffer (Active Motif) and then disrupted with a Dounce homogenizer. The lysates were sonicated to shear the DNA to an average length of 300–500 bp. To prepare input DNA, aliquots were removed and treated with RNase followed by proteinase K and then de-cross-linked. DNAs were purified and quantified. An aliquot of chromatin was precleared with protein A-agarose beads (Life Technologies) and then incubated with an anti-SOX9 antibody (Millipore). The immune complexes were eluted and reverse-cross-linked. ChIP DNAs were purified, and ChIP enrichment was assayed by quantitative PCR (qPCR) in triplicate. The resulting signals were normalized for primer efficiency using qPCR for each primer pair and input DNA to obtain the fold enrichment. The primers were designed to amplify a region covering 2 kb upstream of the transcription start site of the human IGFBP-4 gene, and a control primer pair was designed to amplify a region in a gene desert on chromosome 12. Primers used for ChIP qPCR are as follows: −124 to −189 [5′-GCCTGCTACAGCTTCCAAAC-3′ (forward) and 5′-CCGCCGCATCTGAAAGTC-3′ (reverse)], −404 to −482 [5′-TTCTGTGCCCCTAGCTGTG-3′ (forward) and 5′-GGTGCTTCCAGCCTCAAC-3′ (reverse)], −589 to −671 [5′-TGAGAGGCAAACATCCAAGA-3′ (forward) and 5′-CTTCCCCAAGATGGAATCAG-3′ (reverse)], −807 to −885 [5′-CAGCCTCCAAAAGTGCTGAC-3′ (forward) and 5′-ATTTCCCAGATGGCAGTGAA-3′ (reverse)], −1882 to −1969 [5′-GGAAGAATGGGTGGGAATTA-3′ (forward) and 5′-TTGAGAAGAGCCCTGGAGTG-3′ (reverse)], and control [5′-TGAGCATTCCAGTGATTTATTG-3′ (forward) and 5′-AAGCAGGTAAAGGTCCATATTTC-3′ (reverse)].
Measurement and counts of adenomas and laser capture microdissection.
Sox9-deficient ApcMin/+ mice and their littermate controls (wild-type Sox9;ApcMin/+ mice) were euthanized at 5 mo of age, and intestines from duodenum to distal colon were dissected, fixed in formalin, and embedded in paraffin. Hematoxylin-eosin-stained FFPE sections were used for adenoma measurements and counts. Areas of each adenoma were measured under a microscope using NIS-Elements Imaging software (Nikon, Melville, NY). The total numbers of adenomas per section were counted and averaged. For laser capture microdissection, adenoma tissues were collected from unstained FFPE sections using the ArcturusXT Laser Capture Microdissection System (Life Technologies).
Statistical analysis.
Values are means ± SD. Differences between the two groups were analyzed using an unpaired Student's t-test. P < 0.05 was considered significant.
RESULTS
Igfbp-4 expression is regulated by SOX9 in mouse intestinal epithelium in vivo.
To identify potential SOX9 target genes that regulate proliferation in the intestinal epithelium, we performed microarray analysis using crypt epithelial cells isolated from mice in which Sox9 was inactivated specifically in the intestinal epithelium (Sox9flox/flox;VilCre+/−; GEO/GSE35481). Microarray analysis identified Igfbp-4 among the genes most significantly downregulated in Sox9-deficient mice. To confirm microarray results, we performed qRT-PCR analysis using Sox9-deficient crypt cells. Consistent with the microarray data, Igfbp-4 gene expression was significantly (6-fold, P < 0.01) downregulated in the Sox9-deficient mouse crypt cells compared with the wild-type littermate control samples (Fig. 1A). Similarly, Western blotting showed that IGFBP-4 protein was significantly downregulated in Sox9-deficient mouse crypt epithelial cells (Fig. 1B).
Fig. 1.

Expression levels of SOX9 and insulin-like growth factor binding protein (IGFBP)-4 are coordinately regulated in mouse intestinal epithelium in vivo. A: quantification by real-time RT-PCR of Sox9 and Igfbp-4 mRNA in intestinal epithelial crypt cells from 3-mo-old Sox9-deficient mice and their littermate controls. After normalization with 18S rRNA, results are shown as expression levels relative to expression levels in wild-type mice (n = 3). *P < 0.05; **P < 0.01. B: Western blot for SOX9 and IGFBP-4 of intestinal epithelial crypt cells from Sox9-deficient mice and their littermate controls. C and D: representative images of SOX9 and IGFBP-4 immunostaining of the small intestine (top) and colon (bottom) of a wild-type (left) and a Sox9-deficient (right) mouse. Arrows show representative IGFBP-4-positive cells.
SOX9 and IGFBP-4 colocalize in mouse intestinal epithelium and in primary human CRC.
Immunohistochemistry staining revealed that the expression pattern of IGFBP-4 highly resembled that of SOX9 in the normal mouse small intestine and colon, with both being localized to the lower half of the crypts (Fig. 1, C and D, left), while in Sox9-deficient mice, IGFBP-4 was no longer detectable in the small intestine or colon (Fig. 1, C and D, right).
Costaining experiments using immunofluorescent-labeled antibodies on mouse small intestine clearly indicated that IGFBP-4 was localized in the cytoplasm of crypt epithelial cells and solitary villus cells that expressed SOX9 in the nuclei (Fig. 2, A and B). The cells in the small intestinal villi that expressed SOX9 and IGFBP-4 consisted of <1% of the total villus epithelial cells. Similarly, in the colon, IGFBP-4 was expressed in the cytoplasm of the cells that expressed SOX9 (Fig. 2C). DCLK-1, a tuft cell-specific marker, and IGFBP-4 were coimmunostained in Sox9-deficient and littermate control mouse intestines. IGFBP-4 and DCLK-1 were coexpressed in the solitary cells of the wild-type villus epithelium, whereas DCLK-1-positive cells no longer showed IGFBP-4 expression in Sox9-deficient mice (Fig. 2D). This observation strongly indicates that IGFBP-4 expression is SOX9-dependent.
Fig. 2.

Representative images of immunofluorescence of SOX9 and IGFBP-4 of mouse intestinal crypts (A and C) and villi (B and D). A: higher expression of SOX9 (green) and IGFBP-4 (red) in Paneth cells and lower expression of IGFBP-4 and SOX9 in crypt base columnar cells. B: SOX9 (green) and IGFBP-4 (red) are colocalized in the solitary cells in the small intestinal epithelial villi. C: SOX9 (green) and IGFBP-4 (red) are also localized to the lower half of the crypts in mouse colons. D: IGFBP-4 (red) is observed in doublecortin-like kinase 1 (DCLK-1)-expressing tuft cells (green) in the wild-type mice (top), whereas IGFBP-4 is no longer expressed in DCLK-1-positive cells of Sox9-deficient mice (bottom).
Normal human colon specimens exhibited a similar expression pattern for SOX9 and IGFBP-4 (Fig. 3A). To determine if SOX9 and IGFBP-4 also colocalize in human CRC, we performed immunofluorescence staining on human primary CRC specimens. Five of six specimens were SOX9-positive, and all showed IGFBP-4 expression colocalized with SOX9 (Fig. 3B). Furthermore, the specimen negative for SOX9 also showed significantly less IGFBP-4 immunostaining (Fig. 3C).
Fig. 3.

Representative images of immunofluorescence of SOX9 and IGFBP-4 in human normal colon (A) and primary cancer (B and C) specimens. A: representative images of immunofluorescence staining for SOX9 (green) and IGFBP-4 (red) on human normal colon sections. SOX9 and IGFBP-4 are colocalized in the lower crypt cells. Bottom: higher-magnification images corresponding to insets above. B: immunofluorescence showed colocalization of SOX9 and IGFBP-4 in SOX9-positive human colorectal cancer (CRC). Bottom: higher-magnification images corresponding to insets above. C: IGFBP-4 is significantly downregulated in SOX9-negative human CRC. DAPI, 4′,6-diaminido-2-phenylindole.
SOX9 regulates cell proliferation through IGFBP-4 in human CRC cells.
IGFBP-4 expression has been reported in several cancer cell lines, including most CRC-derived cell lines (15, 28, 35). We measured the expression of IGFBP-4 and SOX9 in CRC cell lines, including Caco-2, DLD1, HT-29, HCT116, Lim6, and LS-174T cells. All cell lines expressed SOX9 and IGFBP-4. Furthermore, the expression level of IGFBP-4 was relatively low in low-SOX9-expressing cells but relatively high in high-SOX9-expressing cells (Fig. 4A). Caco-2 cells have been reported to differentiate to an intestinal phenotype upon reaching confluency, and IGFBP-4 mRNA expression is correlated with reduced cell proliferation (19). We examined the relative expression levels of SOX9 and IGFBP-4 in Caco-2 cells at different time points. Weak expression of IGFBP-4 and SOX9 was detected in undifferentiated Caco-2 cells at day 0, and IGFBP-4 and SOX9 expression coordinately increased with differentiation until day 7, when both showed the highest levels of expression. Subsequently, IGFBP-4 and SOX9 expression decreased by day 12 (Fig. 4B).
Fig. 4.

Manipulation of SOX9 expression by overexpression or knockdown of SOX9 resulted in altered expression of IGFBP-4. A: Western blots of expression levels of SOX9 and IGFBP-4 in CRC cell lines after overnight culture. B: Western blots of expression levels of SOX9 and IGFBP-4 in cell lysates collected from Caco-2 cells on days 0, 2, 7, and 12 of culture. C and D: Western blot analysis of expression levels of SOX9 and IGFBP-4 (left), quantification of immunoblot bands from 3 repetitions of Western blot experiments (middle), and bromodeoxyuridine (BrdU) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay of cell proliferation after transfection with SOX9-pcDNA in Caco-2 cells or SOX9 small interfering RNA [siRNA, control (Ctrl)] in HT-29 cells (right). E: SOX9 siRNA did not affect expression of any other SOX family transcription factors in the intestine as measured by quantitative RT-PCR. Expression of the SOX family of transcription factors that are known to be expressed in the intestine, including SOX2, SOX4, SOX6, SOX9, and SOX10, was measured by real-time PCR from SOX9 siRNA- and control siRNA-transfected (Ctrl) HT-29 cells. F: reduction of cell proliferation in Caco-2 cells was restored following treatment with a neutralizing antibody against IGFBP-4. Overexpression of SOX9 upregulated IGFBP-4 expression and suppressed cell proliferation in human CRC cells. *P < 0.05; **P < 0.01, ***P < 0.001.
To determine if the IGFBP-4 expression levels can be altered by SOX9, we transfected SOX9-pcDNA into Caco-2 (Fig. 4C) or DLD1 (data not shown) cells, which showed relatively lower endogenous SOX9, to overexpress SOX9. We also knocked down SOX9 expression using siRNA in HT-29 and HCT116 cells, which showed higher endogenous SOX9 levels (Fig. 4D and data not shown). We confirmed by qRT-PCR that expression of SOX9, but not other members of the SOX transcription factor gene family, was specifically knocked down by the SOX9 siRNA (Fig. 4E). Western blot analysis confirmed that IGFBP-4 expression was increased 48 h after transfection with SOX9-pcDNA in Caco-2 and DLD1 cells (Fig. 4C and data not shown), while knockdown of SOX9 significantly reduced the expression of IGFBP-4 in HT-29 and HCT116 cells (Fig. 4D and data not shown). Furthermore, overexpression of SOX9 in Caco-2 and DLD1 cells showed a small, but significant, reduction in proliferation (Fig. 4C and data not shown). This reduction of cell proliferation by SOX9 was restored following treatment with a neutralizing antibody against IGFBP-4 (Fig. 4F), suggesting that SOX9 requires IGFBP-4 to regulate proliferation. In contrast, knockdown of SOX9 correlated with increased proliferation in HT-29 and HCT116 cells (Fig. 4D and data not shown).
SOX9 transactivates the IGFBP-4 promoter in human CRC cells.
Sequence analysis of the human, bovine, mouse, and rat IGFBP-4 promoter regions revealed a high degree of homology within a 1-kb region upstream of the transcription start site (72% for human and bovine, 70% for human and rat, and 68% for human and mouse). The mouse and human IGFBP-4 promoters share 79% homology within the region 200 bp upstream of the transcription start site.
To determine if SOX9 transactivates the IGFBP-4 promoter, we cloned the 2-kb human and mouse IGFBP-4 promoters, which contain the high-homology region, into the pGL3 luciferase vector. In Caco-2 and DLD1 cells, luciferase activity of the human and mouse IGFBP-4 reporters was significantly increased in a dose-dependent manner (Fig. 5A and data not shown). To test if this regulation was mediated by the transcriptional function of SOX9, reporter plasmids were cotransfected with SOX9 constructs carrying mutations in the HMG DNA-binding domain (SOX9-HMG-M). The SOX9 plasmid with the mutant HMG domain did not transactivate the IGFBP-4 promoter (Fig. 5B), which indicates that activation was mediated by direct binding of SOX9 to the IGFBP-4 promoter. Indeed, there are five SOX9 consensus sequences, (A/T)(A/T)CAA(A/T)G, in this region of the human IGFBP-4 promoter, allowing for up to two mismatches. To determine the putative SOX9 responsive element, 2-bp mutations were made at each of these sites. When cotransfected with SOX9, only the reporter plasmid with a mutation at positions −162 to −161 showed a significant decrease in luciferase activity (Fig. 5, C and D).
Fig. 5.
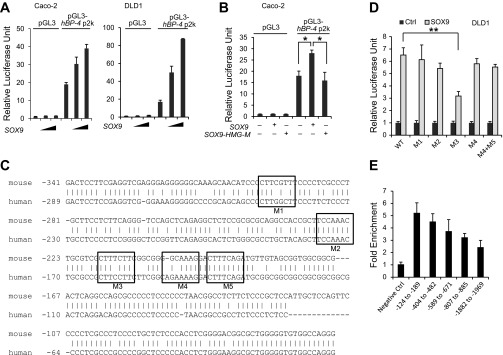
SOX9 transactivates the IGFBP-4 promoter in human CRC cells. A: SOX9 activated the reporter plasmid containing human IGFBP-4 promoter in a dose-dependent manner. B: expression of SOX9 mutant carrying a mutation in the high-mobility group (HMG) domain abrogated activation of the human IGFBP-4 promoter. C: human and mouse IGFBP-4 promoter sequences were aligned by ClustalW2. M1–M5, conserved consensus motifs for SOX9 binding. D: wild-type and mutant pGL3-human IGFBP-4 promoter plasmids were transfected into DLD1 cells with SOX9-pcDNA-expressing vector or an empty pcDNA vector. Activation was significantly decreased with a mutation in M3. E: chromatin immunoprecipitation-quantitative PCR showed enrichment close to the transcription start site.
To further confirm SOX9 binding to the IGFBP-4 promoter, ChIP experiments were performed in HT-29 cells using a SOX9 antibody. qPCR analysis was performed using primer pairs spanning the IGFBP-4 promoter region, and significant enrichment of DNA fragments was observed in the promoter region close to the transcription start site compared with the control primer pair that amplified a region in a gene desert on chromosome 12 (Fig. 5E), with a peak of enrichment overlapping the site confirmed by transient transfection with mutation assays.
Sox9 deficiency in ApcMin/+ mice resulted in increased tumor burden relative to ApcMin/+ control mice.
SOX9 and IGFBP-4 are expressed in most human CRCs, CRC-derived cell lines, and intestinal adenoma cells of ApcMin/+ mice. To confirm the tumor suppressive action of SOX9 in vivo, Sox9-deficient mice were bred with ApcMin/+ mice. Adenoma cells from ApcMin/+ mice showed high expression of SOX9 and IGFBP-4, which were barely detected in adenoma cells from Sox9-deficient ApcMin/+ mice (Fig. 6A). Statistical analyses showed that Sox9-deficient ApcMin/+ mice developed more adenomas than ApcMin/+ control mice (P = 0.03; Fig. 6B, left), and the difference was more significant in the number of large (>1-mm2) adenomas (Fig. 6B, right), while the difference in number of small (≤1-mm2) adenomas was not significant (P = 0.14), suggesting that SOX9 suppresses tumor growth in ApcMin/+ adenomas.
Fig. 6.
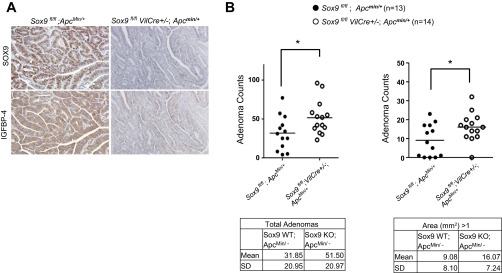
Sox9 deficiency in ApcMin/+ mice resulted in increased tumor burden relative to ApcMin/+ control mice. A: SOX9 and IGFBP-4 were highly expressed in most of the adenoma cells of ApcMin/+ control mice (left), while neither SOX9 nor IGFBP-4 was detectable in Sox9-deficient ApcMin/+ mice (right). B: Sox9 deficiency [Sox9 knockout (KO)] in ApcMin/+ mice resulted in increased total number of adenomas relative to ApcMin/+ wild-type (WT) mice (left). Number of larger (>1 mm2) adenomas significantly increased in Sox9-deficient ApcMin/+ mice relative to their littermate controls (right).
DISCUSSION
The role of SOX9 in cell proliferation in CRC has been the focus of several recent studies. Previous studies using conditional inactivation of Sox9 showed that SOX9 suppresses proliferation in the mouse intestinal epithelium (5, 16, 25). Similarly, gain-of-function and loss-of-function studies in CRC cell lines, as well as in IEC-18 cells, have shown that SOX9 suppresses proliferation and reduces tumorigenicity (14, 16). In this study, we identified IGFBP-4 as one of the SOX9 direct target genes that regulates cell proliferation in the intestinal epithelium. The effects of SOX9 in luciferase assays with IGFBP-4 promoter constructs were relatively modest, while inactivation of Sox9 almost abolished IGFBP-4 expression in mouse intestinal epithelium. We speculate that IGFBP-4 might have enhancer elements in addition to the one identified in the promoter region. In fact, in chondrocytes, SOX9 has been reported to have more than two binding sites on most of its target genes (26).
New reports have suggested that SOX9 may have a proliferation role as well. A recent report demonstrated that SOX9 promotes proliferation and represses the tumor suppressor Ink4a/Arf through activation of Bmi1 (23). Another recent report demonstrated a bimodal role for SOX9 in the intestinal epithelium, where low levels of SOX9 expression support proliferative capacity, while high levels of SOX9 expression suppress proliferation (16). Therefore, the contradictory data regarding the function of SOX9 may be the result of use of different levels of SOX9 in different studies. In fact, the expression level of SOX9 is cell type-specific among mouse intestinal epithelial cells, with the highest level in terminally differentiated Paneth cells and tuft cells and a much lower level in transit-amplifying cells.
Our in vivo data from Sox9-deficient ApcMin/+ mice indicate that SOX9 suppresses tumor growth and SOX9 directly regulates the expression of IGFBP-4. Taken together, the data presented in this report provide strong evidence that SOX9 has an antiproliferative function through direct regulation of IGFBP-4 in CRC. Although Sox9 inactivation in ApcMin/+ mice accelerated tumorigenesis compared with wild-type Sox9 expression, the difference was relatively modest. We speculate that the modest difference could be due to SOX9 support of cancer stem cells through Bmi1 as well as tumor-suppressive target genes at the same time. In fact, the difference was more significant in the number of larger adenomas, while the difference in the number of small adenomas was not significant. This suggests that SOX9 suppresses tumor growth but likely does not suppress tumor initiation. Further studies are required to determine the role of SOX9 in CRC tumor formation.
Our study is the first to demonstrate the loss of function of SOX9 in ApcMin/+ mice. Inactivation of Sox9 resulted in increased numbers of large adenomas, indicating that loss of SOX9 increases tumor growth. It would be of interest to study the correlation between timing of silencing of SOX9 and the outcome of CRC cases, because SOX9 has recently been identified as one of the frequently mutated genes in CRC.
Although the inhibitory regulation of SOX9 on Wnt/β-catenin has been demonstrated in several studies (4, 5, 7), it is likely that SOX9 regulates proliferation in intestinal epithelium through distinct mechanisms, one of which is due to the transcriptional regulation of SOX9 on IGFBP-4. In addition to the inhibitory function of IGFBP-4 on IGF-I and IGF-II, IGFBP-4 has also been identified as an inhibitor of canonical Wnt signaling (36). IGFBP-4 has been shown to physically interact with the Wnt receptor Frizzled 8 (Frz8) and the Wnt coreceptor low-density lipoprotein receptor-related protein 6 (LRP6) and inhibit the binding of Wnt3A to Frz8 and LRP6 during cardiogenesis (36). However, in the CRC cells, where activation of the Wnt/β-catenin pathway is constitutively active, it is likely that the modification of interaction of Wnt3A with Frz8 has no effect on the activity of the Wnt/β-catenin pathway in these cells. This was confirmed through Top-flash reporter assays showing that IGFBP-4 had no effect on the Wnt/β-catenin pathway in human CRC cells (data not shown). We also determined here that the suppression of cell proliferation is at least in part due to SOX9 regulation on IGFBP-4.
Activation of the insulin-IGF pathway is an important risk factor for the development of CRC (29); therefore, elucidating the mechanism of SOX9 involvement in this pathway is critical to the development of treatment strategies for CRC.
GRANTS
This study was supported by a Sara Jordan Research Scholar Award from the American Gastroenterological Association (Y. Mori-Akiyama), National Institute of Diabetes and Digestive and Kidney Diseases Grant P30 DK-56338, and the Department of Pathology and Immunology at Baylor College of Medicine and the Department of Pathology at Texas Children's Hospital.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.S. and C.-I.C. performed the experiments and analyzed the data; T.-A.M. performed the statistical analysis; A.M. provided technical support for histology; Y.M.-A. conceived the project, performed the experiments, analyzed the data, and drafted, edited, and revised the manuscript.
ACKNOWLEDGMENTS
We thank Dr. David Y. Graham for valuable advice and collection of human samples. We thank Drs. Mary K. Estes, Daniel Lacorazza, Stephanie Pangas, and Tor Savidge for critical reading of the manuscript.
REFERENCES
- 1. Abdel-Samad R, Zalzali H, Rammah C, Giraud J, Naudin C, Dupasquier S, Poulat F, Boizet-Bonhoure B, Lumbroso S, Mouzat K, Bonnans C, Pignodel C, Raynaud P, Fort P, Quittau-Prevostel C, Blache P. MiniSOX9, a dominant-negative variant in colon cancer cells. Oncogene 30: 2493– 2503, 2011 [DOI] [PubMed] [Google Scholar]
- 2. Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci USA 101: 6502– 6507, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 16: 2813– 2828, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B. Interactions between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev 18: 1072– 1087, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, Blache P, Jay P. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 178: 635– 648, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet 22: 85– 89, 1999 [DOI] [PubMed] [Google Scholar]
- 7. Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN, Clevers H, Jay P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 166: 37– 47, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Network Comprehensive molecular characterization of human colon, and rectal cancer. Nature 487: 330– 337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chaboissier MC, Kobayashi A, Vidal VI, Lutzkendorf S, van de Kant HJ, Wegner M, de Rooij DG, Behringer RR, Schedl A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 131: 1891– 1901, 2004 [DOI] [PubMed] [Google Scholar]
- 10. Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science 279: 563– 566, 1998 [DOI] [PubMed] [Google Scholar]
- 11. Contois LW, Nugent DP, Caron JM, Cretu A, Tweedie E, Akalu A, Liebes L, Friesel R, Rosen C, Vary C, Brooks PC. Insulin-like growth factor binding protein-4 differentially inhibits growth factor-induced angiogenesis. J Biol Chem 287: 1779– 1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Darido C, Buchert M, Pannequin J, Bastide P, Zalzali H, Mantamadiotis T, Bourgaux JF, Garambois V, Jay P, Blache P, Joubert D, Hollande F. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res 68: 4258– 4268, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Duan C. Specifying the cellular responses to IGF signals: roles of IGF-binding proteins. J Endocrinol 175: 41– 54, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Dupasquier S, Abdel-Samad R, Glazer RI, Bastide P, Jay P, Joubert D, Cavailles V, Blache P, Quittau-Prevostel C. A new mechanism of SOX9 action to regulate PKCα expression in the intestine epithelium. J Cell Sci 122: 2191– 2196, 2009 [DOI] [PubMed] [Google Scholar]
- 15. Durai R, Yang SY, Sales KM, Seifalian AM, Goldspink G, Winslet MC. Increased apoptosis and decreased proliferation of colorectal cancer cells using insulin-like growth factor binding protein-4 gene delivered locally by gene transfer. Colorectal Dis 9: 625– 631, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Formeister EJ, Sionas AL, Lorance DK, Barkley CL, Lee GH, Magness ST. Distinct SOX9 levels differentially mark stem/progenitor populations and enteroendocrine cells of the small intestine epithelium. Am J Physiol Gastrointest Liver Physiol 296: G1108– G1118, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gerbe F, van Es JH, Makrini L, Brulin B, Mellitzer G, Robine S, Romagnolo B, Shroyer NF, Bourgaux JF, Pignodel C, Clevers H, Jay P. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J Cell Biol 192: 767– 780, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, Pollak M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 351: 1393– 1396, 1998 [DOI] [PubMed] [Google Scholar]
- 19. Hoeflich A, Yang Y, Rascher W, Blum WF, Huber S, Koepf G, Kolb HJ, Kiess W. Coordinate expression of insulin-like growth factor II (IGF-II) and IGF-II/mannose-6-phosphate receptor mRNA and stable expression of IGF-I receptor mRNA during differentiation of human colon carcinoma cells (Caco-2). Eur J Endocrinol 135: 49– 59, 1996 [DOI] [PubMed] [Google Scholar]
- 20. Jay P, Berta P, Blache P. Expression of the carcinoembryonic antigen gene is inhibited by SOX9 in human colon carcinoma cells. Cancer Res 65: 2193– 2198, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev 16: 3– 34, 1995 [DOI] [PubMed] [Google Scholar]
- 22. Lu B, Fang Y, Xu J, Wang L, Xu F, Xu E, Huang Q, Lai M. Analysis of SOX9 expression in colorectal cancer. Am J Clin Pathol 130: 897– 904, 2008 [DOI] [PubMed] [Google Scholar]
- 23. Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, Canamero M, Bujanda L, Schedl A, Cheah KS, Skotheim RI, Lothe RA, Lopez de Munain A, Briscoe J, Serrano M, Lovell-Badge R. Oncogenicity of the developmental transcription factor Sox9. Cancer Res 72: 1301– 1315, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci USA 100: 9360– 9365, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, Clevers H, de Crombrugghe B. SOX9 is required for the differentiation of Paneth cells in the intestinal epithelium. Gastroenterology 133: 539– 546, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Oh CD, Maity SN, Lu JF, Zhang J, Liang S, Coustry F, de Crombrugghe B, Yasuda H. Identification of SOX9 interaction sites in the genome of chondrocytes. PLos One 5: e10113, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev 28: 20– 47, 2007 [DOI] [PubMed] [Google Scholar]
- 28. Singh P, Dai B, Dhruva B, Widen SG. Episomal expression of sense and antisense insulin-like growth factor (IGF)-binding protein-4 complementary DNA alters the mitogenic response of a human colon cancer cell line (HT-29) by mechanisms that are independent of and dependent upon IGF-I. Cancer Res 54: 6563– 6570, 1994 [PubMed] [Google Scholar]
- 29. Sridhar SS, Goodwin PJ. Insulin-insulin-like growth factor axis and colon cancer. J Clin Oncol 27: 165– 167, 2009 [DOI] [PubMed] [Google Scholar]
- 30. Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev 17: 1677– 1689, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wetterau LA, Moore MG, Lee KW, Shim ML, Cohen P. Novel aspects of the insulin-like growth factor binding proteins. Mol Genet Metab 68: 161– 181, 1999 [DOI] [PubMed] [Google Scholar]
- 32. Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst 92: 1472– 1489, 2000 [DOI] [PubMed] [Google Scholar]
- 33. Yu H, Spitz MR, Mistry J, Gu J, Hong WK, Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: a case-control analysis. J Natl Cancer Inst 91: 151– 156, 1999 [DOI] [PubMed] [Google Scholar]
- 34. Zalzali H, Naudin C, Bastide P, Quittau-Prevostel C, Yaghi C, Poulat F, Jay P, Blache P. CEACAM1, a SOX9 direct transcriptional target identified in the colon epithelium. Oncogene 27: 7131– 7138, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Zhou R, Diehl D, Hoeflich A, Lahm H, Wolf E. IGF-binding protein-4: biochemical characteristics and functional consequences. J Endocrinol 178: 177– 193, 2003 [DOI] [PubMed] [Google Scholar]
- 36. Zhu W, Shiojima I, Ito Y, Li Z, Ikeda H, Yoshida M, Naito AT, Nishi J, Ueno H, Umezawa A, Minamino T, Nagai T, Kikuchi A, Asashima M, Komuro I. IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 454: 345– 349, 2008 [DOI] [PubMed] [Google Scholar]