

Abstract
The barrier function of the esophageal epithelium is a major defense against gastroesophageal reflux disease. Previous studies have shown that reflux damage is reflected in a decrease in transepithelial electrical resistance associated with tight junction alterations in the esophageal epithelium. To develop novel therapies, it is critical to understand the molecular mechanisms whereby contact with a refluxate impairs esophageal barrier function. In this study, surgical models of duodenal and mixed reflux were developed in mice. Mouse esophageal epithelium was analyzed by gene microarray. Gene set enrichment analysis showed upregulation of inflammation-related gene sets and the NF-κB pathway due to reflux. Significance analysis of microarrays revealed upregulation of NF-κB target genes. Overexpression of NF-κB subunits (p50 and p65) and NF-κB target genes (matrix metalloproteinases-3 and -9, IL-1β, IL-6, and IL-8) confirmed activation of the NF-κB pathway in the esophageal epithelium. In addition, real-time PCR, Western blotting, and immunohistochemical staining also showed downregulation and mislocalization of claudins-1 and -4. In a second animal experiment, treatment with an NF-κB inhibitor, BAY 11-7085 (20 mg·kg−1·day−1 ip for 10 days), counteracted the effects of duodenal and mixed reflux on epithelial resistance and NF-κB-regulated cytokines. We conclude that gastroesophageal reflux activates the NF-κB pathway and impairs esophageal barrier function in mice and that targeting the NF-κB pathway may strengthen esophageal barrier function against reflux.
Keywords: mouse model, gastroesophageal reflux, NF-κB, barrier function, tight junction
gastroesophageal reflux disease (GERD) is a chronic disorder caused by prolonged exposure of the distal esophagus to gastric or gastroduodenal contents (21). Prolonged exposure to these noxious refluxates results in disease by impairment of the intrinsic defenses within the esophageal epithelium, particularly those responsible for barrier function (29). GERD significantly impacts patient quality of life and may lead to long-term complications, such as Barrett's esophagus and esophageal adenocarcinoma. Approximately 10–20% of people in the Western world have GERD, with at least weekly heartburn and/or acid regurgitation (8). Current therapy of GERD relies predominantly on the use of acid-suppressant medications, such as proton pump inhibitors, but response to these medications is less than optimal. For this reason, novel treatments remain desirable, and to develop them, a clearer understanding of the molecular mechanisms responsible for GERD-induced damage to the esophageal epithelium is needed.
The impairment of esophageal barrier function by gastroesophageal reflux has been extensively investigated. Endoscopic esophageal biopsy specimens of GERD patients are characterized by dilated intercellular spaces (DIS) between esophageal epithelial cells and higher permeability to hydrogen ions than tissue from healthy subjects (6, 41). In vitro experiments show a decrease in transepithelial electrical resistance (TEER) and an increase in paracellular permeability in bile acid-treated human esophageal epithelium and acid-treated esophageal-like squamous epithelium (7, 33). Animal studies also demonstrate a decrease in TEER in acidified pepsin-treated rabbit esophageal epithelial segments, as well as in rabbit esophageal epithelium perfused in vivo (16, 42).
Traditionally, GERD is believed to be a caustic injury produced by gastric or gastroduodenal contents. On the basis of human pathology and a rabbit model of acid-induced reflux disease, it was shown that the refluxate initially produces injury to the esophageal epithelial junctions, resulting in an increase in paracellular permeability and DIS. Then acid penetrates the intercellular spaces in sufficient quantity to acidify the area and, therefore, gain access to the acid-permeable basolateral membrane. It is acid entry into the esophageal cytoplasm across the basolateral membrane that is believed to initiate a cascade that results in cell necrosis, inflammation, and erosion (30, 31). However, this traditional view has been challenged recently using a rat model in which the duodenum is connected directly to the esophagus. This rat model of gastroduodenal reflux was used to show that reflux induced an increase in the chemokine IL-8 in the esophageal epithelium and that infiltration of inflammatory cells took place prior to the erosions on the surface of the esophageal epithelium (38). This suggests that inflammation is an important mediator of erosions, a finding previously established in a rabbit model of esophagitis by Naya et al. (25). Furthermore, Yamaguchi and colleagues (43) found that epithelial keratinocytes secreted IL-8 in a cell culture model and a rat model of esophagitis, suggesting that gastroesophageal reflux may activate inflammatory pathways and then impair esophageal barrier function.
To understand how gastroesophageal reflux attenuates esophageal barrier function, surgical models were developed in mice to mimic duodenal and mixed reflux. Activation of the NF-κB pathway was assessed as a causative factor leading to esophageal barrier dysfunction.
METHODS
Animals and surgical procedures.
Wild-type C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animal experiments were approved by the Institutional Animal Care and Use Committee of North Carolina Central University (protocol no. XC-12-03-2008). Eight-week-old wild-type mice were housed five per cage, given laboratory chow and water ad libitum, and maintained on a 12:12-h light-dark cycle. The mice received anesthetics premixed in normal saline (80 mg/kg ketamine and 12 mg/kg xylazine ip). All surgeries were performed through an upper midline incision. 1) In the mixed reflux model, two 5-mm incisions were made on the esophagus and the duodenum and then anastomosed with accurate mucosal-to-mucosal opposition (Fig. 1A). 2) In the duodenal reflux model, the whole stomach was removed after ligation of the gastroesophageal junction and the pylorus subsequent to the procedure described for the mixed reflux study (Fig. 1B). The abdominal cavity was washed and closed with 6-0 silk sutures. Control mice received anesthesia and were subjected to mock surgery in which only a skin incision was made and then sutured.
Fig. 1.

Surgical models of gastroesophageal reflux in mice: mixed reflux (A) and duodenal reflux (B).
In the first animal experiment, 15 mice of each group were euthanized 4 wk after surgery. Samples of the esophageal epithelium of three mice were snap-frozen and stored in liquid nitrogen for gene microarray and real-time PCR. Samples of eight mice were frozen and used for Western blotting and ELISA. For histopathological analysis, the whole esophagus of four mice was opened longitudinally and fixed in 10% buffered formalin.
In the second animal experiment, we aimed to test the efficacy of NF-κB inhibition. Mice with mixed or duodenal reflux were treated with BAY 11-7085 (20 mg·kg−1·day−1 ip; Santa Cruz Biotechnology, Santa Cruz, CA) for 3 days before and 7 days after surgery and euthanized 7 days after surgery. As a potent IκK inhibitor, BAY 11-7085 has been safely used in vivo at this dose (22). The esophageal epithelium was harvested and prepared for TEER measurement and ELISA (IL-1β, IL-6, and IL-8). Eight mice were studied in each group: four mice were used for TEER analysis and four for ELISA.
TEER.
Esophageal epithelial tissues for chamber studies were immersed in ice-cold oxygenated Ringer solution and immediately transported to the laboratory for mounting mucosal-side-up in mini-Ussing chambers (19) with 0.0314-cm2 Lucite rings with a 2-mm-diameter aperture. The samples were bathed on both sides with 5 ml of normal Ringer solution when gassed with 95% O2-5% CO2 at 37°C. Two sets of electrodes connected the solutions in the chamber to voltage current clamps (model MC6, Physiologic Instruments, San Diego, CA) that permitted direct recording of the transmural electrical potential difference (PD) and determination of short-circuit current (Isc) by passage of current. Total electrical resistance (RT) was calculated using Ohm's law, where PD = Isc × RT. After equilibration for 30 min, basal electrical readings of PD, Isc, and RT were obtained every 15 min for 2 h.
RNA isolation and microarray analysis.
Total RNA was extracted from individual frozen esophageal epithelium with an RNeasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA). The quality of RNA samples was checked using gel electrophoresis, and concentrations were measured by spectrophotometry. RNA quality (RNA integrity number >7) was further checked with a Bioanalyzer (Agilent Technologies, Santa Clara, CA) at the Genomics Core Facility at the Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill.
Microarray data were collected from two-channel mouse microarrays (4X44K, Agilent). After hybridization, the arrays were scanned using a GenePix 4000B scanner (Axon Instruments, Foster City, CA). The images were analyzed by GenePix Pro 5.0 software (Axon Instruments). Data preprocessing was carried out using the University of North Carolina MicroArray Database for quality filtering and data normalization. Gene expression values were quantified by the two-based logarithmic ratio of red channel intensity (mean) vs. green channel intensity (mean) followed by Lowess normalization to remove the intensity-dependent dye bias. Agilent array data were extracted on the probe level. For probes spotted multiple times, the mean expression value was computed and retained. All probe sequences were mapped using BLAT against the National Center for Biotechnology Information database and annotated with Gene Symbol. In case multiple probes were targeted on the same gene (with the same gene symbol), these data were collapsed onto Gene Symbol, and mean values were computed as the gene expression value. Preprocessed data were used to construct a series of data matrix files for targeted downstream analyses. For a given data matrix, the rows were excluded if >40% of values were missing. The rest of the missing data were imputed with a K-nearest-neighbor (k = 9) approach. Differentially expressed genes were obtained from two-class significance analysis of microarrays (SAM) in Excel with the median number of false positives <1. Gene set enrichment analysis (GSEA) was carried out as an add-in in Excel. Curated gene sets in three major categories, canonical pathway (CP; 880 gene sets), gene ontology (C5, 1,454 gene sets), and transcription factor (615 gene sets), were downloaded from the GSEA web portal and used in this study (http://www.broadinstitute.org/gsea/index.jsp). One thousand permutations were applied to generate a null distribution for statistical testing, and significantly enriched gene sets were obtained at a false discovery rate cutoff of 0.5. The microarray data have been submitted to the Gene Expression Omnibus (GEO) database (GSE 39629).
Real-time PCR.
cDNA was prepared from total RNA using an Advantage RT-for-PCR kit (Clontech, Mountain View, CA). Real-time PCR was performed with TaqMan primers [claudin (Cldn)-1, Cldn4] using a real-time PCR system (model 7500, Applied Biosystems, Foster City, CA) under standard PCR conditions. 18S rRNA was used as an internal control. ΔCt value was calculated after duplicate PCR of each sample. ΔΔCt values were calculated and used to determine fold change in expression.
Western blotting.
The total proteins were prepared by homogenization of tissues in 20 volumes of a 50 mM HEPES buffer (pH 7.4) with 1% Triton X-100, 0.05% SDS, 0.2% sodium deoxycholate, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, and Protease Inhibitor Cocktail (Sigma-Aldrich, St. Louis, MO). Cell debris was removed by centrifugation at 5,000 rpm. An aliquot of cleared lysate was kept for protein quantitation using the bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL). Protein samples were mixed with Laemmli buffer at 100°C for 2 min, separated by PAGE, and transferred to a polyvinylidene difluoride membrane. After it was blocked with 0.5% nonfat dry milk, the membrane was probed with a mouse anti-Cldn1 monoclonal antibody (3 μg/ml; catalog no. 37-4900, Invitrogen, Camarillo, CA) or a rabbit anti-Cldn4 polyclonal antibody (3 μg/ml; catalog no. 36-4800, Invitrogen) in Tris-Tween-buffered saline-0.1% Tween at 4°C for 1 h. The membrane was washed three times and then incubated with a horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody. Immunoreactivity was visualized by application of horseradish peroxidase enhanced chemiluminescence substrate (Pierce Biotechnology) and immediately exposing the membranes to X-ray film. To verify equal loading of samples, the blots were stripped and reprobed with a rabbit anti-GAPDH monoclonal antibody (1:2,000 dilution; catalog no. 2118, Cell Signaling Technology, Boston, MA).
ELISA.
The protein samples were prepared after homogenization and centrifugation at full speed. An aliquot was used for the quantification of total proteins by bicinchoninic acid assay. Measurement of IL-1β and IL-6 followed the instructions of a mouse cytokine/chemokine magnetic bead panel kit (EMD Millipore, Billerica, MA), while measurement of IL-8 followed the instructions of a mouse IL-8 ELISA kit (MyBiosource, San Diego, CA). Levels of IL-1β and IL-6 were expressed as picograms per 100 μg of total protein and the level of IL-8 as picograms per 1 μg of total protein.
Immunohistochemical staining.
The deparaffinized sections were submerged in methanol containing 0.3% hydrogen peroxide for 15 min at room temperature to inhibit endogenous peroxidase activity. Antigen retrieval was done prior to incubation with a rabbit anti-NF-κB p50 polyclonal antibody (1:100 dilution; ab7971, Abcam), a rabbit anti-NF-κB p65 polyclonal antibody (1:500 dilution; ab7970, Abcam), a rabbit anti-matrix metalloproteinase (MMP)-3 polyclonal antibody (1:40 dilution; ab52915, Abcam), a rabbit anti-MMP-9 polyclonal antibody (1:2,000 dilution; ab38898, Abcam), a rabbit anti-Cldn1 polyclonal antibody (1:200 dilution; B6327, LSBio, Seattle, WA), and a rabbit anti-Cldn4 polyclonal antibody (1:25 dilution; B2370, LSBio) overnight at 4°C. Tissue sections were washed again in PBS and incubated with peroxidase-conjugated secondary antibodies for 30 min at 37°C. Detection of the antibody complex was done using the streptavidin-peroxidase reaction kit with 3,3′-diaminobenzidine as a chromogen (ABC kit, Vector Labs, Burlingame, CA).
Statistical evaluation.
Values are means ± SE. Each experiment was repeated twice, and representative results are shown. All other values were subjected to two-tailed Student's t-test.
RESULTS
Gastroesophageal reflux activates the NF-κB pathway in mouse esophageal epithelium.
To understand the molecular mechanisms of the impairment of esophageal barrier function during gastroesophageal reflux, gene microarray was performed to identify differentially expressed genes and gene sets (see Excel S1 and Excel S2 in Supplemental Material for this article, available online at the Journal website). At 4 wk after surgery, GSEA-CP analysis showed that many signaling pathways were activated by reflux, and many of these pathways were inflammatory signaling pathways. Nine and 11 inflammatory CPs were enriched in esophageal samples with duodenal and mixed reflux, respectively. Consistent with the activated inflammatory CPs, inflammation-associated gene sets were also found to be enriched by GSEA gene ontology analysis. Thirteen and 14 of such gene sets were enriched in esophageal samples with duodenal and mixed reflux, respectively. NF-κB, a well-known transcription factor involved in inflammation and proliferation (5, 36), was one of the activated transcription factors in esophageal epithelium with duodenal or mixed reflux based on GSEA transcription factor analysis (Table 1).
Table 1.
Enrichment of selected gene sets and known NF-κB target genes in esophageal epithelium with duodenal or mixed reflux compared with control mice
| Duodenal Reflux | Mixed Reflux | |
|---|---|---|
| Canonical pathway | CP89: cytokine-cytokine receptor interaction | CP89: cytokine-cytokine receptor interaction |
| CP89: cytokine-cytokine receptor interaction | CP89: cytokine-cytokine receptor interaction | |
| CP122: TLR signaling pathway | CP122: TLR signaling pathway | |
| CP237: cytokine pathway | CP129: TCR signaling pathway | |
| CP238: inflammation pathway | CP237: cytokine pathway | |
| CP283: IL-10 pathway | CP238: inflammation pathway | |
| CP702: signaling in immunity | CP283: IL-10 pathway | |
| CP872: MMP-cytokine connection | CP408: activated TLR4 signaling | |
| CP446: chemokine receptors bind chemokine | CP838: TNF pathway | |
| CP391: TNFR2 pathway | CP872: MMP-cytokine connection | |
| CP312: NF-κB pathway | ||
| Gene ontology | GO320: regulation of cytokine secretion | GO335: positive regulation of immune system process |
| GO513: cytokine production | GO419: positive regulation of cytokine biosynthetic process | |
| GO593: inflammation response | GO513: cytokine production | |
| GO672: regulation of cytokine production | GO578: leukocyte activation | |
| GO975: cytokine secretion | GO584: adaptive immune response | |
| GO1233: chemokine activity | GO672: regulation of cytokine production | |
| GO1268: chemokine receptor binding | GO815: T cell activation | |
| GO500: regulation of T cell activation | GO882: regulation of lymphocyte activation | |
| GO578: leukocyte activation | GO918: innate immune response | |
| GO583: adaptive immune response | GO926: lymphocyte activation | |
| GO918: innate immune response | GO1034: positive regulation of cytokine production | |
| GO714: positive regulation of T cell activation | GO1147: IL binding | |
| GO882: regulation of lymphocyte activation | GO1422: cytokine binding | |
| GO1443: cytokine activity | ||
| Transcription factor | TF30: NF-κB | TF30: NF-κB |
| Known NF-κB target genes | ||
| Upregulated | Cxcl5, Cxcl1, Apoc3, Ccl4, Sele, Il12b, Ltf, Mmp3, Lcn2, Il6, Csf3, Il1b, Cd3 g, Saa3, Il11, Gad1, Afp, Ptgs2, Self, Pthlh, Slfn2, Pglyrp1, Lta, Ifng, Dio2, Ccl20, Rdh7, Cd38, Tnfaip3, Hif1a, Tnfsf13b, Btk, Tacr1, Cxcl9, Klra1, Il23a, Tnf, Nfkbi2, Mx1, Bir1, Pian, Cd80, Igfbp1, Saa1, Tff3 | Il1b, Ccl4 |
| Downregulated | Bdnf, Igfbp2, Sh3bgrl, Hoxa9, Fgfr, BclII, Bmp4, Ptgis, Gclc | — |
CP, canonical pathway; GO, gene ontology; TF, transcription factor; TLR, Toll-like receptor; TCR, T cell receptor; MMP, matrix metalloproteinase; TNFR2, TNF receptor type 2.
With SAM, we further identified individual genes differentially expressed in esophageal epithelium with duodenal or mixed reflux. Comparing our SAM data with the NF-κB target gene list (see Excel S2 in Supplemental Material), we found that 45 and 2 NF-κB target genes were upregulated in the esophageal epithelium with duodenal and mixed reflux at 4 wk after surgery, respectively. Such genes included chemokines (e.g., Cxcl1, Cxcl5, Cxcl9, Ccl4, and Ccl20), cytokines (e.g., IL-1β, IL-6, IL-11, IL-12α/β, IL-23α, and TNF-α), and MMP-3 and MMP-9 (Table 1). These data suggested that gastroesophageal reflux activated the NF-κB pathway in mouse esophageal epithelium.
To validate NF-κB activation in the esophageal epithelium, we examined expression of NF-κB subunits (p50 and p65) and NF-κB target genes (MMP-3, MMP-9, IL-1β, IL-6, and IL-8) with immunohistochemical staining and ELISA. NF-κB p50 and p65 were rarely expressed in normal esophageal epithelium but were overexpressed in the nucleus and cytoplasm of the esophageal epithelium with duodenal and mixed reflux. MMP-3 was not expressed in the esophageal epithelium of control mice. Its expression was upregulated in the cytoplasm of esophageal epithelial cells with duodenal and mixed reflux. A significant increase of MMP-9-positive cells was observed in the submucosa of the esophageal epithelium with duodenal and mixed reflux (Fig. 2A). Duodenal reflux induced a dramatic increase of all three cytokines compared with control, while mixed reflux significantly increased the levels of IL-6 and IL-8 (Fig. 2B).
Fig. 2.
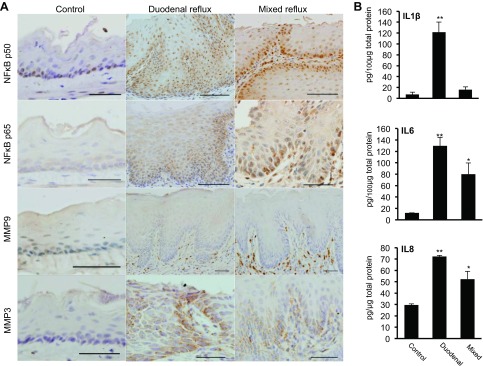
Overexpression of NF-κB subunits (p50 and p65) and NF-κB target genes [matrix metalloproteinase (MMP)-3, MMP-9, IL-1β, IL-6, and IL-8] in esophageal epithelium at 4 wk after duodenal or mixed reflux. A: immunohistochemical staining for NF-κB p50, NF-κB p65, MMP-3, and MMP-9. B: ELISA of IL-1β, IL-6, and IL-8. Values are means ± SE. *P < 0.05, **P < 0.01 vs. control.
Gastroesophageal reflux causes alterations of tight junction genes in mouse esophageal epithelium.
Previous studies clearly demonstrated that acid, bile acids, and pepsin disrupt squamous epithelial barrier function by modulating expression and localization of tight junction proteins (7, 33). With real-time PCR, we confirmed downregulation of Cldn1 and Cldn4 mRNA in the esophageal epithelium with mixed or duodenal reflux compared with control. We focused on Cldn1 and Cldn4, because they are major claudins in the squamous epithelium of human esophagus (19) (Fig. 3A). Western blotting also showed downregulation of Cldn1 and Cldn4 in the esophageal epithelium with mixed or duodenal reflux (Fig. 3, B and C).
Fig. 3.
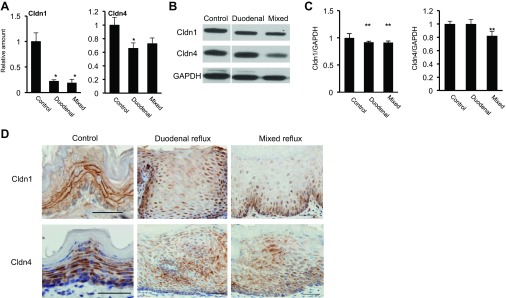
Downregulation and mislocalization of claudins (Cldn) in esophageal epithelium at 4 wk after duodenal or mixed reflux. A: real-time PCR of Cldn1 and Cldn4. Values are means ± SE. *P < 0.05 vs. control. B: Western blots of Cldn1 and Cldn4. C: quantitation of Cldn1 and Cldn4 Western blots. **P < 0.01 vs. control. D: immunohistochemical staining for Cldn1 and Cldn4.
Cldn1 and Cldn4 were predominantly localized on the cell membrane of basal, suprabasal, and superficial epithelial cells of the esophageal epithelium. Increased cytoplasmic staining and decreased membrane staining of Cldn1 and Cldn4 were observed in the esophageal epithelium with duodenal or mixed reflux, suggesting that duodenal and mixed reflux cause mislocalization of Cldn1 and Cldn4 and, thus, impair esophageal barrier function (Fig. 3D).
Inhibition of NF-κB counteracted reflux-induced impairment of esophageal barrier function.
To examine the potential cause-and-effect relationship between NF-κB activation and barrier dysfunction, we treated mice with duodenal or mixed reflux with a NF-κB inhibitor. A decrease in TEER was observed in the esophageal epithelium with mixed or duodenal reflux at 7 days after surgery. BAY 11-7085 (20 mg·kg−1·day−1 ip for 10 days) significantly counteracted the decrease in TEER in the esophageal epithelium with mixed reflux (P < 0.01). In case of duodenal reflux, BAY 11-7085 also counteracted the decrease in TEER, but the difference was not statistically significant (Fig. 4A).
Fig. 4.
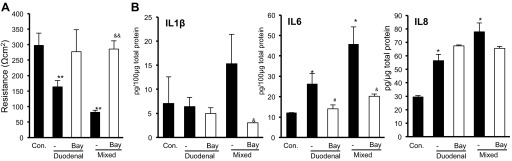
Effects of an NF-κB inhibitor, BAY 11-7085 (Bay), on transepithelial electrical resistance (A) and NF-κB-regulated cytokines (B) in esophageal epithelium with duodenal or mixed reflux at 7 days after surgery. Values are means ± SE. *P < 0.05, **P < 0.01 vs. control (Con). #P < 0.05 vs. duodenal reflux. &P < 0.05, &&P < 0.01 vs. mixed reflux.
Tissue levels of IL-1β, IL-6, and IL-8 were determined as an indicator of NF-κB activity. In the esophageal epithelium with duodenal reflux, BAY 11-7085 significantly reduced the levels of IL-6. In the esophageal epithelium with mixed reflux, BAY 11-7085 significantly reduced the levels of IL-1β and IL-6. IL-8 was not significantly modulated by NF-κB inhibition (Fig. 4B).
DISCUSSION
With surgically induced duodenal and mixed reflux models in mice, our study provided an alternative insight into the mechanism by which the NF-κB pathway plays an integral role in gastroesophageal reflux-induced impairment of esophageal barrier function in addition to direct damage of esophageal barrier function by refluxates. More importantly, treatment with an NF-κB inhibitor suppressed NF-κB-regulated cytokines and inhibited damage to esophageal barrier function in vivo.
NF-κB was activated in the esophageal mucosa at an early stage in GERD. It is known that NF-κB positively regulates the expression of IL-1β, IL-6, and IL-8 in immune cells and epithelial cells, which in turn directly or indirectly activates NF-κB via signaling pathways (23). IL-8 and IL-1β have been shown to be upregulated, even in endoscopy-negative patients (11, 14, 15, 20, 26, 28). In vitro experiments also showed that exposure of human esophageal epithelial cells to acid or bile acid activated the NF-κB pathway and upregulated IL-6 and IL-8 (10, 12, 17, 35) as a function of the degree of acidity and exposure time (13). In this study, activation of the NF-κB pathway was observed in the esophageal epithelium with duodenal or mixed reflux (Table 1, Fig. 2).
In addition to these NF-κB-regulated cytokines, gastroesophageal reflux also upregulated many NF-κB target genes, including MMP-3 and MMP-9 (Table 1). MMP-3 and MMP-9 were overexpressed in epithelial cells and inflammatory cells, respectively (Fig. 2A). Previous work on in vitro and in vivo models showed that ectopic expression of MMP-3 in mammary epithelial cells resulted in a loss of E-cadherin and catenins, which are critical adherens junction genes essential for epithelial barrier function (24, 40). Activation of MMP-9-expressing neutrophils also led to higher permeability of bovine retinal pigment epithelium via downregulation of tight junction proteins (44). In fact, cleavage of E-cadherin by a metalloproteinase contributed to the impairment of esophageal barrier function in GERD patients (18).
While gastroesophageal reflux activated the NF-κB pathway in mouse esophageal epithelium, it also caused downregulation and mislocalization of Cldn1 and Cldn4 (Fig. 3), the major claudins in the squamous epithelium of human esophagus (19). This is consistent with in vitro experiments showing that acid, bile salts, and pepsin disrupt esophageal barrier function through the loss or mislocalization of Cldn1, Cldn4, occludin, and zonula occludens-1 (7). Similar findings were also observed in a rat model of GERD (4, 27). These data suggested that NF-κB activation may impair the epithelial barrier function via deregulation of tight junctions (1–3, 39). To test this hypothesis, we treated mice with duodenal or mixed reflux with an NF-κB inhibitor in a short-term proof-of-concept experiment. As expected, BAY 11-7085 not only suppressed NF-κB activity in the esophagus but also protected the barrier function (Fig. 4).
Our data on the mouse models and previous studies on the rabbit model and human patients suggested that impairment of esophageal barrier function during gastroesophageal reflux may result from combined actions of chemical injury and NF-κB activation (Fig. 5). Noxious chemicals (e.g., acid and pepsin) in the gastroesophageal refluxate may immediately and directly attack and damage the apical junction complex to produce DIS and reduce TEER in the esophageal epithelium. High concentrations and long exposure of chemicals lead to epithelial edema and necrosis, subsequently triggering an inflammatory cascade. On the other hand, refluxate stimulates esophageal epithelial cells by activating the NF-κB pathway at the early stage. As a result, chemokines (e.g., IL-8 and IL-1β) are secreted to promote inflammation. Inflammatory cytokines may further activate NF-κB in the esophageal epithelium. Over time, these cytokines downregulate and mislocalize tight junction proteins (e.g., Cldn1 and Cldn4). Impairment of epithelial barrier function, mediated by direct chemical injury or NF-κB activation, would further exacerbate epithelial inflammation, evolving into a vicious cycle. It has been shown that deficiency of tight junction proteins (e.g., Cldn7 and occludin) results in chronic gastrointestinal inflammation, and deficiency of an adherens junction protein (p120 catenin) produced chronic dermatitis (9, 34, 37). In all these situations, NF-κB activation plays a critical role in epithelial inflammation.
Fig. 5.

Proposed mechanism of esophageal barrier dysfunction due to gastroesophageal reflux. Gastroesophageal reflux impairs esophageal barrier function through direct chemical injury (e.g., acid, bile, and enzymes) and subsequent tissue damage. Reflux activates NF-κB in esophageal epithelial cells at the early stage and triggers an inflammatory cascade. Persistent inflammation causes permanent damage to cell-cell junctions. As a result, gastroesophageal reflux disease (GERD) will develop, with the histopathology of esophageal tissue injury and inflammation. Therefore, the NF-κB pathway may be potentially targeted for GERD therapy. TEER, transepithelial electrical resistance; DIS, dilated intercellular spaces.
Our experiment with BAY 11-7085 was a proof-of-concept experiment with one dose and one time point. Further studies are needed to test multiple doses, later time points, and multiple inhibitors of NF-κB and its downstream effectors (e.g., MMPs). With the surgical models we developed in mice, genetically modified strains may also be used to further validate the role of NF-κB activation in impairment of esophageal barrier function by gastroesophageal reflux. The mouse esophagus is lined by a fully keratinized epithelium that is more sensitive to duodenal than gastric refluxate. Gastric reflux in mouse esophagus does not produce strong inflammation (data not shown), unlike gastric acid exposure in the rabbit model and acid reflux in human patients (32). Because of these differences between mouse and human esophagus, mouse models have limitations, and further studies are needed before mouse data can be translated into human studies. Nevertheless, our study clearly suggests targeting the NF-κB pathway as a potential GERD therapy.
GRANTS
This study was supported by National Cancer Institute Grant U54 CA-156735 and North Carolina Biotechnology Center Grant 2011-MRG-1101.
DISCLOSURES
X. Chen received financial support as a research grant from Takeda Pharmaceuticals North America, Inc. R. C. Orlando recently received research grants from Astra Zeneca and Takeda Pharmaceuticals North America, Inc. N. J. Shaheen recently received research grants from BÂRRX Medical, Inc., and Takeda Pharmaceuticals North America, Inc.
AUTHOR CONTRIBUTIONS
Y.F., H.C., Y.H., Z.D., and W.T. performed the experiments; Y.F. and H.C. analyzed the data; Y.F. prepared the figures; Y.F. and J.H. drafted the manuscript; W.T., N.J.S., R.C.O., and X.C. edited and revised the manuscript; R.C.O. and X.C. approved the final version of the manuscript; X.C. is responsible for conception and design of the research; X.C. interpreted the results of the experiments.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge excellent microarray service by Dr. Yan Shi and her staff at the Genomics Core Facility, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill. The authors thank Dr. Carlton W. Anderson (Center for Gastrointestinal Biology and Disease ImmunoTech Core, University of North Carolina at Chapel Hill).
REFERENCES
- 1.Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1β-induced increase in intestinal epithelial tight junction permeability. J Immunol 180: 5653–5661, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Sadi R, Ye D, Said HM, Ma TY. IL-1β-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-κB pathway. Am J Pathol 177: 2310–2322, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Sadi RM, Ma TY. IL-1β causes an increase in intestinal epithelial tight junction permeability. J Immunol 178: 4641–4649, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asaoka D, Miwa H, Hirai S, Ohkawa A, Kurosawa A, Kawabe M, Hojo M, Nagahara A, Minoo T, Ohkura R, Ohkusa T, Sato N. Altered localization and expression of tight-junction proteins in a rat model with chronic acid reflux esophagitis. J Gastroenterol 40: 781–790, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25: 280–288, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Carlsson R, Fandriks L, Jonsson C, Lundell L, Orlando RC. Is the esophageal squamous epithelial barrier function impaired in patients with gastroesophageal reflux disease? Scand J Gastroenterol 34: 454–458, 1999 [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Oshima T, Shan J, Fukui H, Watari J, Miwa H. Bile salts disrupt human esophageal squamous epithelial barrier function by modulating tight junction proteins. Am J Physiol Gastrointest Liver Physiol 303: G199–G208, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Dent J, El-Serag HB, Wallander MA, Johansson S. Epidemiology of gastro-oesophageal reflux disease: a systematic review. Gut 54: 710–717, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, Cao J, Chen YH. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology 142: 305–315, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duggan SP, Behan FM, Kirca M, Smith S, Reynolds JV, Long A, Kelleher D. An integrative genomic approach in oesophageal cells identifies TRB3 as a bile acid responsive gene, downregulated in Barrett's oesophagus, which regulates NF-κB activation and cytokine levels. Carcinogenesis 31: 936–945, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald RC, Onwuegbusi BA, Bajaj-Elliott M, Saeed IT, Burnham WR, Farthing MJ. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: immunological determinants. Gut 50: 451–459, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldman A, Chen HD, Roesly HB, Hill KA, Tome ME, Dvorak B, Bernstein H, Dvorak K. Characterization of squamous esophageal cells resistant to bile acids at acidic pH: implication for Barrett's esophagus pathogenesis. Am J Physiol Gastrointest Liver Physiol 300: G292–G302, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green NH, Huang Q, Corfe BM, Bury JP, MacNeil S. NF-κB is activated in oesophageal fibroblasts in response to a paracrine signal generated by acid-exposed primary oesophageal squamous cells. Int J Exp Pathol 92: 345–356, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isomoto H, Nishi Y, Kohno S. CXC receptor 1 is overexpressed in endoscopy-negative gastroesophageal reflux disease. Scand J Gastroenterol 40: 231–232, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Isomoto H, Saenko VA, Kanazawa Y, Nishi Y, Ohtsuru A, Inoue K, Akazawa Y, Takeshima F, Omagari K, Miyazaki M, Mizuta Y, Murata I, Yamashita S, Kohno S. Enhanced expression of interleukin-8 and activation of nuclear factor κB in endoscopy-negative gastroesophageal reflux disease. Am J Gastroenterol 99: 589–597, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Jacobson I, Poorkhalkali N, Jonsson-Rylander AC, Orlando RC. Effect of acid perfusion on passive electrophysiological properties of rabbit esophagus in vivo. Dig Dis Sci 47: 1369–1380, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Jenkins GJ, Cronin J, Alhamdani A, Rawat N, D'Souza F, Thomas T, Eltahir Z, Griffiths AP, Baxter JN. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-κB activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis 23: 399–405, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Jovov B, Que J, Tobey NA, Djukic Z, Hogan BL, Orlando RC. Role of E-cadherin in the pathogenesis of gastroesophageal reflux disease. Am J Gastroenterol 106: 1039–1047, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jovov B, Van Itallie CM, Shaheen NJ, Carson JL, Gambling TM, Anderson JM, Orlando RC. Claudin-18: a dominant tight junction protein in Barrett's esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol 293: G1106–G1113, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Kanazawa Y, Isomoto H, Wen CY, Wang AP, Saenko VA, Ohtsuru A, Takeshima F, Omagari K, Mizuta Y, Murata I, Yamashita S, Kohno S. Impact of endoscopically minimal involvement on IL-8 mRNA expression in esophageal mucosa of patients with non-erosive reflux disease. World J Gastroenterol 9: 2801–2804, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kandulski A, Malfertheiner P. Gastroesophageal reflux disease—from reflux episodes to mucosal inflammation. Nat Rev Gastroenterol Hepatol 9: 15–22, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Kim SR, Lee KS, Park SJ, Min KH, Lee MH, Lee KA, Bartov O, Atlas D, Lee YC. A novel dithiol amide CB3 attenuates allergic airway disease through negative regulation of p38 mitogen-activated protein kinase. Am J Respir Crit Care Med 183: 1015–1024, 2011 [DOI] [PubMed] [Google Scholar]
- 23.Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancer's inflammatory roots. Clin Cancer Res 17: 6125–6129, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol 139: 1861–1872, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naya MJ, Pereboom D, Ortego J, Alda JO, Lanas A. Superoxide anions produced by inflammatory cells play an important part in the pathogenesis of acid and pepsin induced oesophagitis in rabbits. Gut 40: 175–181, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Riordan JM, Abdel-latif MM, Ravi N, McNamara D, Byrne PJ, McDonald GS, Keeling PW, Kelleher D, Reynolds JV. Proinflammatory cytokine and nuclear factor κB expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol 100: 1257–1264, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Oguro M, Koike M, Ueno T, Asaoka D, Mori H, Nagahara A, Uchiyama Y, Watanabe S. Dissociation and dispersion of claudin-3 from the tight junction could be one of the most sensitive indicators of reflux esophagitis in a rat model of the disease. J Gastroenterol 46: 629–638, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Oh TY, Lee JS, Ahn BO, Cho H, Kim WB, Kim YB, Surh YJ, Cho SW, Hahm KB. Oxidative damages are critical in pathogenesis of reflux esophagitis: implication of antioxidants in its treatment. Free Radic Biol Med 30: 905–915, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Orlando RC. The integrity of the esophageal mucosa. Balance between offensive and defensive mechanisms. Best Pract Res Clin Gastroenterol 24: 873–882, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orlando RC. Pathophysiology of gastroesophageal reflux disease. J Clin Gastroenterol 42: 584–588, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Orlando RC. Pathophysiology of gastroesophageal reflux disease: esophageal epithelial resistance. In: The Esophagus (5th ed.), edited by Castell DO, Richter JE. Chichester, UK: Wiley, 2012 [Google Scholar]
- 32.Orlando RC, Powell DW, Carney CN. Pathophysiology of acute acid injury in rabbit esophageal epithelium. J Clin Invest 68: 286–293, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oshima T, Koseki J, Chen X, Matsumoto T, Miwa H. Acid modulates the squamous epithelial barrier function by modulating the localization of claudins in the superficial layers. Lab Invest 92: 22–31, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Perez-Moreno M, Song W, Pasolli HA, Williams SE, Fuchs E. Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci USA 105: 15399–15404, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rafiee P, Nelson VM, Manley S, Wellner M, Floer M, Binion DG, Shaker R. Effect of curcumin on acidic pH-induced expression of IL-6 and IL-8 in human esophageal epithelial cells (HET-1A): role of PKC, MAPKs, and NF-κB. Am J Physiol Gastrointest Liver Physiol 296: G388–G398, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Rayet B, Gelinas C. Aberrant rel/NFκB genes and activity in human cancer. Oncogene 18: 6938–6947, 1999 [DOI] [PubMed] [Google Scholar]
- 37.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131–4142, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Souza RF, Huo X, Mittal V, Schuler CM, Carmack SW, Zhang HY, Zhang X, Yu C, Hormi-Carver K, Genta RM, Spechler SJ. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 137: 1776–1784, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem 286: 31263–31271, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol 125: 681–693, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tobey NA, Carson JL, Alkiek RA, Orlando RC. Dilated intercellular spaces: a morphological feature of acid reflux—damaged human esophageal epithelium. Gastroenterology 111: 1200–1205, 1996 [DOI] [PubMed] [Google Scholar]
- 42.Tobey NA, Hosseini SS, Argote CM, Dobrucali AM, Awayda MS, Orlando RC. Dilated intercellular spaces and shunt permeability in nonerosive acid-damaged esophageal epithelium. Am J Gastroenterol 99: 13–22, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi T, Yoshida N, Tomatsuri N, Takayama R, Katada K, Takagi T, Ichikawa H, Naito Y, Okanoue T, Yoshikawa T. Cytokine-induced neutrophil accumulation in the pathogenesis of acute reflux esophagitis in rats. Int J Mol Med 16: 71–77, 2005 [PubMed] [Google Scholar]
- 44.Zhou J, He S, Zhang N, Spee C, Zhou P, Ryan SJ, Kannan R, Hinton DR. Neutrophils compromise retinal pigment epithelial barrier integrity. J Biomed Biotechnol 2010: 289360, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.