

Abstract
Stroke and circulatory arrest cause interferences in blood flow to the brain that result in considerable tissue damage. The primary method to reduce or prevent neurologic damage to patients suffering from brain ischemia is prompt restoration of blood flow to the ischemic tissue. However, paradoxically, restoration of blood flow causes additional damage and exacerbates neurocognitive deficits among patients who suffer a brain ischemic event. Mitochondria play a critical role in reperfusion injury by producing excessive reactive oxygen species (ROS) thereby damaging cellular components, and initiating cell death. In this review, we summarize our current understanding of the mechanisms of mitochondrial ROS generation during reperfusion, and specifically, the role the mitochondrial membrane potential plays in the pathology of cerebral ischemia/reperfusion. Additionally, we propose a temporal model of ROS generation in which post-translational modifications of key oxidative phosphorylation proteins (OxPhos) caused by ischemia, induce a hyperactive state upon reintroduction of oxygen. Hyperactive OxPhos generates high mitochondrial membrane potentials, a condition known to generate excessive ROS. Such a state would lead to a ‘burst’ of ROS upon reperfusion, thereby causing structural and functional damage to the mitochondria and inducing cell death signalling that eventually culminate in tissue damage. Finally, we propose that strategies aimed at modulating this maladaptive hyperpolarization of the mitochondrial membrane potential may be a novel therapeutic intervention and present specific studies demonstrating the cytoprotective effect of this treatment modality.
Keywords: Brain, Reactive Oxygen Species, Ischemia, Reperfusion, Mitochondria, Oxidative Phosphorylation
1. Background: Neuropathology of Reperfusion Injury
Brain ischemia/reperfusion results in extensive injury and is a substantial medical burden because of the ensuing morbidity and mortality. In adults, ischemic insults to the brain typically result from stroke (caused by either thrombotic occlusion or rupture of a blood vessel) or cardiac arrest while, in infants, cerebral ischemia is initiated by complications during labor and delivery, resulting in neonatal hypoxic-ischemic encephalopathy. In both cohorts, restoring blood flow and thus reestablishing nutrient and oxygen delivery to the ischemic brain will undoubtedly salvage neurons. However, reperfusion itself causes additional, substantial brain damage termed “reperfusion injury”. The impact of cerebral ischemia/reperfusion injury on patient mortality is sizable irrespective of age or etiology.
1.1 Stroke
Stroke is the third leading cause of death and disability among Americans (Lloyd-Jones et al. 2010). Strokes are most commonly ischemic in origin, caused by vascular obstruction in the cerebral circulation. If diagnosed in a timely manner, ischemic stroke can be reversed by administration of thrombolytic agents or, alternatively, the obstruction can be physically removed. Neurons that lie distal to the obstructed vessel, relying exclusively on blood supply from this vessel, die from the prolonged complete ischemia. These neurons comprise the infarcted region of the brain that is termed the “ischemic core”. Such neurons never regain function upon restoration of blood flow, and are dead prior to any window of therapeutic intervention. Of greater clinical interest are the populations of neurons that die in a delayed manner after reperfusion is initiated. These neurons, surrounding the ischemic core - referred to as the “penumbra” - are in part perfused by collateral blood flow (i.e., are not fully reliant on blood flow from the obstructed vessel), and are therefore more resistant to ischemic damage. Although neurons of the penumbra do not succumb to the initial ischemia-induced cell death, they go on to die during reperfusion in a delayed manner, resembling an apoptotic pathway. The delay in death of these cells offers a window for therapeutic intervention, thus it is critical that treatment of the penumbra be considered prior to recanalization of the obstructed vessel.
1.2 Cardiac Arrest/Resuscitation
Another leading cause of brain ischemia/reperfusion injury is cardiac arrest followed by resuscitation. In contrast to a focal brain insult caused by stroke, cardiac arrest results in complete ischemia throughout the entire brain. The brain is an extremely sensitive organ, hence even short durations of global ischemia (beyond 10 minutes) can result in debilitating neurologic deficits (Krause et al. 1986). This sensitivity to ischemia may result from reliance on OxPhos for energy production, a concept we will address in detail in subsequent sections.
Successful resuscitation can rapidly restore blood flow and oxygen delivery to the body, including the brain. While approximately 70,000 patients are resuscitated from cardiac arrest each year, only ∼10-35% of resuscitated patients survive to hospital discharge, likely as result of the severe brain damage caused by cerebral ischemia (Bloom et al. 2007; Krause et al. 1986; Nichol et al. 2008). Whereas stroke results in a cerebral infarct, resuscitation from cardiac arrest results in neuronal death in select cell populations that are most sensitive to ischemic injury. The most sensitive populations of neurons, the CA1 hippocampal neurons (Kumar et al. 1987; Jenkins et al. 1981), die during the first days of reperfusion (Ito et al. 1975; Kirino and Sano 1984). The specific biochemical events that result in delayed neuronal death continue to be elucidated; however, overwhelming evidence has identified ROS generation as a key damaging event that leads to death of neurons (Pulsinelli et al. 1997; Hayashi et al. 2003; Piantadosi and Zhang 1996; Sugawara and Chan 2003).
1.3 Neonatal Hypoxic-Ischemic Encephalopathy
Ischemic insults to the infant brain cause hypoxic-ischemic encephalopathy (HIE), which leads to long-term neurocognitive deficits including cerebral palsy and epilepsy (Al-Macki et al. 2009). With an occurrence of approximately 2-4 per 1000 full-term in-hospital deliveries, HIE is a serious medical concern. The consequences of HIE are more severe among low birth weight and premature newborns (Vannucci 2000; Volpe 1992). Various antepartum causes of HIE have been identified including maternal hypotension and fetal growth restriction, yet, the most prevalent cause is prolapse or compression of the umbilical cord and placenta abruption (Badawi et al. 1998; Sie et al. 2000; Cowan et al. 2003). There is extensive heterogeneity in neuropathology following HIE, primarily due to variation in the etiology and severity of the hypoxic/ischemic events (Ferriero 2004). However, as in the adult, reperfusion undoubtedly contributes significantly to the overall pathologic progression of HIE and thus is of therapeutic interest.
2. A Mitochondrial Perspective on Reperfusion Injury
Mitochondria have long been known to play a critical role in the pathogenesis of cerebral ischemia/reperfusion injury, via ROS generation, mitochondrial failure or dysfunction, and mitochondrial (type II) apoptosis. In fact, during brain ischemia/reperfusion evidence exists for the three above deleterious events occurring within the mitochondria (see (Chan 2001; Fiskum et al. 1999). In this review, we present a common link between the three above events in mitochondrial pathologies seen during stroke, cardiac arrest, and HIE In Section 3 we extend this concept to unify these three events into a novel paradigm that positions OxPhos as a central linchpin in the initiation and execution of cell death caused by ischemia/reperfusion. However, before presenting this model of reperfusion injury, it is critical that we discuss: 1) how the OxPhos system functions; 2) the mechanism by which OxPhos generates and controls the mitochondrial membrane potential (ΔΨm); 3) control of OxPhos by phosphorylation; and finally 4) the role of the ΔΨm in ROS generation.
2.1 The Electron Transport Chain and Oxidative Phosphorylation
The mitochondrial electrochemical gradient or protonmotive force (Δpm) is generated and utilized by the OxPhos system, composed of the electron transport chain (ETC) and ATP synthase. A primary function of the ETC is to execute the transfer of electrons to the final electron acceptor, O2. The molecular machinery that comprise the ETC include two major sites for electron entry, complex I (NADH dehydrogenase), and complex II (succinate dehydrogenase). Electrons donated to either of these protein complexes are transferred to complex III (bc1 complex) via the non-protein electron carrier ubiquinone. Electrons are transferred from complex III to complex IV (Cytochrome c Oxidase, CcO) via the electron carrier cytochrome c (Cytc). CcO catalyses the final, and proposed rate limiting step in electron transfer; the donation of electrons to O2, allowing conversion of O2 and H+ to H2O. The energy stored in the electrons is stepwise extracted by complexes I, III, and IV, which couple electron transfer along the ETC to pumping of H+ across the inner mitochondrial membrane. This pumping of protons across the inner mitochondrial membrane constitutes the largest contributing force of the mitochondrial membrane potential (ΔΨm), representing the overall charge difference across the inner mitochondrial membrane (see Section 2.2). In addition, these chemical protons that are taken up from the matrix site contribute to the generation of Δpm. Finally, Δpm is utilized by ATP synthase (Complex V), which drives the conversion of ADP and Pi to ATP (Fig. 1, Normal Oxidative Phosphorylation). This process provides the vast majority (90%) of ATP in the brain under normal conditions.
Figure 1. Progression of Ischemia/reperfusion Injury.
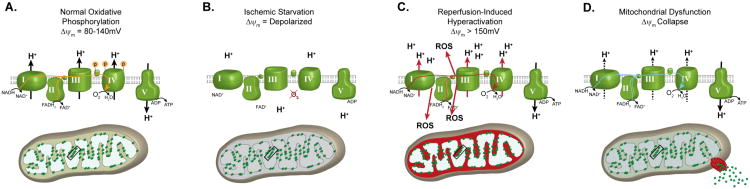
(A) Under normal, non-stressed conditions, OxPhos components are phosphorylated (as illustrated for Cytc and CcO), promoting controlled electron transfer and maintaining the ΔΨm in the 80-140mV range. This respiratory state is conducive to maximal ATP production and minimal ROS generation. (B) Ischemia induces a starvation state were the ETC cannot function due to lack of O2. Dephosphorylation of OxPhos during ischemia renders these enzymes ‘primed’ for hyperactivity, however they cannot operate due to lack of the terminal substrate for respiration, O2. (C) Reintroduction of O2 with reperfusion initiates electron transfer, proton pumping and ATP synthesis. However, in this hyperactive (dephosphorylated) state, OxPhos hyperpolarizes the ΔΨm, causing an exponential increase in ROS generation at complexes I and/or III. (D) This burst in ROS can act as a signal for triggering apoptosis. In addition, damage caused by ROS induces a mitochondrial dysfunction state, with reduced electron transfer kinetics and reduced ΔΨm levels, which results in energetic failure.
Under non-stressed conditions, the transfer of electrons is a tightly regulated process. In fact, the vast majority of electrons that are donated to the ETC complete the entire reaction, culminating in reduction of O2. However, a small percentage of electrons escape the ETC and can react with O2 to form superoxide (O2-), a potent ROS. The specific sites of ROS generation along the ETC are complexes I and III. Although CcO (complex IV) produces several radical intermediates during the reduction of O2, no electrons are allowed to escape; as a result CcO does not directly produce ROS. Under normal conditions, endogenous antioxidant systems are sufficient to scavenge the modest amounts of ROS generated and prevent cellular damage. However, under pathologic conditions antioxidants become overwhelmed or exhausted, allowing the unopposed and uncontrolled production of ROS and resultant ROS-initiated damage to cellular proteins, lipids, nucleic acids, and polysaccharides in an indiscriminate fashion (Stadtman and Levine 2000; Richter and Frei 1988; Kaur and Halliwell 1994; LeDoux et al. 1999; Rubbo et al. 1994). To fully understand how ROS generation occurs and how this is controlled in a normal physiologic context, an appreciation of Δpm and its primary component, the mitochondrial membrane potential ΔΨm, is required.
2.2 The Proton Motive Force and Mitochondrial Membrane Potential
Δpm consists of two components, 1) an electrical constituent, simply referred to as the mitochondrial membrane potential ΔΨm, and 2) a chemical constituent, the pH difference across the inner mitochondrial membrane. Their relationship is defined as Δpm = ΔΨm - 59 ΔpH. The electrical component, ΔΨm, represents the major portion of Δpm in higher organisms.
In the traditional view, Δpm (and ΔΨm) are determined by two basic components, substrate availability and respiratory control, which both act on the OxPhos complexes. The most basic means of mitochondrial OxPhos regulation is dependent on both availability of substrate (e.g., NADH, O2, ADP, Pi) and its product, ATP. ATP and ADP are allosteric inhibitors and activators of CcO, respectively, and this control mechanism was proposed to adjust ETC activity to energy demand (Arnold and Kadenbach 1999; Kadenbach et al. 2010). Another major OxPhos regulatory mechanism is provided by Δpm itself, and is called respiratory control, as was demonstrated in isolated mitochondria more than five decades ago (Chance and Williams 1955). Respiratory control is a mechanism by which Δpm causes inhibition of the ETC proton pumps when the proton gradient exceeds a threshold value, preventing further proton pumping at high Δpm levels. When ATP synthase converts ADP to ATP by utilizing the proton gradient, the reduction in Δpm allows the proton pumps (i.e., complexes I, III, and IV) to resume electron transfer and to pump protons across the inner membrane. In resting mitochondria, when the vast majority of ADP has been converted into ATP, synthesis of ATP slows and Δpm increases, inhibiting the proton pumps and thus mitochondrial respiration. This feedback mechanism pairs the ETC activity to ΔΨm and serves to maintain ΔΨm at physiologic levels of 80-140mV - a range in which ATP production is efficient and ROS generation is minimal.
Due to the difficulty of measuring absolute Δpm in intact cells, most publications report ΔΨm values, which constitute the majority of Δpm. ΔΨm can be monitored in living cells using voltage-dependent fluorescent probes such as the rhodamine dye TMRE (tetramethyl-rhodamine ethyl ester), and changes in fluorescence indicate relative changes in ΔΨm. Absolute ΔΨm levels in millivolts can also be determined in isolated mitochondria by measuring the distribution of a membrane permeable cation such as tetraphenylphosphonium (TPP) with a TPP-sensitive electrode. In addition, absolute mV values for ΔΨm can be determined in live cells by monitoring the redox states of the redox centers in bc1 complex, thus allowing the precise calculation of ΔΨm (Kim et al. 2012).
Since ΔΨm could be measured readily, studies investigating ΔΨm revealed an important difference of ΔΨm levels observed in isolated mitochondria versus intact cells. In isolated mammalian mitochondria from liver and brain under state 4 conditions, ΔΨm values were measured ≥150 mV, often exceeding 200 mV (Nicholls 1974; Labajova et al. 2006; Cossarizza et al. 1996; Barger et al. 2003; Shears and Kirk 1984; Brand et al. 1988; da Silva et al. 1998; Moreira et al. 2001). In contrast, the majority of studies performed in a more physiological context with a variety of intact mammalian cells or even intact organs showed lower ΔΨm values in the range of 80 to 140 mV (Wan et al. 1993; Zhang et al. 2001; Brand and Felber 1984; Backus et al. 1993; Porteous et al. 1998), with few studies reporting higher values between 140 – 161 mV (Nicholls 2006; Hoek et al. 1980; Brand and Felber 1984; Porteous et al. 1998; Nobes et al. 1990; Cortese 1999). This discrepancy may be explained by differences in the regulation of OxPhos activity in higher organisms.
Respiratory control has traditionally been viewed as a key regulator of OxPhos. While this may be correct for OxPhos systems in bacteria, it appears that, in eukaryotes, additional regulatory mechanisms are in place. This idea is further supported by the fact that OxPhos enzymes are more complex in higher organisms. For example, CcO from bacteria contains 2 to 4 subunits whereas the mammalian enzyme is composed of 13 subunits per monomer and functions as a dimer, suggesting divergence with enhanced regulation (Iwata et al. 1995; Tsukihara et al. 1996). Although some differences between studies may be explained by different experimental conditions and the use of cells from different species and tissues, the emerging picture is that ΔΨm values in isolated mitochondria are higher compared to those in intact cells. Explanations for this discrepancy, and the consequences of high ΔΨm values, will be discussed in the next two sections.
2.3 OxPhos is Regulated by Reversible Phosphorylation
Higher ΔΨm values observed in isolated mitochondria compared to intact cells suggest that the isolation procedure per se may induce modifications resulting in readings above the true physiologic range. Importantly, all mammalian OxPhos complexes are phosphorylated in vivo (reviewed in (Hüttemann et al. 2007), and we propose that these phosphorylations may be lost during traditional mitochondria isolation. A recent study analyzing mitochondrial morphology and function showed that the structure of isolated mitochondria is clearly different compared to the morphology found in vivo (Picard et al. 2011). The authors further demonstrated a ∼2-fold increase in CcO activity. Calcium is a buffer component used in some traditional protocols to purify mitochondria, and is a highly potent physiological second messenger and activator of mitochondrial function (Robb-Gaspers et al. 1998). Calcium was shown to trigger dephosphorylation of most mitochondrial proteins (Hopper et al. 2006), which may be mediated by calcium-dependent phosphatases as we and others have postulated (Hüttemann et al. 2007; Lee et al. 2005; Bender and Kadenbach 2000). A similar scenario likely takes place during ischemic stress, and this will be the focus of Section 3.
We propose that phosphorylation of OxPhos complexes is a critical regulatory mechanism in higher organisms to maintain healthy respiration rates and to prevent hyperpolarization of ΔΨm. Using novel methods of mitochondrial and OxPhos protein isolation that preserve protein phosphorylation sites (Lee et al. 2009), we and others found that Cytc and CcO were reversibly phosphorylated at multiple residues (Hüttemann et al. 2008; Huttemann et al. 2012; Helling et al. 2012). Moreover, phosphorylation of these proteins altered their electron transfer kinetics and affected allosteric regulation by ATP and ADP (Hüttemann et al. 2008). Phosphorylation of Cytc at either Tyr48 and Tyr97, caused reduced reaction rates with CcO, and is proposed to lead to normal physiologic electron transfer rates (Lee et al. 2006; Yu et al. 2008a; Pecina et al. 2010). In all tissue types investigated, Cytc was normally present in both the phosphorylated and dephosphorylated state. Upon cellular stress (specifically, cerebral ischemia), the enzyme is rapidly dephosphorylated (Sanderson et al. 2012). Interestingly, activation of cell signaling cascades that promote cell survival, such as insulin signaling (Sanderson et al. 2008), induce Cytc phosphorylation (Sanderson et al. 2012). We also identified a residue on CcO that is reversibly phosphorylated (Tyr304), leading to inhibition of CcO (Lee et al. 2005). Thus, dephosphorylation of CcO results in higher basal respiration.
We further propose that mitochondrial isolation procedures or cellular stress, including ischemia, alter the physiological phosphorylation state of the OxPhos complexes. This concept is supported by a study demonstrating hypoxia-induced changes in CcO phosphorylation in the heart (Prabu et al. 2006). In our model, phosphorylation of OxPhos proteins induce a healthy respiratory state where ΔΨm values >140 mV inhibit further proton pumping, maintaining the 80-140 mV range where ROS production in minimal. In contrast, cellular stress in vivo and isolation of mitochondria in vitro causes changes and/or dephosphorylations of ETC complexes promoting maximal activity, and ΔΨm only inhibits further proton pumping at very high ΔΨm values; thus, in this state ΔΨm is hyperpolarized. Support of our model is provided by studies showing that phosphorylation of CcO and Cytc, as found in vivo, leads to partial inhibition and thus healthy cell respiration (Lee et al. 2005; Lee et al. 2006; Yu et al. 2008b).
These studies demonstrate that stressful stimuli, such as ischemia, can result in changes in phosphorylation pattern of the OxPhos complexes, leading to ROS generation. Conversely, survival signaling promotes phosphorylation and ‘controlled’ electron transfer rates. Our novel findings suggest a mechanism by which cell signaling cascades can regulate the overall basal activity rate of OxPhos (Huttemann et al. 2012). Similar regulatory mechanisms may be discovered on other OxPhos complexes and functionally studied in the future. It is our hypothesis that mitochondria in intact cells under healthy conditions do not function at maximal capacity. This is reasonable because: 1) maximal rates of ATP synthesis by ATP synthase occur at ΔΨm=100-120 mV (Kaim and Dimroth 1999); thus a further increase in ΔΨm will not result in more ATP production, and 2) as discussed in Section 2.4, high ΔΨm levels cause excessive ROS production.
2.4 Mitochondrial Membrane Potential Controls ROS Production
Under normal conditions, over 90% of oxygen is fully reduced to H2O by CcO and only a small number of electrons ‘leak’ and lead to partial reduction of O2 to superoxide. This ROS production takes place proximal to CcO, at complexes I and III, which release superoxide on the matrix and intermembrane space sides, respectively (St-Pierre et al. 2002b). In complex I, two major sites of electron escape have been proposed, the flavoprotein component of electron entry into complex I (Han et al. 2003) or the iron-sulfur cluster (Kushnareva et al. 2002). For complex III, ROS is produced by inhibition of electron transfer through the Q cycle (Liu 1999). Electron transfer from the cytochrome bL heme to the bH heme is inhibited at high ΔΨm, resulting in ubisemiquinone radical generation (Rottenberg et al. 2009a). Ubisemiquinone generated near the intermembrane space then reduces O2 to form superoxide.
Electron leak occurs at Complex I, Complex III, or both, depending on the type of stressful stimuli and cell type, however one common mechanism exists. It has been clearly demonstrated that ROS generation is dependent on ΔΨm. Specifically: 1) the maintenance of physiologically optimal ΔΨm values, not exceeding 140 mV, prevents generation of ROS, while providing the full capability to produce ATP (Kaim and Dimroth 1999); 2) although there are some conditions where ROS can be generated at low ΔΨm levels through different mechanisms (Suski et al. 2012), it is generally accepted that pathophysiologic levels of ROS are produced at high ΔΨm values; and 3) hyperpolarization of ΔΨm (exceeding 140 mV) causes an exponential increase in ROS generation at both complexes I and III (Liu 1999; Starkov and Fiskum 2003; Liu 2010; Korshunov et al. 1997). It was also shown that the ΔΨm component alone, and not ΔpH of the proton motive force, determines ROS production at complex III (Rottenberg et al. 2009b). High ΔΨm levels extend the half-life of reaction intermediates of electron transfer at sites capable of single electron reduction of O2, thus allowing electron escape.
Once partially reduced to superoxide, this oxygen radical reacts with other molecules such as H2O or H+ to generate even more reactive species H2O2, HO2, and OH. In addition, superoxide interacts with NO to form equally cytotoxic reactive nitrogen species. ROS generated in the mitochondria can freely cross mitochondrial membranes or exit via mitochondrial channels such as VDAC and, once released, can cause oxidative damage throughout the cell. Of note, ROS react quickly (half-life of seconds to nanoseconds) and irreversibly to damage cell constituents.
An important question remains: why do mitochondria have an excess capacity to generate higher ΔΨm levels with potentially disastrous consequences for the cell? One reason is that mitochondria must have the capacity to adapt to varying energy demands. However, a more plausible explanation is their involvement in mitochondrial (type II) apoptosis. Numerous studies have demonstrated that induction of apoptosis, which is accompanied by stress signaling and can involve: 1) excessive calcium release; 2) transient hyperpolarization of ΔΨm; and 3) a burst in the production of ROS, which has been proposed as a key signal for committing a cell to apoptosis (reviewed in (Kadenbach et al. 2004). Accordingly, in Sections 3 and 4, we will integrate this mitochondrial-centric sequence of events leading to cellular demise into a model of ischemia/reperfusion injury, and discuss the concept that modulation of ΔΨm may provide a novel strategy to attenuate brain damage caused by ischemia/reperfusion.
3. Model of Ischemia/Reperfusion Injury
The preceding sections have provided insight regarding the role of mitochondria in cell death caused by brain ischemia/reperfusion, and lead us to propose a model that focuses on changes of the phosphorylation state of mitochondrial OxPhos and subsequent ΔΨm hyperpolarization (Fig 1). This model predicts that ischemic alterations of mitochondrial OxPhos primes the mitochondria for reperfusion-induced ΔΨm hyperpolarization, an associated burst in mitochondrial ROS generation and loss of mitochondrial function, and subsequent delayed neuronal death. This progression is simplified into four main states that summarize the induction, progression, and execution of cell death during brain ischemia/reperfusion: 1) ischemic starvation; 2) reperfusion-induced hyperactivation; 3) mitochondrial dysfunction; and 4) delayed neuronal death (Fig 1).
3.1 Ischemic-Starvation State: Ischemic OxPhos Dephosphorylation and the Role of Calcium
A unique feature of the brain is that it is almost exclusively dependent on oxidative phosphorylation to generate energy. Therefore it is necessary to have a constant supply of oxygen to sustain functionality. As discussed in previous sections, even under normal conditions, oxidative phosphorylation results in low-level production of ROS which immediately react with antioxidants and do not cause measurable cellular damage. Brain ischemia of even short durations (on the order of seconds-minutes) causes cessation of electron transport, as OxPhos cannot proceed under anoxic conditions. Electron stalling occurs when the rate of entry of electrons into complex I exceeds the rate of transit through the slowest step of the chain. During ischemia, limiting electron transit through complex IV causes upstream build-up of electrons at complexes I and III, thus leading to reduced ETC.
Without electron transfer and proton pumping across the inner mitochondrial membrane, the proton gradient quickly dissipates, thereby disabling Δpm-driven ATP generation by ATP synthase. Ischemia has been found to cause depolarization of ΔΨm following simulated ischemia in vitro (Abramov et al. 2007) and in vivo during experimental stroke (Liu and Murphy 2009). If ischemia persists, this eventually leads to ATP depletion and failure of all energy-dependent processes in the mitochondria and throughout the cell (Folbergrova et al. 1997; Katsura et al. 1993). Depletion of ATP during ischemia would not allow ATP synthase to maintain ΔΨm by operating in the reverse mode via ATP hydrolysis, a mechanism that operates under certain conditions such as in some cancer cells (Domenis et al. 2011), where it is driven by high ATP flux through glycolysis. Of particular importance to ischemia/reperfusion injury is the equilibration of Ca2+ across the plasma membrane and subsequent accumulation of Ca2+ in the cytosol. At high cytosolic [Ca2+], mitochondria actively sequester Ca2+ to prevent pathologic increases in cytosolic [Ca2+]. However, under the condition of ischemic depolarization, intramitochondrial [Ca2+] increases to pathologic levels with evidence (by electron microscopy) of severe mitochondrial swelling and accompanying hallmarks of cell death (Puka-Sundvall et al. 2000).
Calcium is a potent activation signal for mitochondrial phosphatases. For example, the calcium-dependent Ser/Thr-phosphatase, calcineurin, can dephosphorylate proteins within the mitochondria (Ankarcrona et al. 1996). Indeed, Ca2+ accumulation in the mitochondria induces dephosphorylation of multiple mitochondrial proteins (Hopper et al. 2006; Hüttemann et al. 2008). Moreover, it has been demonstrated that increased mitochondrial Ca2+ is a potent activation signal for mitochondrial respiration, leading to increased respiration and excessive ROS generation. This scenario is consistent with that suggested by McCormack and Denton who postulated that the main role of increased mitochondrial Ca2+ is to stimulate ATP production by activating enzymes involved in metabolism (McCormack and Denton 1993). The effect of Ca2+ on mitochondrial respiration does not appear to be due to a direct action of Ca2+ on ETC components, suggesting an intermediate step exists that could be activated by Ca2+. The recent discoveries that dephosphorylation of CcO and Cytc lead to increased respiration rates provide a potential explanation for these early observations of the effect of Ca2+ on mitochondrial metabolism.
Our proposed model represents a pathologic alteration as a response to an imbalance between energy availability and energy demand. Under conditions of mild hypoxia and inadequate ATP, energy demand would trigger Ca2+ signaling to increase mitochondrial respiration to increase ATP production in order to correct the deficiencies (Balaban 2002). In contrast, under ischemic conditions, attempts to increase OxPhos activity in response to inadequate ATP are futile, as the final electron acceptor, O2, is not present. One can speculate that this feed-forward mechanism caused by ischemia would eventually promote maximal activation of OxPhos. Indeed, others have questioned how a normal physiologic stimulus to increase energy production can lead to a pathologic increase in ROS generation (Brookes et al. 2004). When this process is viewed in the context of OxPhos dephosphorylation inducing subsequent hyperactivation of OxPhos, one can appreciate how ischemia could promote a mitochondrial state where substantial ROS can be generated upon reperfusion.
3.2 Reperfusion-Induced Hyperactivation State: OxPhos Hyperactivity, ΔΨm Hyperpolarization, and ROS Generation
It is obvious that reperfusion of the ischemic brain is necessary for any attempt to salvage tissue. However, as we previously discussed, reperfusion per se contributes significantly to tissue damage. From the perspective of ischemic mitochondria, reperfusion is necessary to restore the terminal substrate for OxPhos and nutrients to reinitiate cellular respiration. However, ischemia promotes a mitochondrial state that is conducive to hyperactive electron transfer upon reperfusion (Fig. 2, Reperfusion-Induced Hyperactivation).
Figure 2. Mechanism of ROS generation during reperfusion.
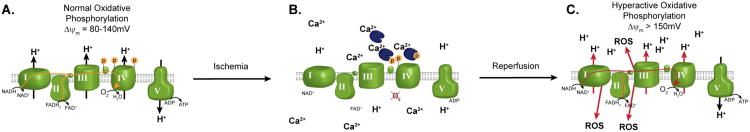
During extended brain ischemia, increased intramitochondrial Ca2+ activates phosphatases that dephosphorylate OxPhos components, as shown for Cytc and CcO in B. This promotes a state of OxPhos hyperactivity; however, because O2 is absent electron transport cannot proceed. (C) Upon induction of reperfusion, OxPhos is allowed to operate at maximal activity, generating high ΔΨm levels, which in turn promotes ROS generation.
According to our model, ischemia evokes an increase in intermitochondrial Ca2+ (Zaidan and Sims 1994; Kristian et al. 2007; Puka-Sundvall et al. 2000), causing activation of mitochondrial phosphatases and dephosphorylation of OxPhos complexes (Hopper et al. 2006), most notably Cytc and CcO (Fig. 2). In addition to the effect on overall respiratory rate, dephosphorylation of CcO also leads to loss of allosteric inhibition by ATP (Bender and Kadenbach 2000; Lee et al. 2005; Hüttemann et al. 2008; Lee et al. 2006; Yu et al. 2008b). Increased OxPhos activity alone could hyperpolarize ΔΨm when reperfusion is initiated, and the loss of allosteric inhibition by ATP would further exacerbate this hyperpolarization. This effect could essentially ‘reset’ respiratory control to a higher level, leading to loss of feedback inhibition between OxPhos and ΔΨm at the normal 120-140mV range. Under these conditions, hyperpolarization could be exacerbated and persist longer than otherwise possible without OxPhos dephosphorylation.
Enhanced and prolonged ΔΨm hyperpolarization would have dramatic consequences. Liu et al. provided compelling evidence of the exponential nature of the relationship between ROS and ΔΨm (reviewed in (Liu 1999). When ΔΨm exceeds 140 mV the exponential nature becomes clear, resulting in a 70-90% increase in ROS generation with a modest 10 mV increase in ΔΨm (Liu 1999; Starkov and Fiskum 2003). Experimental measurements of ΔΨm in this elevated range are plausible. In fact, as we previously discussed, traditional mitochondrial isolation methods that do not take into account preservation of OxPhos phosphorylation often result in ΔΨm above 200 mV (Nicholls 1974; Labajova et al. 2006; Cossarizza et al. 1996; Barger et al. 2003; Shears and Kirk 1984; Brand et al. 1988; da Silva et al. 1998; Moreira et al. 2001). These studies suggest that OxPhos dephosphorylation during ischemia would have profound consequences when reperfusion is initiated.
During the initial moments of reperfusion, OxPhos dephosphorylation would serve to promote rapid reestablishment of ΔΨm and restoration of cellular ATP levels. Indeed, following reversal of brain ischemia, ΔΨm is restored to contol levels within 1 minute, and cellular ATP levels are restored in less than 15 minutes (Liu and Murphy 2009). However, if normal respiratory control mechanims are lost (including loss of alosteric inhibtion by ATP), ΔΨm would surpass normal levels, resulting in pathologic ΔΨm hyperpolarization. In this regard, Liu et al demonstrated that rapid restoration of ΔΨm is quickly followed by hyperpolarization of ΔΨm. A study in neuronal cell culture exposed to simulated ischemia/reperfusion injury demonstrated ΔΨm hyperpolaization during the initial reperfusion stages (Iijima et al. 2006). Moreover, inhibition of ΔΨm hyperpolarization by blocking complex I has been shown to prevent a stress-induced ROS burst and subsequent cell death (Choi et al. 2009). Finally, this concept was extended by Starkov and Fiskum, who reported that mitochondria isolated from brain do indeed have an exponential relationship between ΔΨm and ROS when assayed in vitro (Starkov and Fiskum 2003). These studies suggests a pathologic condition where reperfusion results in ΔΨm hyperpolarization which subsequently causes a significant ROS burst upon reperfusion.
Additional support for the OxPhos paradigm is provided by evidence demonstrating that the majority of ROS generation after brain ischemia occurs immediately upon reflow. For example, in the setting of global brain ischemia, ROS generation is most pronounced during the first 15 minutes of reperfusion (Kunimatsu et al. 2011). Moreover, the predominant source of these ROS are the mitochondrial OxPhos complexes (Fabian et al. 1995; Piantadosi and Zhang 1996; Kudin et al. 2008) and, in brain, escape of electrons from complex I appears to be responsible for most of the ROS produced (Barja and Herrero 1998; Barja 1999; Kudin et al. 2008; St-Pierre et al. 2002a).
As discussed in Section 2.4, the pivotal event that drives excessive electron escape and generation of ROS in vivo is ΔΨm hyperpolarization. These data imply that interventions aimed at regulating ΔΨm and preventing hyperpolarization may serve to attenuate ROS generation. Of particular interest is the dephosphorylation and hyperactivation of CcO, as CcO is the proposed rate-limiting step in OxPhos. Alternatively, direct targeting of ΔΨm (for example, by using mitochondrial membrane uncoupling agents) may provide a feasible approach. However, before considering modulation of ΔΨm as a therapeutic strategy (see Section 4), we review the cytotoxic consequences of ROS generation.
3.3 Mitochondrial Dysfunction
Early studies into mitochondrial function following brain ischemia/reperfusion injury found that, by 30 minutes of reperfusion, mitochondrial respiration is dramatically decreased in cell populations that are destined to die (Sims and Pulsinelli 1987; Sims 1991). More recent reports have demonstrated that global brain ischemia/reperfusion leads to a reduction in complex I and CcO activity at later stages of reperfusion. This respiratory inhibition occurs within the first hour of reperfusion for CcO, and progresses more slowly for Complex I (i.e., maximal inhibition by 24 hours of reperfusion (Racay et al. 2009)). This loss of respiratory function is accompanied by a reduction and eventual collapse of ΔΨm, leading to cell death. Interestingly, while traditional studies have demonstrated reduced OxPhos activity during later reperfusion, recent evidence by Chomova and colleagues has shown that, in the early minutes of reflow, OxPhos activities are increased over control levels (Chomova et al. 2012). These studies suggest that our current understanding of the responses of OxPhos to ischemia/reperfusion may be confounded by inappropriate mitochondrial extraction techniques employed in older reports.
Mitochondrial dysfunction during reperfusion has often been attributed to ROS-induced damage of mitochondrial components (Zhang et al. 1990; Kushnareva et al. 2002). The resulting OxPhos hypoactivation state (Fig 1, Mitochondrial Dysfunction) result in impaired proton pumping and reduced electron transfer kinetics. The mitochondrial switch from a hyperactive to dysfunctional hypoactive state has been attributed to oxidative damage of OxPhos complexes and/or oxidative damage to lipids important to OxPhos function. In support of this concept, direct oxidative damage of mitochondria has been shown to be involved in cellular damage following brain ischemia/reperfusion (Murakami et al. 1998; Friberg et al. 2002).
A critical mitochondrial lipid target of ROS damage is cardiolipin. This is a unique phospholipid in that the majority of cardiolipin is found in the inner mitochondrial membrane where it is in tight association with OxPhos components. Cardiolipin plays a critical role in membrane insertion and function of Cytc, CcO, and other OxPhos complexes (Shinzawa-Itoh et al. 2007; Kagan et al. 2009), and there is growing evidence that cardiolipin plays a pivotal role in the regulation of mitochondrial bioenergetics (Kim et al. 2011; Robinson 1993). In fact, alterations in the structure and/or content of this phospholipid are responsible for mitochondrial dysfunction in a variety of pathological settings (Petrosillo et al. 2011; Paradies et al. 2010; Petrosillo et al. 2009; Petrosillo et al. 2004); this concept is illustrated by the fact that disruption in the association of CcO with cardiolipin was accompanied by a ∼50% reduction in activity of the enzyme (Robinson 1993). Upon peroxidation, cardiolipin undergoes redistribution to the outer mitochondrial membrane (Garcia Fernandez et al. 2002) where it is required for release of apoptotic proteins from mitochondria into the cytosol (Kagan et al. 2005). These effects could contribute to decreased CcO activity, impaired mitochondrial respiration, and mitochondrial failure.
Eventually, these pathologic alterations in mitochondrial function affect cellular functions and eventually lead to cell death. Alterations in mitochondrial function are potent signals for induction of cell death cascades. Additionally, ROS has been implicated in directly activating cell death cascades. For example, under conditions of persistent impaired respiration, mitochondrial (type II) apoptosis is induced, with the release of apoptogenic factors (including Cytc) from the mitochondria purportedly serving as the final and irreversible trigger of neuronal death.
3.4 Delayed Neuronal Death: An Apoptotic-Like Phenotype
Cell death is often classified as apoptotic or necrotic, however, following cerebral ischemia/reperfusion, cell death often proceeds in a manner distinct from traditional apoptosis or necrosis. Morphologic and biochemical analysis indicate that both necrotic and apoptotic events occur simultaneously (Puka-Sundvall et al. 2000; Northington et al. 2001), and evidence linking various pathways suggests that a degree of crosstalk exists that results in cell death occurring in a spectrum between apoptosis and necrosis (Leist and Jaattela 2001). The most common form of delayed, ischemia/reperfusion-induced neuronal cell death, and the type of insult that is the focus of our proposed mechanism, is characterized by an apoptotic-like phenotype. While all the specific characteristics of apoptosis may not be present, a key step – specifically, the release of apoptogenic factors from the mitochondria – appears critical in the initiation of cell death cascades (Hetz et al. 2005; Sanderson et al. 2008; Cao et al. 2007; Sugawara et al. 1999).
Many mechanisms have been proposed to explain the release of Cytc from mitochondria. Historically, it was hypothesized that mitochondria simply swell and burst, thereby releasing their contents into the cytosol. More recently, a large body of work has focused on the Bcl-2 family of proteins and their interactions as important regulators of mitochondrial permeability to Cytc. Of the Bcl-2 family, the primary candidates proposed to be involved in outer membrane permeabilization appear to be Bax and Bak; these proteins purportedly interact directly with mitochondria to promote the release of Cytc and other apoptogenic proteins (e.g. AIF, Smac/Diablo) (Cheng et al. 2001; Kuwana and Newmeyer 2003). In addition, other investigators have focused on elucidating the formation of so-called ‘permeability transition pores’ that would facilitate Cytc release.
The caveat in all of these studies is that they are based on the premise that Cytc and other apoptogenic proteins are free within the mitochondria and, thus, could be released after pore formation or alterations in mitochondrial permeability. However, there is evidence that release of Cytc is a two-step process, involving 1) the release of proteins usually tethered to inner membrane by cardiolipin, combined with 2) pore formation (Ott et al. 2002). Indeed, Cytc is among the proteins shown to be tethered to the inner mitochondrial membrane by cardiolipin (Berezhna et al. 2003; Kagan et al. 2005). Exposure of mitochondria to ROS induces peroxidation followed by oxidation of cardiolipin, thereby releasing Cytc into the intermitochondrial space (Ott et al. 2002). Subsequently, upon pore formation or alterations in permeability, the liberated Cytc is free to be released into the cytosol where it promotes formation of the apoptosome (a complex containing Cytc, caspase 9, and Apaf-1) that activates caspase 3. The apoptotic pathways activated following ischemia/reperfusion converge on caspase-3, the downstream cysteine protease, leading to the cleavage of hundreds of potential substrates within the brain and thus executing programmed cell death (Cohen 1997). Indeed, activation of caspase-3, -8, and -9 have all been demonstrated to increase in the brain following ischemia/reperfusion (Blomgren et al. 2001; Cheng et al. 1998; Northington et al. 2001; Zhu et al. 2004).
The aforementioned sequence of events identify multiple points of possible therapeutic intervention that, if appropriately targeted, could stop the progression of delayed neuronal cell death and thereby salvage tissue from ischemia/reperfusion injury. For example, intervening at the level of apoptosis (including prevention of mitochondrial outer membrane pore formation, cardiolipin peroxidation, or caspase activation), should have therapeutic benefits. However, attempts to prevent apoptotic cell death have revealed that multiple concurrent and redundant cell death pathways can be activated, making specific targeting of individual mediators or single pathways of apoptosis difficult or ineffective. Therefore, a focus on upstream targets (that is, molecular events that precede Cytc release) may yield a more logical and robust therapeutic approach to neuroprotection.
4. Intervention at OxPhos or ΔΨm as a Potent Method of Neuroprotection
Targeting ROS to reduce or prevent brain ischemia/reperfusion injury is one potential strategy to target an early cell death signal. However, this method has been met with numerous clinical failures. To understand this seemingly surprising lack of success, we must consider the traditional methodology for treatment of oxidative damage.
Current studies have shown that during reperfusion, ROS production exceeds the availability of endogenous antioxidants. Accordingly, previous attempts to design treatment modalities have focused on bolstering cellular antioxidant defenses to correct this imbalance. This strategy is primarily based on laboratory studies demonstrating robust neuroprotection in transgenic animals designed to overexpress endogenous mitochondrial antioxidants (Chan et al. 1998; Murakami et al. 1998; Fujimura et al. 2000; Christophe and Nicolas 2006). These studies have provided substantial mechanistic insight into the pivotal role of ROS in cerebral ischemia/reperfusion injury. However, attempts to translate this concept and administer ROS scavengers as a clinical therapy have been futile. This discrepancy could be explained by the multitude of potential pitfalls inherent in delivery of pharmacologic scavengers and antioxidants to the brain during reperfusion, including difficulties in delivery, rapid reaction kinetics due to the short half-life of ROS, multiple ROS generation sites, limited cellular drug uptake, and failure to cross the blood-brain barrier (Niizuma et al. 2009; Chan et al. 1993). Despite improvements in drug formulation and delivery, the efficacy of antioxidant strategies seen in animal studies has not been realized in human trials (Chan et al. 1987; He et al. 1993; Shuaib et al. 2007; Weigl et al. 2005).
As an alternative to this antioxidant approach, we propose that interventions designed to prevent ROS generation (rather than scavenge ROS) will avoid many of the confounding issues associated with traditional scavenging techniques. In this regard, hyperpolarization of ΔΨm is a critical regulatory step in multiple pathologies, including early reperfusion of multiple tissues (Iijima et al. 2003; Starkov and Fiskum 2003; Liu and Murphy 2009). Moreover, targeting of hyperpolarization has been shown to be a cytoprotective (Pandya et al. 2009; Brennan et al. 2006; Iijima et al. 2006).
4.1 Uncoupling of Mitochondrial Membrane Potential
Cells express mitochondrial proteins, i.e., uncoupling proteins (UCPs) that allow H+ to cross the inner mitochondrial membrane down the proton gradient. This bypasses ATP synthase and thus does not produce ATP by utilizing the proton gradient. The physiologic role of these proton channels is typically associated with heat generation. Recent studies have, however, found that UCPs have additional functions in the cell, including stabilizing the ΔΨm, thereby limiting electron escape and thus partial reduction of O2 (Liu 2010). Interestingly, when UCPs were investigated in tissues where heat generation is not a primary function (such as brain), it was found that these proteins render the brain resistant to ischemia/reperfusion injury. For example, Haines et al. demonstrated that knockout of uncoupling protein 2 (UCP2) resulted in dramatically larger infarcts after stroke (Haines et al. 2010). The converse was also true: overexpression of UCP2 was associated with a decrease in ischemia/reperfusion-induced brain damage, ROS generation, and induction of apoptosis after stroke (Mattiasson et al. 2003). Finally, pivotal data from Teshima et al. demonstrated that increased expression of UCP2 prevented ROS-induced cell death by stabilizing ΔΨm (Teshima et al. 2003). These findings suggest that uncoupling proteins may play an important role in mitochondria by stabilizing ΔΨm to prevent hyperpolarization and ROS production under stress. As a result, these proteins may represent a potent target for therapeutic intervention.
In addition to the use of UCPs to stabilize the ΔΨm, exogenous chemicals that allow protons to cross the inner mitochondrial membrane have also been tested to prevent ischemia/reperfusion injury. Proton ionophores (agents that allow proton leak across the inner membrane), have been shown to be effective in multiple disease states. For example, mild mitochondrial membrane uncoupling reduced ΔΨm hyperpolarization, prevented ROS, and reduced cell death in models of myocardial ischemia (Brennan et al. 2006), traumatic brain injury (Pandya et al. 2009), and peroxide-induced neuronal damage (Choi et al. 2009).
It is important to note that small concentrations of mitochondrial membrane uncoupling agents are profoundly protective whereas, in contrast, higher concentrations exacerbate cellular damage (Han et al. 2009). These studies are consistent with our proposed mechanism of ischemia/reperfusion injury: mild membrane uncoupling will prevent the hyperpolarization of ΔΨm during stressful conditions, while complete uncoupling would allow excessive proton escape across the inner membrane and result in an energetic crisis. This biphasic effect makes the use of these compounds potentially dangerous, as overdose of an uncoupling agent could dramatically compromise the ability to produce energy through OxPhos. These compounds would also need to be present in the mitochondria during the time window of ΔΨm hyperpolarization. As the majority of studies suggest that this phenomenon occurs during the early seconds-minutes of reperfusion (Kunimatsu et al. 2011), delivery to tissue prior to reperfusion may pose a therapeutic barrier. However, as discussed in the following section, there are potential methods capable of attenuating ΔΨm hyperpolarization that do not require pharmacologic delivery.
4.2 Ischemic Preconditioning
Several studies have demonstrated that ΔΨm hyperpolarization during early reperfusion is critical event in the generation of mitochondrial ROS. For example, Liu and Murphy utilized a customized laser scanning confocal microscope together with ΔΨm-specific fluorescent probes for real-time analysis of ΔΨm following relief of ischemia in a mouse model of stroke (Liu and Murphy 2009) and found that hyperpolarization of ΔΨm was evident immediately following reflow. Moreover, the authors demonstrated that application of a robust neuroprotective strategy, ischemic preconditioning, prevented ΔΨm hyperpolarization and dramatically reduced the extent of neurologic injury.
Of specific interest is the mechanism by which preconditioning can prevent ΔΨm hyperpolarization at the onset of reperfusion. Dave et al. demonstrated that brief episodes of antecedent preconditioning ischemia triggered the activation of PKCε and upregulated mitochondrial survival signaling (Dave et al. 2008). Preconditioning applied prior to global brain ischemia provided multiple beneficial effects to the mitochondria, including phosphorylation of multiple OxPhos complexes, reduction of ROS generation, preservation of mitochondrial respiration during late reperfusion (the mitochondrial dysfunction phase) – events that culminated in decreased Cytc release, the putative trigger for apoptosis (Dave et al. 2008). One could postulate that stimulation of cell signaling pathways that enhance phosphorylation of OxPhos complexes and limit Cytc release could promote controlled respiration and stabilize ΔΨm. Alternatively, a sub-lethal ischemic event could induce OxPhos dephosphorylation on a small scale, generate small amounts of ROS, and thereby stimulating survival signaling responsible for maintaining phosphorylation. If the activation of these kinases were increased during the subsequent ‘lethal’ ischemic event, this could provide protection from ΔΨm hyperpolarization and ROS generation. In addition, preconditioning could induce the expression of UCPs, thereby maintaining lower basal ΔΨm levels and limiting hyperpolarization. Indeed, Liu et al demonstrated that preconditioning does increase UCP2 expression in brain (Liu et al. 2009). However, whether this increase in expression in UCP2 contributes to preconditioning-induced neuroprotection – and, the precise mechanism by which preconditioning modulates ΔΨm – remains unknown.
4.3 Induction of Cell Signaling to Induce OxPhos Phosphorylation
Our current knowledge of Cytc and CcO suggests that the primary role of the phosphorylation sites discovered to date is to sustain controlled respiratory rates and thereby prevent ΔΨm hyperpolarization and ROS generation (Yu et al. 2008b; Sanderson et al. 2012; Samavati et al. 2008; Pecina et al. 2010; Lee et al. 2006; Lee et al. 2005). Accordingly, it stands to reason that induction of cell signaling cascades that induce phosphorylation or prevent dephosphorylation would provide some protective effect. We have shown that phosphorylation of Cytc at Tyr97 can be induced by insulin (Sanderson et al. 2012). Moreover, insulin treatment was found to be neuroprotective in a model of global brain ischemia/reperfusion by preventing the release of Cytc from mitochondria and inducing the upregulation of PI3K and other cell survival signaling cascades (Sanderson et al. 2008; Sanderson et al. 2009). Whether phosphorylation of Cytc at Tyr97 contributes to insulin-induced neuroprotection by stabilizing ΔΨm is a focus of current investigation by our group.
5. Conclusions
In this review, we present an overarching, mitochondrial-centric hypothesis describing the mechanisms that underlie mitochondrial ROS generation and cell death induced by reperfusion of ischemic brain tissue. There is evidence to support this injury mechanism in multiple scenarios of cerebral ischemia/reperfusion injury including stroke, cardiac arrest followed by resuscitation, and hypoxic-ischemic damage. The crux of the hypothesis is that ΔΨm hyperpolarization and the ensuing ROS burst cause oxidative damage that precedes apoptosis in these pathologies. Specifically, we propose that: 1) ischemia induces dephosphorylation of OxPhos, thereby 2) priming the mitochondria for hyperactive electron transport and ΔΨm hyperpolarization upon reperfusion, 3) initiating a burst of ROS which overwhelms endogenous antioxidant systems that 4), damages cellular components and 5) culminates in the initiation of apoptotic-like cell death cascades. Most notably, we propose that stabilization of ΔΨm during early reperfusion provides a novel strategy to limit ROS generation and attenuate ischemia/reperfusion induced to brain.
Acknowledgments
This work was supported by the Department of Emergency Medicine, the Cardiovascular Research Institute, Wayne State University, Detroit, and grant GM089900 from the National Institutes of Health.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27(5):1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Macki N, Miller SP, Hall N, Shevell M. The spectrum of abnormal neurologic outcomes subsequent to term intrapartum asphyxia. Pediatric neurology. 2009;41(6):399–405. doi: 10.1016/j.pediatrneurol.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 1996;394(3):321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- Arnold S, Kadenbach B. The intramitochondrial ATP/ADP-ratio controls cytochrome c oxidase activity allosterically. FEBS Lett. 1999;443(2):105–108. doi: 10.1016/s0014-5793(98)01694-9. [DOI] [PubMed] [Google Scholar]
- Backus M, Piwnica-Worms D, Hockett D, Kronauge J, Lieberman M, Ingram P, LeFurgey A. Microprobe analysis of Tc-MIBI in heart cells: calculation of mitochondrial membrane potential. Am J Physiol. 1993;265(1 Pt 1):C178–187. doi: 10.1152/ajpcell.1993.265.1.C178. [DOI] [PubMed] [Google Scholar]
- Badawi N, Kurinczuk JJ, Keogh JM, Alessandri LM, O'Sullivan F, Burton PR, Pemberton PJ, Stanley FJ. Intrapartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ. 1998;317(7172):1554–1558. doi: 10.1136/bmj.317.7172.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. Journal of molecular and cellular cardiology. 2002;34(10):1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- Barger JL, Brand MD, Barnes BM, Boyer BB. Tissue-specific depression of mitochondrial proton leak and substrate oxidation in hibernating arctic ground squirrels. Am J Physiol Regul Integr Comp Physiol. 2003;284(5):R1306–1313. doi: 10.1152/ajpregu.00579.2002. [DOI] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31(4):347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- Barja G, Herrero A. Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J Bioenerg Biomembr. 1998;30(3):235–243. doi: 10.1023/a:1020592719405. [DOI] [PubMed] [Google Scholar]
- Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466(1):130–134. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- Berezhna S, Wohlrab H, Champion PM. Resonance Raman investigations of cytochrome c conformational change upon interaction with the membranes of intact and Ca2+-exposed mitochondria. Biochemistry. 2003;42(20):6149–6158. doi: 10.1021/bi027387y. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276(13):10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Bloom HL, Shukrullah I, Cuellar JR, Lloyd MS, Dudley SC, Jr, Zafari AM. Long-term survival after successful inhospital cardiac arrest resuscitation. American heart journal. 2007;153(5):831–836. doi: 10.1016/j.ahj.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Felber SM. Membrane potential of mitochondria in intact lymphocytes during early mitogenic stimulation. Biochem J. 1984;217(2):453–459. doi: 10.1042/bj2170453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Hafner RP, Brown GC. Control of respiration in non-phosphorylating mitochondria is shared between the proton leak and the respiratory chain. Biochem J. 1988;255(2):535–539. [PMC free article] [PubMed] [Google Scholar]
- Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovascular research. 2006;72(2):313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American journal of physiology. 2004;287(4):C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27(35):9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21(1):2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18(20):8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH, Kinouchi H, Epstein CJ, Carlson E, Chen SF, Imaizumi S, Yang GY. Role of superoxide dismutase in ischemic brain injury: reduction of edema and infarction in transgenic mice following focal cerebral ischemia. Progress in brain research. 1993;96:97–104. doi: 10.1016/s0079-6123(08)63260-4. [DOI] [PubMed] [Google Scholar]
- Chan PH, Longar S, Fishman RA. Protective effects of liposome-entrapped superoxide dismutase on posttraumatic brain edema. Annals of neurology. 1987;21(6):540–547. doi: 10.1002/ana.410210604. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. The Journal of biological chemistry. 1955;217(1):383–393. [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Molecular cell. 2001;8(3):705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. The Journal of clinical investigation. 1998;101(9):1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Kim J, Kim GW, Choi C. Oxidative stress-induced necrotic cell death via mitochondira dependent burst of reactive oxygen species. Current neurovascular research. 2009;6(4):213–222. doi: 10.2174/156720209789630375. [DOI] [PubMed] [Google Scholar]
- Chomova M, Tatarkova Z, Dobrota D, Racay P. Ischemia-Induced Inhibition of Mitochondrial Complex I in Rat Brain: Effect of Permeabilization Method and Electron Acceptor. Neurochem Res. 2012 doi: 10.1007/s11064-011-0689-6. [DOI] [PubMed] [Google Scholar]
- Christophe M, Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Current pharmaceutical design. 2006;12(6):739–757. doi: 10.2174/138161206775474242. [DOI] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. The Biochemical journal. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese JD. Rat liver GTP-binding proteins mediate changes in mitochondrial membrane potential and organelle fusion. Am J Physiol. 1999;276(3 Pt 1):C611–620. doi: 10.1152/ajpcell.1999.276.3.C611. [DOI] [PubMed] [Google Scholar]
- Cossarizza A, Ceccarelli D, Masini A. Functional heterogeneity of an isolated mitochondrial population revealed by cytofluorometric analysis at the single organelle level. Exp Cell Res. 1996;222(1):84–94. doi: 10.1006/excr.1996.0011. [DOI] [PubMed] [Google Scholar]
- Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, Meiners LC, Dubowitz LM, de Vries LS. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361(9359):736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- da Silva EM, Soares AM, Moreno AJ. The use of the mitochondrial transmembrane electric potential as an effective biosensor in ecotoxicological research. Chemosphere. 1998;36(10):2375–2390. doi: 10.1016/s0045-6535(97)10206-5. [DOI] [PubMed] [Google Scholar]
- Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, Perez-Pinzon MA. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J Neurosci. 2008;28(16):4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenis R, Comelli M, Bisetto E, Mavelli I. Mitochondrial bioenergetic profile and responses to metabolic inhibition in human hepatocarcinoma cell lines with distinct differentiation characteristics. J Bioenerg Biomembr. 2011;43(5):493–505. doi: 10.1007/s10863-011-9380-5. [DOI] [PubMed] [Google Scholar]
- Fabian RH, DeWitt DS, Kent TA. In vivo detection of superoxide anion production by the brain using a cytochrome c electrode. J Cereb Blood Flow Metab. 1995;15(2):242–247. doi: 10.1038/jcbfm.1995.30. [DOI] [PubMed] [Google Scholar]
- Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351(19):1985–1995. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19(4):351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Li PA, Uchino H, Smith ML, Siesjo BK. Changes in the bioenergetic state of rat hippocampus during 2.5 min of ischemia, and prevention of cell damage by cyclosporin A in hyperglycemic subjects. Experimental brain research Experimentelle Hirnforschung Experimentation cerebrale. 1997;114(1):44–50. doi: 10.1007/pl00005622. [DOI] [PubMed] [Google Scholar]
- Friberg H, Wieloch T, Castilho RF. Mitochondrial oxidative stress after global brain ischemia in rats. Neuroscience letters. 2002;334(2):111–114. doi: 10.1016/s0304-3940(02)01116-3. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J Neurosci. 2000;20(8):2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research. 2002;13(9):449–455. [PubMed] [Google Scholar]
- Haines BA, Mehta SL, Pratt SM, Warden CH, Li PA. Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30(11):1825–1833. doi: 10.1038/jcbfm.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol. 2003;64(5):1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- Han YH, Kim SH, Kim SZ, Park WH. Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an O2(*-) generator induces apoptosis via the depletion of intracellular GSH contents in Calu-6 cells. Lung Cancer. 2009;63(2):201–209. doi: 10.1016/j.lungcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Nishi T, Maier CM, Kinouchi H, Chan PH. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J Cereb Blood Flow Metab. 2003;23(10):1117–1128. doi: 10.1097/01.WCB.0000089600.87125.AD. [DOI] [PubMed] [Google Scholar]
- He YY, Hsu CY, Ezrin AM, Miller MS. Polyethylene glycol-conjugated superoxide dismutase in focal cerebral ischemia-reperfusion. The American journal of physiology. 1993;265(1 Pt 2):H252–256. doi: 10.1152/ajpheart.1993.265.1.H252. [DOI] [PubMed] [Google Scholar]
- Helling S, Huttemann M, Ramzan R, Kim SH, Lee I, Muller T, Langenfeld E, Meyer HE, Kadenbach B, Vogt S, Marcus K. Multiple phosphorylations of cytochrome c oxidase and their functions. Proteomics. 2012;12(7):950–959. doi: 10.1002/pmic.201100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC, Antonsson B. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. The Journal of biological chemistry. 2005;280(52):42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- Hoek JB, Nicholls DG, Williamson JR. Determination of the mitochondrial protonmotive force in isolated hepatocytes. J Biol Chem. 1980;255(4):1458–1464. [PubMed] [Google Scholar]
- Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45(8):2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I. Regulation of mitochondrial respiration and apoptosis through cell signaling: Cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012;1817(4):598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, Doan JW. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J Bioenerg Biomembr. 2008;40(5):445–456. doi: 10.1007/s10863-008-9169-3. [DOI] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Samavati L, Yu H, Doan JW. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochimica et biophysica acta. 2007;1773:1701–1720. doi: 10.1016/j.bbamcr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Iijima T, Mishima T, Akagawa K, Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain Res. 2003;993(1-2):140–145. doi: 10.1016/j.brainres.2003.09.041. [DOI] [PubMed] [Google Scholar]
- Iijima T, Mishima T, Akagawa K, Iwao Y. Neuroprotective effect of propofol on necrosis and apoptosis following oxygen-glucose deprivation--relationship between mitochondrial membrane potential and mode of death. Brain research. 2006;1099(1):25–32. doi: 10.1016/j.brainres.2006.04.117. [DOI] [PubMed] [Google Scholar]
- Ito U, Spatz M, Walker JT, Jr, Klatzo I. Experimental cerebral ischemia in mongolian gerbils. I. Light microscopic observations. Acta neuropathologica. 1975;32(3):209–223. doi: 10.1007/BF00696570. [DOI] [PubMed] [Google Scholar]
- Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376(6542):660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- Jenkins LW, Povlishock JT, Lewelt W, Miller JD, Becker DP. The role of postischemic recirculation in the development of ischemic neuronal injury following complete cerebral ischemia. Acta neuropathologica. 1981;55(3):205–220. doi: 10.1007/BF00691320. [DOI] [PubMed] [Google Scholar]
- Kadenbach B, Arnold S, Lee I, Hüttemann M. The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochim Biophys Acta. 2004;1655(1-3):400–408. doi: 10.1016/j.bbabio.2003.06.005. [DOI] [PubMed] [Google Scholar]
- Kadenbach B, Ramzan R, Wen L, Vogt S. New extension of the Mitchell Theory for oxidative phosphorylation in mitochondria of living organisms. Biochimica et biophysica acta. 2010;1800(3):205–212. doi: 10.1016/j.bbagen.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, Greenberger JS, Pitt B, Shvedova AA, Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free radical biology & medicine. 2009;46(11):1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature chemical biology. 2005;1(4):223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Kaim G, Dimroth P. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. Embo J. 1999;18(15):4118–4127. doi: 10.1093/emboj/18.15.4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura K, Rodriguez de Turco EB, Folbergrova J, Bazan NG, Siesjo BK. Coupling among energy failure, loss of ion homeostasis, and phospholipase A2 and C activation during ischemia. Journal of neurochemistry. 1993;61(5):1677–1684. doi: 10.1111/j.1471-4159.1993.tb09803.x. [DOI] [PubMed] [Google Scholar]
- Kaur H, Halliwell B. Aromatic hydroxylation of phenylalanine as an assay for hydroxyl radicals. Measurement of hydroxyl radical formation from ozone and in blood from premature babies using improved HPLC methodology. Analytical biochemistry. 1994;220(1):11–15. doi: 10.1006/abio.1994.1291. [DOI] [PubMed] [Google Scholar]
- Kim J, Minkler PE, Salomon RG, Anderson VE, Hoppel CL. Cardiolipin: characterization of distinct oxidized molecular species. Journal of lipid research. 2011;52(1):125–135. doi: 10.1194/jlr.M010520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Ripple MO, Springett R. Measurement of the Mitochondrial Membran Potential and pH Gradient from the Redox Poise of the Hemes of the bc1 Complex. The Biochemical journal. 2012 doi: 10.1016/j.bpj.2012.02.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta neuropathologica. 1984;62(3):201–208. doi: 10.1007/BF00691853. [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- Krause GS, Kumar K, White BC, Aust SD, Wiegenstein JG. Ischemia, resuscitation, and reperfusion: mechanisms of tissue injury and prospects for protection. American heart journal. 1986;111(4):768–780. doi: 10.1016/0002-8703(86)90114-6. [DOI] [PubMed] [Google Scholar]
- Kristian T, Pivovarova NB, Fiskum G, Andrews SB. Calcium-induced precipitate formation in brain mitochondria: composition, calcium capacity, and retention. Journal of neurochemistry. 2007;102(4):1346–1356. doi: 10.1111/j.1471-4159.2007.04626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudin AP, Malinska D, Kunz WS. Sites of generation of reactive oxygen species in homogenates of brain tissue determined with the use of respiratory substrates and inhibitors. Biochim Biophys Acta. 2008;1777(7-8):689–695. doi: 10.1016/j.bbabio.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Kumar K, Goosmann M, Krause GS, Nayini NR, Estrada R, Hoehner TJ, White BC, Koestner A. Ultrastructural and ionic studies in global ischemic dog brain. Acta neuropathologica. 1987;73(4):393–399. doi: 10.1007/BF00688266. [DOI] [PubMed] [Google Scholar]
- Kunimatsu T, Kobayashi K, Yamashita A, Yamamoto T, Lee MC. Cerebral reactive oxygen species assessed by electron spin resonance spectroscopy in the initial stage of ischemia-reperfusion are not associated with hypothermic neuroprotection. Journal of clinical neuroscience: official journal of the Neurosurgical Society of Australasia. 2011;18(4):545–548. doi: 10.1016/j.jocn.2010.07.140. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002;368(Pt 2):545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Current opinion in cell biology. 2003;15(6):691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Labajova A, Vojtiskova A, Krivakova P, Kofranek J, Drahota Z, Houstek J. Evaluation of mitochondrial membrane potential using a computerized device with a tetraphenylphosphonium-selective electrode. Analytical biochemistry. 2006;353(1):37–42. doi: 10.1016/j.ab.2006.03.032. [DOI] [PubMed] [Google Scholar]
- LeDoux SP, Driggers WJ, Hollensworth BS, Wilson GL. Repair of alkylation and oxidative damage in mitochondrial DNA. Mutation research. 1999;434(3):149–159. doi: 10.1016/s0921-8777(99)00026-9. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Huttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280(7):6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Samavati L, Pecina P, Pecinova A, Hüttemann M. Isolation of regulatory-competent, phosphorylated cytochrome c oxidase. Methods Enzymol. 2009;345 doi: 10.1016/S0076-6879(09)05011-3. in press. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Yu K, Doan JW, Grossman LI, Hüttemann M. New prospects for an old enzyme: mammalian cytochrome c is tyrosine-phosphorylated in vivo. Biochemistry. 2006;45(30):9121–9128. doi: 10.1021/bi060585v. [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nature reviews Molecular cell biology. 2001;2(8):589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- Liu RR, Murphy TH. Reversible cyclosporin A-sensitive mitochondrial depolarization occurs within minutes of stroke onset in mouse somatosensory cortex in vivo: a two-photon imaging study. J Biol Chem. 2009;284(52):36109–36117. doi: 10.1074/jbc.M109.055301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SS. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain--superoxide generating and cycling mechanisms in mitochondria. J Bioenerg Biomembr. 1999;31(4):367–376. doi: 10.1023/a:1018650103259. [DOI] [PubMed] [Google Scholar]
- Liu SS. Mitochondrial Q cycle-derived superoxide and chemiosmotic bioenergetics. Annals of the New York Academy of Sciences. 2010;1201:84–95. doi: 10.1111/j.1749-6632.2010.05632.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen L, Xu X, Vicaut E, Sercombe R. Both ischemic preconditioning and ghrelin administration protect hippocampus from ischemia/reperfusion and upregulate uncoupling protein-2. BMC physiology. 2009;9:17. doi: 10.1186/1472-6793-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121(7):e46, e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nature medicine. 2003;9(8):1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Denton RM. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochemical Society transactions. 1993;21((Pt 3) (3)):793–799. doi: 10.1042/bst0210793. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Santos MS, Moreno A, Oliveira C. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci Rep. 2001;21(6):789–800. doi: 10.1023/a:1015536808304. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18(1):205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA: the journal of the American Medical Association. 2008;300(12):1423–1431. doi: 10.1001/jama.300.12.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur J Biochem. 1974;50(1):305–315. doi: 10.1111/j.1432-1033.1974.tb03899.x. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J Biol Chem. 2006;281(21):14864–14874. doi: 10.1074/jbc.M510916200. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. Journal of neurochemistry. 2009;109(1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Brown GC, Olive PN, Brand MD. Non-ohmic proton conductance of the mitochondrial inner membrane in hepatocytes. J Biol Chem. 1990;265(22):12903–12909. [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early Neurodegeneration after Hypoxia-Ischemia in Neonatal Rat Is Necrosis while Delayed Neuronal Death Is Apoptosis. Neurobiol Dis. 2001;8(2):207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Sullivan PG. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Experimental neurology. 2009;218(2):381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free radical biology & medicine. 2010;48(10):1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- Pecina P, Borisenko GG, Belikova NA, Tyurina YY, Pecinova A, Lee I, Samhan-Arias AK, Przyklenk K, Kagan VE, Hüttemann M. Phosphomimetic substitution of cytochrome C tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry. 2010;49(31):6705–6714. doi: 10.1021/bi100486s. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Di Venosa N, Moro N, Colantuono G, Paradies V, Tiravanti E, Federici A, Ruggiero FM, Paradies G. In vivo hyperoxic preconditioning protects against rat-heart ischemia/reperfusion injury by inhibiting mitochondrial permeability transition pore opening and cytochrome c release. Free radical biology & medicine. 2011;50(3):477–483. doi: 10.1016/j.freeradbiomed.2010.11.030. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Matera M, Moro N, Ruggiero FM, Paradies G. Mitochondrial complex I dysfunction in rat heart with aging: critical role of reactive oxygen species and cardiolipin. Free radical biology & medicine. 2009;46(1):88–94. doi: 10.1016/j.freeradbiomed.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: role of cardiolipin. J Biol Chem. 2004;279(51):53103–53108. doi: 10.1074/jbc.M407500200. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke; a journal of cerebral circulation. 1996;27(2):327–331. doi: 10.1161/01.str.27.2.327. discussion 332. [DOI] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MM, Romestaing C, Hepple RT. Mitochondrial Structure and Function Are Disrupted by Standard Isolation Methods. PLoS One. 2011;6(3):e18317. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteous WK, James AM, Sheard PW, Porteous CM, Packer MA, Hyslop SJ, Melton JV, Pang CY, Wei YH, Murphy MP. Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur J Biochem. 1998;257(1):192–201. doi: 10.1046/j.1432-1327.1998.2570192.x. [DOI] [PubMed] [Google Scholar]
- Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. The Journal of biological chemistry. 2006;281(4):2061–2070. doi: 10.1074/jbc.M507741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puka-Sundvall M, Gajkowska B, Cholewinski M, Blomgren K, Lazarewicz JW, Hagberg H. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain research Developmental brain research. 2000;125(1-2):31–41. doi: 10.1016/s0165-3806(00)00110-3. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Jacewicz M, Levy DE, Petito CK, Plum F. Ischemic brain injury and the therapeutic window. Ann N Y Acad Sci. 1997;835:187–193. doi: 10.1111/j.1749-6632.1997.tb48629.x. [DOI] [PubMed] [Google Scholar]
- Racay P, Tatarkova Z, Chomova M, Hatok J, Kaplan P, Dobrota D. Mitochondrial calcium transport and mitochondrial dysfunction after global brain ischemia in rat hippocampus. Neurochem Res. 2009;34(8):1469–1478. doi: 10.1007/s11064-009-9934-7. [DOI] [PubMed] [Google Scholar]
- Richter C, Frei B. Ca2+ release from mitochondria induced by prooxidants. Free radical biology & medicine. 1988;4(6):365–375. doi: 10.1016/0891-5849(88)90088-3. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. Embo J. 1998;17(17):4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NC. Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr. 1993;25(2):153–163. doi: 10.1007/BF00762857. [DOI] [PubMed] [Google Scholar]
- Rottenberg H, Covian R, Trumpower BL. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. The Journal of biological chemistry. 2009a;284(29):19203–19210. doi: 10.1074/jbc.M109.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, Covian R, Trumpower BL. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem. 2009b;284(29):19203–19210. doi: 10.1074/jbc.M109.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269(42):26066–26075. [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Hüttemann M. Tumor necrosis factor α inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008;283(30):21134–21144. doi: 10.1074/jbc.M801954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Kumar R, Murariu-Dobrin AC, Page AB, Krause GS, Sullivan JM. Insulin activates the PI3K-Akt survival pathway in vulnerable neurons following global brain ischemia. Neurological research. 2009;31(9):947–958. doi: 10.1179/174313209X382449. [DOI] [PubMed] [Google Scholar]
- Sanderson TH, Kumar R, Sullivan JM, Krause GS. Insulin blocks cytochrome c release in the reperfused brain through PI3-K signaling and by promoting Bax/Bcl-XL binding. Journal of neurochemistry. 2008;106(3):1248–1258. doi: 10.1111/j.1471-4159.2008.05473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Lee I, Pecina P, Kumar R, Tousignant RN, Yu K, Mahapatra G, Varughese A, Salomon AR, Hüttemann M. Cytochrome c is tyrosine 97 phosphorylated by neuroprotective insulin treatment. Journal of neuroscience research Pending. 2012 doi: 10.1371/journal.pone.0078627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB, Kirk CJ. Characterization of a rapid cellular-fractionation technique for hepatocytes. Application in the measurement of mitochondrial membrane potential in situ. Biochem J. 1984;219(2):375–382. doi: 10.1042/bj2190375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. Embo J. 2007;26(6):1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357(6):562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- Sie LT, van der Knaap MS, Oosting J, de Vries LS, Lafeber HN, Valk J. MR patterns of hypoxic-ischemic brain damage after prenatal, perinatal or postnatal asphyxia. Neuropediatrics. 2000;31(3):128–136. doi: 10.1055/s-2000-7496. [DOI] [PubMed] [Google Scholar]
- Sims NR. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. Journal of neurochemistry. 1991;56(6):1836–1844. doi: 10.1111/j.1471-4159.1991.tb03438.x. [DOI] [PubMed] [Google Scholar]
- Sims NR, Pulsinelli WA. Altered mitochondrial respiration in selectively vulnerable brain subregions following transient forebrain ischemia in the rat. Journal of neurochemistry. 1987;49(5):1367–1374. doi: 10.1111/j.1471-4159.1987.tb01001.x. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002a;277(47):44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002b;277(47):44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Levine RL. Protein oxidation. Annals of the New York Academy of Sciences. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. Journal of neurochemistry. 2003;86(5):1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxidants & redox signaling. 2003;5(5):597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19(22):RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR. Relation Between Mitochondrial Membrane Potential and ROS Formation. Methods Mol Biol. 2012;810:183–205. doi: 10.1007/978-1-61779-382-0_12. [DOI] [PubMed] [Google Scholar]
- Teshima Y, Akao M, Jones SP, Marban E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circulation research. 2003;93(3):192–200. doi: 10.1161/01.RES.0000085581.60197.4D. [DOI] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272(5265):1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Vannucci RC. Hypoxic-ischemic encephalopathy. American journal of perinatology. 2000;17(3):113–120. doi: 10.1055/s-2000-9293. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Brain injury in the premature infant--current concepts of pathogenesis and prevention. Biology of the neonate. 1992;62(4):231–242. doi: 10.1159/000243876. [DOI] [PubMed] [Google Scholar]
- Wan B, Doumen C, Duszynski J, Salama G, Vary TC, LaNoue KF. Effects of cardiac work on electrical potential gradient across mitochondrial membrane in perfused rat hearts. Am J Physiol. 1993;265(2 Pt 2):H453–460. doi: 10.1152/ajpheart.1993.265.2.H453. [DOI] [PubMed] [Google Scholar]
- Weigl M, Tenze G, Steinlechner B, Skhirtladze K, Reining G, Bernardo M, Pedicelli E, Dworschak M. A systematic review of currently available pharmacological neuroprotective agents as a sole intervention before anticipated or induced cardiac arrest. Resuscitation. 2005;65(1):21–39. doi: 10.1016/j.resuscitation.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Yu H, Lee I, Salomon AR, Yu K, Huttemann M. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim Biophys Acta. 2008a;1777(7-8):1066–1071. doi: 10.1016/j.bbabio.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lee I, Salomon AR, Yu K, Hüttemann M. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochimica et biophysica acta. 2008b;1777(7-8):1066–1071. doi: 10.1016/j.bbabio.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidan E, Sims NR. The calcium content of mitochondria from brain subregions following short-term forebrain ischemia and recirculation in the rat. Journal of neurochemistry. 1994;63(5):1812–1819. doi: 10.1046/j.1471-4159.1994.63051812.x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Huang HM, Carson RC, Mahmood J, Thomas HM, Gibson GE. Assessment of membrane potentials of mitochondrial populations in living cells. Anal Biochem. 2001;298(2):170–180. doi: 10.1006/abio.2001.5348. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. The Journal of biological chemistry. 1990;265(27):16330–16336. [PubMed] [Google Scholar]
- Zhu C, Wang X, Cheng X, Qiu L, Xu F, Simbruner G, Blomgren K. Post-ischemic hypothermia-induced tissue protection and diminished apoptosis after neonatal cerebral hypoxia-ischemia. Brain research. 2004;996(1):67–75. doi: 10.1016/j.brainres.2003.10.013. [DOI] [PubMed] [Google Scholar]