

Abstract
The spectrum of DNA damage caused by reactive oxygen species includes a wide variety of modifications of purine and pyrimidine bases. Among these modified bases, 7,8-dihydro-8-oxoguanine (8-oxoG) is an important mutagenic lesion. Base excision repair is a critical mechanism for preventing mutations by removing the oxidative lesion from the DNA. That the spontaneous mutation frequency of the Escherichia coli mutT mutant is much higher than that of the mutM or mutY mutant indicates a significant potential for mutation due to 8-oxoG incorporation opposite A and G during DNA replication. In fact, the removal of A and G in such a situation by MutY protein would fix rather than prevent mutation. This suggests the need for differential removal of 8-oxoG when incorporated into DNA, versus being generated in situ. In this study we demonstrate that E.coli Nth protein (endonuclease III) has an 8-oxoG DNA glycosylase/AP lyase activity which removes 8-oxoG preferentially from 8-oxoG/G mispairs. The MutM and Nei proteins are also capable of removing 8-oxoG from mispairs. The frequency of spontaneous G:C→C:G transversions was significantly increased in E.coli CC103mutMnthnei mutants compared with wild-type, mutM, nth, nei, mutMnei, mutMnth and nthnei strains. From these results it is concluded that Nth protein, together with the MutM and Nei proteins, is involved in the repair of 8-oxoG when it is incorporated opposite G. Furthermore, we found that human hNTH1 protein, a homolog of E.coli Nth protein, has similar DNA glycosylase/AP lyase activity that removes 8-oxoG from 8-oxoG/G mispairs.
INTRODUCTION
Reactive oxygen species (ROS) are generated in living cells during normal cellular metabolism as well as by exogenous stimuli such as ionizing radiation and various chemical oxidants (1). A prominent target for oxidation in cells is the DNA. The spectrum of ionizing radiation- and ROS-caused damage to DNA is broad and includes a wide variety of modifications to purine and pyrimidine bases (2). 7,8-Dihydro-8-oxoguanine (8-oxoG) is an important mutagenic lesion produced by ROS in DNA and causes G:C→T:A transversions (3,4). Base excision repair is an important mechanism for preventing such transversions by removing the oxidative lesion from the DNA (3,5,6).
In Escherichia coli two DNA glycosylases are involved in the 8-oxoG repair process. MutM protein (formamidopyrimidine-DNA glycosylase) removes 8-oxoG preferentially from 8-oxoG/C pairs in duplex DNA in which in situ oxidation has caused formation of 8-oxoG (3–7). Unrepaired 8-oxoG can form a mutagenic 8-oxoG/A mispair during subsequent DNA replication (3,5,8). MutY protein removes A from the 8-oxoG/A pair and thus provides a second opportunity to prevent mutations (5,6,9). Mutations of the mutM or mutY gene increase G:C→T:A transversions (5,9–11). G:C→C:G transversions are also increased in E.coli mutY mutants, suggesting that the 8-oxoG/G mispair is also formed during DNA replication (12).
MutT is a ubiquitous 8-oxo-dGTPase which prevents the incorporation of 8-oxo-dGTP into DNA by degrading the oxidized form of dGTP generated in the nucleotide pool (13). A considerable amount of 8-oxo-dGTP appears to be generated spontaneously in the nucleotide pool, since the spontaneous mutation frequency of E.coli mutT mutants is much higher than that of mutM or mutY mutants (14). 8-oxo-dGTP can be incorporated opposite A and G during DNA replication, which implies a significant potential for mutation due to 8-oxoG. MutY protein removes G from 8-oxoG/G as well as A from 8-oxoG/A mispairs (12). However, the removal of A and G in such a situation by MutY protein would fix rather than prevent mutations. Therefore, differential removal of 8-oxoG when incorporated into nascent DNA appears to be needed. Recently Hazra et al. (15) found that E.coli Nei protein (endonuclease VIII) could excise 8-oxoG from 8-oxoG/A and 8-oxoG/G mispair-containing oligonucleotides. However, it remains uncertain and it is important to clarify whether the Nei protein plays a role in the prevention of mutations in vivo and whether E.coli has an additional enzyme(s) to remove 8-oxoG in the nascent strand.
In Saccharomyces cerevisiae and mammalian cells the OGG1 proteins that have been identified are 8-oxoG DNA glycosylase/AP lyases (16,17). Recently a second 8-oxoG DNA glycosylase (OGG2) has been purified from S.cerevisiae and human cells (18–20). These OGG2 proteins remove 8-oxoG from 8-oxoG/A and 8-oxoG/G mispairs in vitro (18–20). The S.cerevisiae OGG2 (Ntg1) gene encodes a protein with significant homology to Nth protein (endonuclease III) of E.coli (18). The yeast enzyme has similar substrate specificity to E.coli Nth protein: thymine glycol, 5-hydroxyuracil and 5-hydroxycytosine (20). These findings led us to investigate whether E.coli has proteins equivalent to yeast Ntg1.
In this report we show that E.coli Nth protein has an 8-oxoG DNA glycosylase/AP lyase activity which removes 8-oxoG preferentially from 8-oxoG/G mispairs. MutM and Nei proteins are also capable of removing 8-oxoG from such mispairs. The frequency of spontaneous G:C→C:G transversions was significantly increased in E.coli CC103mutMnthnei mutants compared with wild-type, mutM, nth, nei, mutMnei, mutMnth and nthnei strains. From these results it is concluded that in E.coli Nth protein, together with the MutM and Nei proteins, is involved in the repair pathway for 8-oxoG/G mispairs. Furthermore, we found that human hNTH1 protein, a structural and functional homolog of E.coli Nth protein (5,6,21,22), has similar DNA glycosylase/AP lyase activity that could remove 8-oxoG from 8-oxoG/G mispairs.
MATERIALS AND METHODS
Bacteria, plasmids and media
Bacterial strains used in this study were derivatives of E.coli K12. Strains KSR1 (nth::Cm), KSR2 (nei::Kan), KSR3 (mutM::Tet) and KSR7 (mutM::Tet nth::Cm nei::Kan) were derivatives of CSH26 (23). CC103 is a derivative of P90C [ara Δ(lac proB)XIII] with the F′lac proAB episome carrying a lacZ– mutation affecting residue 461 in β-galactosidase (24). CC103 derivatives with different DNA repair capacities were constructed using P1 transduction according to Miller (25) with a slight modification. Strain BL21 (dcm ompT hsdS gal) was also used.
LB broth and glucose minimal medium were used to culture bacteria throughout this study. When necessary, ampicillin (50 µg/ml), kanamycin (50 µg/ml), tetracycline (25 µg/ml) or chloramphenicol (30 µg/ml) was added to the medium.
Enzymes and chemicals
Tetracycline hydrochloride, kanamycin, chloramphenicol, dithiothreitol (DTT), bovine serum albumin (BSA) and NaBH4 were purchased from Wako Pure Chemicals (Osaka, Japan). Ampicillin was obtained from Meiji Seika (Tokyo, Japan). T4 polynucleotide kinase was obtained from New England Biolabs (Beverly, MA). Plasmids pGEX-4T-1 and pGEX-4T-3, glutathione–Sepharose 4B and thrombin were purchased from Amersham Pharmacia Biotech (Uppsala, Sweden). Restriction enzymes were obtained from Takara Shuzo (Kyoto, Japan) and Toyobo (Osaka, Japan). [γ-32P]ATP (>148 TBq/mmol) was obtained from ICN Biomedicals (Costa Mesa, CA). The BCA Protein Assay Kit was obtained from Pierce (Rockford, IL).
Spontaneous mutation assay
The reversion to Lac+ was assayed as previously described (12). In brief, single colonies of E.coli with different repair capacities were inoculated into glucose minimal medium and cultured at 37°C to stationary phase. Aliquots of 0.1 ml of each cell suspension were plated onto lactose minimal plates containing 0.2% lactose and then incubated at 37°C for up to 18 days. The normal plating density was ∼2 × 108 cells/plate. The mutation frequency was determined as Lac+ revertants/108 viable cells.
Preparation of substrate duplex DNA
The oligonucleotide containing an 8-oxoG was obtained from Trevigen (Gaithersburg, MD). Other oligonucleotides complementary to the 8-oxoG-containing oligonucleotide but with A, G, C and T opposite 8-oxoG were obtained from Biologica (Nagoya, Japan). The sequences of the oligonucleotides used (Seq. 1–Seq. 8) are shown in Figure 1. The oligonucleotide containing 8-oxoG was labeled at the 5′-end with [γ-32P]ATP using T4 polynucleotide kinase and annealed with the complementary strands as previously described (12,26).
Figure 1.

The sequences of oligonucleotides used. O represents 8-oxoG.
Expression and purification of E.coli Nth protein
The plasmid expressing glutathione S-transferase (GST)–Nth fusion protein was constructed as previously described (23). The sequences were checked to verify that no mutations had been introduced by PCR. The resulting plasmid was named pGEX-4T-1-Nth. Escherichia coli BL21 was transformed with pGEX-4T-1-Nth. The overnight culture was diluted 50-fold with fresh LB medium and grown at 37°C until the optical density at 600 nm reached 0.4. After addition of isopropyl-β-d-galactopyranoside (IPTG) at 0.1 mM the culture was incubated at 37°C for 6.5 h. All subsequent procedures were carried out at 4°C. The cells were pelleted, resuspended in phosphate-buffered saline (PBS), pH 7.2, containing 0.2% Triton X-100 and then sonicated on ice. After centrifugation of the cell lysate at 15 000 g for 30 min the supernatant was applied to a glutathione–Sepharose 4B column. The column was washed with 20 vol of PBS and the GST–Nth fusion protein was eluted with 15 mM glutathione in 50 mM Tris–HCl pH 9.6, followed by dialysis against buffer A (20 mM Tris–HCl pH 8.0, 2 mM EDTA, 6.7 mM 2-mercaptoethanol, 0.02% Triton X-100, 300 mM NaCl). The GST–Nth fusion protein was then cleaved with thrombin by incubation for 18 h at 4°C and dialyzed against PBS. The purified Nth protein was stored at –80°C until use. Proteins were analyzed by SDS–PAGE. The concentration of protein was determined using a BCA Protein Assay Kit (Pierce) with BSA as the standard.
Purification of human hNTH1 protein
Plasmid containing human hNTH1 cDNA (27) was digested with BamHI and EcoRI, after which it was subcloned into BamHI- and EcoRI-digested plasmid vector pGEX-4T-3; the resultant plasmid was named pGEX-4T-3-hNTH1. Expression and purification of hNTH1 protein were carried out by essentially the same methods as described above for E.coli Nth protein.
Trapping assay for 8-oxoG-containing oligonucleotides
The DNA trapping assay was carried out at 37°C for 60 min in a reaction mixture (10 µl) containing 10 fmol of labeled duplex oligonucleotides with purified Nth protein or hNTH1 protein (1–40 pmol) in 25 mM Tris–HCl pH 7.5, 1 mM DTT and 2.5 mM EDTA in the presence of 100 mM NaBH4. The reaction was terminated by heating the reaction mixture at 95°C for 5 min. Trapped complexes were separated by SDS–PAGE. The gels were dried and autoradiographed using Fuji RX film at –80°C.
Nicking assay for 8-oxoG-containing oligonucleotides
The DNA nicking assay was performed at 37°C for 60 min in a reaction mixture (10 µl) containing 20 fmol of 32P-labeled duplex oligonucleotides and purified Nth protein or hNTH1 protein in 25 mM Tris–HCl pH 7.5, 2.5 mM EDTA, 1 mM DTT, 50 mM NaCl and 5% glycerol. The reaction was terminated by addition of a stop solution (95% formamide, 0.1% BPB, 0.1% xylene cyanol and 20 mM EDTA). After heating at 95°C for 5 min the samples were cooled to room temperature and then applied to 20% polyacrylamide gels in 90 mM Tris–borate pH 8.3, containing 7 M urea and 2 mM EDTA. After electrophoresis at 1300 V the gels were dried and autoradiographed using Fuji RX film at –80°C.
RESULTS
Frequencies of spontaneous G:C→C:G transversions in E.coli CC103 with various DNA repair capacities
It is well known that MutM protein removes 8-oxoG, which is formed by in situ oxidation of G, preferentially from 8-oxoG/C pairs in duplex DNA (5–7). In addition, this enzyme can remove 8-oxoG from 8-oxoG/G mispairs in vitro. Furthermore, Hazra et al. (15) recently found a novel 8-oxoG DNA glycosylase that is active on 8-oxoG/G and 8-oxoG/A mispairs and identified the enzyme as E.coli Nei protein. Therefore, we examined whether the mutM and nei mutations affect the frequency of Lac+ reversion in E.coli CC103. In this strain only G:C→C:G transversions can restore the wild-type codon at 461 in the lacZ gene (24). Escherichia coli CC103 cells with different DNA repair capacities were plated on lactose minimal plates, followed by incubation at 37°C for up to 18 days. There was only a slight increase in the mutation frequency in the mutMnei mutant compared with the wild-type strain (Fig. 2). On the other hand, more Lac+ revertants accumulated in the CC103mutMnthnei strain than in wild-type CC103 and CC103mutMnei strains during incubation (Fig. 2). Mutations of nth, mutM, nei, nthnei and mutMnth did not affect the frequency of spontaneous G:C→C:G transversions (Table 1).
Figure 2.

Accumulation of Lac+ revertants in E.coli CC103, CC103mutMnei and CC103mutMnthnei. Single colonies of E.coli CC103 (open circles), CC103mutMnei (filled circles) and CC103mutMnthnei (filled squares) were inoculated into glucose minimal medium and cultured at 37°C for 18 h. Aliquots of 0.1 ml of each cell suspension were plated on lactose minimal agar plates and incubated at 37°C for up to 18 days. Mutation frequency was calculated as Lac+ revertants/108 viable cells. Each value represents the mean of at least three experiments.
Table 1. Frequencies of spontaneous mutation to Lac+ in E.coli CC103 with different DNA repair capacities .
| Strain |
Lac+ revertants/108 cellsa |
| CC103 | 1 |
| CC103mutM | 2 |
| CC103nth | 2 |
| CC103nei | 1 |
| CC103mutMnth | 3 |
| CC103mutMnei | 3 |
| CC103nthnei | 5 |
| CC103mutMnthnei | 38 |
aEach value represents the mean of three independent experiments.
Trapping assays of Nth protein with the 8-oxoG/G oligonucleotide
We carried out the borohydride trapping assay to detect proteins with DNA glycosylase/AP lyase activities for 8-oxoG in E.coli extracts. This assay is based on the fact that DNA glycosylases/AP lyases form a Schiff base intermediate that can be trapped by NaBH4 to generate enzyme–DNA crosslinks (23,28,29). Cell extracts prepared from E.coli wild-type and various DNA repair-defective mutants were incubated with duplex oligonucleotides containing 8-oxoG/G mispairs in the presence of 100 mM NaBH4. Trapped DNA–protein complexes were analyzed by SDS–PAGE. One shifted band of trapped complex was detected in extracts from the mutMnei mutant (data not shown). Introduction of the nth mutation into the mutant resulted in complete loss of the shifted band. Other DNA repair mutations tested (23) did not affect formation of the DNA–protein complex. These results suggested that the trapped complex consisted of 8-oxoG/G-containing oligonucleotides and Nth protein.
Next, we purified Nth protein from E.coli cells to examine whether the enzyme is directly trapped by 8-oxoG-containing oligonucleotides and whether it can cleave 8-oxoG-containing DNA. The entire open reading frame of the nth gene was amplified by PCR and subcloned into pGEX-4T-1 to obtain a GST–Nth fusion protein (23). The fusion protein was expressed in E.coli BL21/pGEX-4T-1-Nth with IPTG induction and purified by means of glutathione–Sepharose 4B column chromatography. Purified Nth protein gave a single band with an apparent molecular mass of 23.6 kDa on a SDS–polyacrylamide gel (data not shown).
The trapping assay revealed that the purified Nth protein formed a complex with 8-oxoG/G and 8-oxoG/A but not with 8-oxoG/C or G:C-containing oligonucleotides (Fig. 3).
Figure 3.

Borohydride trapping analysis of purified Nth protein. Duplex oligonucleotide substrates (10 fmol) containing G:C (Seq. 2/Seq. 6), 8-oxoG/C (Seq. 1/Seq. 6), 8-oxoG/G (Seq. 1/Seq. 3), 8-oxoG/A (Seq. 1/Seq. 4) and 8-oxoG/T (Seq. 1/Seq. 5) were incubated with the purified GST–Nth or Nth protein (20 pmol) in the presence of 100 mM NaBH4 at 37°C for 30 min. The shifted band (trapped complex) and free oligonucleotide are indicated. Lanes 1–3, G:C; lanes 4–6, 8-oxoG/C; lanes 7–9, 8-oxoG/G; lanes 10–12, 8-oxoG/A; lanes 13–15, 8-oxoG/T. Lanes 1, 4, 7, 10 and 13, no protein; lanes 2, 5, 8, 11 and 14, GST–Nth protein; lanes 3, 6, 9, 12 and 15, Nth protein.
Cleavage of 8-oxoG-containing duplex DNA by purified Nth protein
The Nth protein is a DNA glycosylase/AP lyase that catalyzes both cleavage of the glycosylic bond to release damaged bases and incision of the phosphodiester backbone at the resulting AP site via a β-elimination reaction (5,6,30). To provide more evidence that Nth protein indeed has a DNA glycosylase/AP lyase activity that removes 8-oxoG from 8-oxoG/G mispair-containing oligonucleotides, a DNA nicking assay was performed. Duplex oligonucleotides containing 8-oxoG/G, 8-oxoG/A, 8-oxoG/T or 8-oxoG/C mispairs were incubated with the protein at 37°C for 60 min, followed by product analysis by PAGE. The results are shown in Figures 4 and 5. Nth protein strongly preferred the 8-oxoG/G substrate, followed by 8-oxoG/A, but catalyzed no detectable 8-oxoG/C- or 8-oxoG/T-specific strand cleavage. Incubation of unlabeled 8-oxoG-containing duplex oligonucleotide annealed with 5′-labeled complementary strands with Nth protein produced no radioactive cleavage products with Nth protein. Thus, the strand incision activity of Nth protein was specific for the 8-oxoG-containing strand in the duplex oligonucleotide. In all cases the incision product results from a β-elimination reaction. These results indicated that 8-oxoG is recognized and removed by Nth protein as a natural substrate. The activity of Nth protein in cleaving the 8-oxoG/G-containing oligonucleotide was about one-quarter of that for cleaving thymine glycol-containing oligonucleotides.
Figure 4.
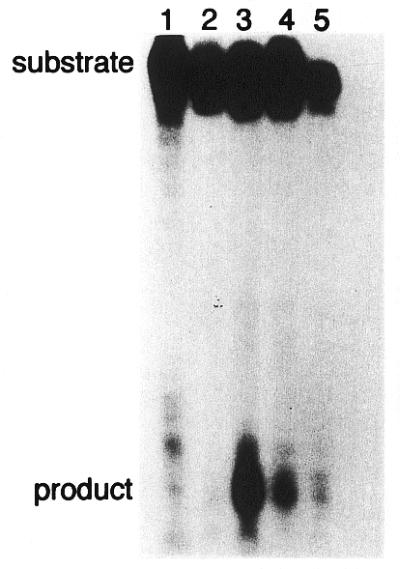
Cleavage of 24mer duplex oligonucleotides containing 8-oxoG by Nth protein. The duplex oligonucleotides (20 fmol) containing G:C (lane 1), 8-oxoG/C (lane 2), 8-oxoG/G (lane 3), 8-oxoG/A (lane 4) or 8-oxoG/T (lane 5) were incubated at 37°C for 15 min with purified Nth protein (20 pmol). The products were separated by denaturing 20% PAGE in gels containing urea.
Figure 5.
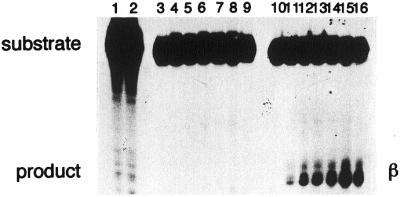
Concentration dependence of cleavage of 24mer duplex oligonucleotides containing 8-oxoG by purified Nth protein. The duplex oligonucleotides (20 fmol) containing G:C (lanes 1 and 2), 8-oxoG/C (lanes 3–9) or 8-oxoG/G (lanes 10–16) were incubated at 37°C for 15 min with purified Nth protein (20 pmol). The products were separated by denaturing 20% PAGE in gels containing urea. Nth protein was added at 0 (lanes 1, 3 and 10), 1 (lanes 4 and 11), 2 (lanes 5 and 12), 5 (lanes 6 and 13), 10 (lanes 7 and 14), 20 (lanes 8 and 15) and 40 pmol (lanes 9 and 16).
Trapping and cleavage assays with purified hNTH1 protein
The endonuclease III (Nth) family of DNA glycosylases constitutes a highly conserved class of repair enzymes identified in bacteria, yeast and mammalian cells (21,22,31,32). The human hNTH1 enzyme has properties similar to E.coli Nth (5,6,21,22,31,32). Therefore, it was of interest to know whether the human enzyme has an 8-oxoG DNA glycosylase/AP lyase activity like E.coli Nth protein. A plasmid carrying the human hNTH1 cDNA was constructed and introduced into E.coli BL21 and extracts from the cells were tested to determine whether or not they contained 8-oxoG DNA glycosylase/AP lyase activity. The results are shown in Figure 6. There was a dense band produced by trapping of the protein in extracts from E.coli KSR7/pGEX-4T-3-hNTH1 to duplex oligonucleotides containing 8-oxoG/G mispairs (Fig. 6, lane 1). Purified hNTH1 protein was efficiently trapped to the oligonucleotides. The substrate specificity of hNTH1 protein was similar to that of E.coli Nth protein (Fig. 6, lanes 2–9). The incision product resulted from a β-elimination reaction.
Figure 6.
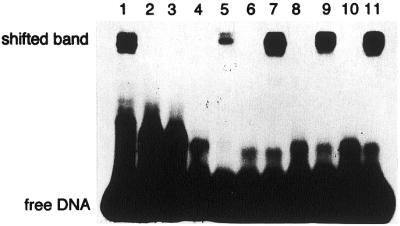
Borohydride trapping analysis of purified hNTH1 protein. Duplex oligonucleotide substrates (10 fmol) containing G:C (lanes 2 and 3), 8-oxoG/T (lanes 4 and 5), 8-oxoG/G (lanes 1, 6 and 7), 8-oxoG/A (lanes 8 and 9) or 8-oxoG/C (lanes 10 and 11) pairs were incubated with crude extracts or purified hNTH1 protein (20 pmol) in the presence of 100 mM NaBH4 at 37°C for 30 min. Lane 1, with extracts from E.coli KSR7 (mutMnthnei) carrying pGEX-4T-3-hNTH1; lanes 2, 4, 6, 8 and 10, without protein; lanes 3, 5, 7, 9 and 11, with purified hNTH1 protein.
Furthermore, purified hNTH1 protein was shown to preferentially cleave duplex oligonucleotides containing an 8-oxoG/G mispair at the lesion site (Fig. 7, lanes 5 and 6), whereas no detectable cleavage occurred with 8-oxoG/C or G:C-containing oligonucleotides (Fig. 7, lanes 1–4). These results indicated that human hNTH1 protein could also recognize and repair 8-oxoG/G mispairs in DNA by removing the 8-oxoG. The activity of hNTH1 protein in cleaving the 8-oxoG/G-containing oligonucleotide was about one-tenth of that for cleaving thymine glycol-containing oligonucleotides.
Figure 7.
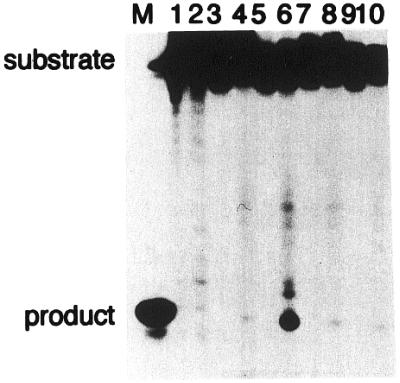
Cleavage of 24mer duplex oligonucleotides containing 8-oxoG by purified hNTH1 protein. Duplex oligonucleotides (20 fmol) containing G:C (lanes 1 and 2), 8-oxoG/C (lanes 3 and 4), 8-oxoG/G (lanes 5 and 6), 8-oxoG/A (lanes 7 and 8) or 8-oxoG/T (lanes 9 and 10) were incubated at 37°C for 15 min with purified hNTH1 protein (20 pmol). The products were separated by denaturing 20% PAGE in gels containing urea.
DISCUSSION
It is not surprising that Nth protein possesses 8-oxoG DNA glycosylase activity. Several recent studies have shown that MutM, Nth and Nei DNA glycosylases show overlap in their substrate specificity. Nth protein was first identified as an activity in E.coli, which introduced strand incision in X-irradiated and heavily UV-irradiated DNA (33,34). This enzyme was characterized as a DNA glycosylase specific for oxidized thymine products such as thymine glycol and urea (5,6). Recently a wide variety of additional substrates have been identified for Nth protein. They include methyl tartonylurea, 5-hydroxy-5-methylhydantoin, 5,6-dihydrothymine, 5-hydro-6-hydrothymine, 5-hydroxy-6-hydrouracil, 5,6-dihydrouracil, uracil glycol, 6-hydroxy-5,6-dihydrocytidine, 5-hydroxycytidine, 5-hydroxyuracil and formamidopyrimidine (5,6). Recently, using defined oligonucleotide substrates, we demonstrated that Nth protein has an activity that recognizes and repairs 5-formyluracil (23). A common feature of the identified Nth substrates is that they are no longer aromatic and planar in the pyrimidine ring (6). However, the active site of Nth protein must be able to accommodate this wide variety of substrates, including 8-oxoG. It will be of interest to compare the structures of the active site pocket of Nth protein when bound to thymine glycol and 8-oxoG.
MutM protein is a major E.coli enzyme responsible for the removal of formamidopyrimidine and 8-oxoG in DNA (5,7,35). A variety of other substrates for MutM protein have been identified. MutM recognizes and removes 7,8-dihydro-8-oxoguanine, 7,8-dihydro-8-oxoadenine, 2,6-diamino-4-hydroxy-5-formamidopyrimidine, 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine and 4,6-diamino-5-formamidopyrimidine (36). Recently, 5-hydroxycytosine and 5-hydroxyuracil have been found to be released by MutM protein (35,36). Furthermore, we have found that 5-formyluracil is also a substrate of MutM protein (23). It is possible that MutM protein is able to accommodate these small oxidized pyrimidines within its active site since it might be designed for larger oxidized purines (36). Nei protein shares several catalytic activities with MutM protein, including cleavage of DNA by β,δ-elimination (37,38). Nei protein recognizes premutagenic pyrimidine lesions such as 5-hydroxycytosine, 5-hydroxyuracil and uracil glycol. This protein also recognizes 5-formyluracil in DNA (32). Recently Blaisdell et al. (39) and Hazra et al. (15) have shown that Nei protein has 8-oxoG DNA glycosylase activity. These facts give insight into the common mechanism of recognition of the substrate by these enzymes.
In this study we found that Nth protein preferentially removes 8-oxoG paired with G and shows only a weak activity for 8-oxoG/A mispairs (Figs 4–6). It had no significant activity for 8-oxoG/C pairs. The most highly preferred substrate of Nei protein is 8-oxoG paired with G or A (15). In addition, 8-oxoG/C and 8-oxoG/G pairs are substrates of MutM protein (5–7). Although these enzymes have different substrate preferences, they might be back-up enzymes to repair 8-oxoG/G mispairs formed during DNA replication. This possibility was supported by the fact that the frequency of Lac+ reversion due to G:C→C:G transversions was markedly increased in the E.coli CC103mutMnthnei mutant (Fig. 2 and Table 1).
Oxidatively damaged bases such as 8-oxoG in general arise by the following two processes: 8-oxoG is generated in DNA in situ by attack of a hydroxyl radical on G (7) or is incorporated into DNA from ROS-induced 8-oxo-dGTP in the nucleotide pool (13). MutM protein removes 8-oxoG preferentially from the 8-oxoG/C pair in duplex DNA, which results from in situ oxidation of G (7). Unrepaired 8-oxoG can form the mutagenic 8-oxoG/A and 8-oxoG/G pairs during subsequent DNA replication (8,12). MutY protein removes A and G from the 8-oxoG/A and 8-oxoG/G pairs, respectively, and thus provides a second opportunity to prevent mutations (9,12). On the other hand, 8-oxoG can be incorporated into nascent DNA, opposite A and G in the template, from the 8-oxo-dGTP pool when MutT fails to destroy 8-oxoG-dGTP (13). The misincorporation of 8-oxo-dGTP would increase the frequency of A:T→C:G and G:C→C:G transversions (14).
The significant increase in G:C→T:A and G:C→C:G transversions in E.coli and human cells after ROS treatment suggests that such mutations could result from 8-oxoG/A and 8-oxoG/G pairing during DNA replication (5,9–12). We have presented in the Introduction the argument that E.coli has an additional 8-oxoG repair enzyme that removes the lesion when incorporated in nascent DNA and present as an 8-oxoG/A or 8-oxoG/G mispair. The Nth and Nei proteins fulfill the criteria for such an enzyme. The 8-oxoG incorporated opposite G is removed by Nth protein (Figs 3–5) and Nei protein excises 8-oxoG paired with A and G (15,39). The primary requirements for Nth and MutY should be that they act specifically on the nascent strand. This specificity for nascent DNA repair is strongly reminiscent of the mismatch repair process in E.coli and mammalian cells (40,41) and suggests a common mechanism for strand discrimination in mismatch repair and 8-oxoG excision repair.
It is interesting that both human hNTH1 and E.coli Nth proteins prefer 8-oxoG/G to 8-oxoG/A and other 8-oxoG pairs (Figs 6 and 7). It was shown earlier that 8-oxoG can pair with G as a 6-enolate 8-keto tautomer (42). Furthermore, MutY protein was shown to excise not only A but G when paired with 8-oxoG (12). Therefore, it is possible that the 8-oxoG/G pair could occur in vivo and be subject to repair by Nth or Nei protein.
Base excision repair is highly conserved from bacteria to humans. The endonuclease III (Nth) family of DNA glycosylases constitutes a highly conserved class of repair enzymes found in bacteria, yeast, invertebrates and mammals (5,6,21,22,31,32). The eukaryotic enzymes have properties similar to those of Nth protein. It will be of interest to know whether the substrate range of the eukaryotic enzymes also includes 8-oxoG. In this study we have characterized hNTH1 protein further and shown that the purified enzyme has efficient 8-oxoG DNA glycosylase activity similar to that found for E.coli Nth. Bauche and Laval (43) and Senturker et al. (44) have reported that the radiation-resistant bacterium Deinococcus radiodurans possesses two proteins that excise oxidized purines, including Fapy and 8-oxoG, one of which is endowed with thymine glycol DNA glycosylase activity. These data and our results might provide a ubiquitous mechanism in all organisms: OGG1 functions as a major 8-oxoG DNA glycosylase and OGG2 functions as a minor and alternative 8-oxoG repair enzyme. The presence of OGG2 activities in both yeast and humans suggests that it functions in the mechanism for anti-mutagenic processing in all eukaryotes. In an OGG2-defective S.cerevisiae strain A:T→C:G transversions indeed increased (18).
Hazra et al. (15) identified Nei protein as an OGG2 that excised 8-oxoG paired with G and A in E.coli. Additionally, they have observed another 8-oxoG DNA glycosylase/AP lyase activity in crude cell extracts of E.coli mutMnei mutants, suggesting that there are two OGG2 activities in E.coli. Their third 8-oxoG DNA glycosylase/AP lyase might correspond to Nth.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to express our thanks to Drs J.H.Miller (University of California, Los Angeles), K.Yamamoto (Tohoku University, Sendai) and S.Yasuda (National Institute of Genetics, Mishima) for kindly supplying plasmids and E.coli strains. This study was partly supported by grants from the Ministry of Education, Science, Sports and Culture of Japan and from the Nissan Science Foundation (to Q.-M.Z.).
References
- 1.Ames B.N., Shigenaga,M.K. and Hagen,T.M. (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl Acad. Sci. USA, 90, 7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seeberg E., Eide,L. and Bjoras,M. (1995) The base excision repair pathways. Trends Biochem. Sci., 20, 391–397. [DOI] [PubMed] [Google Scholar]
- 3.Michaels M.L. and Miller,J.H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol., 174, 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grollman A.P. and Moriya,M. (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet., 9, 246–249. [DOI] [PubMed] [Google Scholar]
- 5.Wallace S.S. (1997) Oxidative damage to DNA and its repair. In Scandalios,J.G. (ed.), Oxidative Stress and the Molecular Biology of Antioxidant Defenses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 49–81.
- 6.McCulough A.K., Dodson,M.L. and Lloyd,R.S. (1999) Initiation of base excision repair: glycosylase mechanisms and structures. Annu. Rev. Biochem., 68, 255–285. [DOI] [PubMed] [Google Scholar]
- 7.Michaels M.L., Pham,L., Cruz,C. and Miller,J.H. (1991) MutM, a protein that prevents G:C→T:A transversions, is formamidopyrimidine-DNA glycosylase. Nucleic Acids Res., 19, 3629–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 9.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine. Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nghiem Y., Cabrera,M., Cupples,C.G. and Miller,J.H. (1988) The mutY gene: a mutator locus in Escherichia coli that generates G:C→T:A transversions. Proc. Natl Acad. Sci. USA, 85, 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wood M.L., Esteve,M., Morningstar,M.L., Kuziemko,G.M. and Essigmann,J.M. (1992) Genetic effects of oxidative DNA damage: comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucleic Acids Res., 20, 6023–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q.-M., Ishikawa,N., Nakahara,T. and Yonei,S. (1998) Escherichia coli MutY protein has a guanine-DNA glycosylase that acts on 7,8-dihydro-8-oxoguanine:guanine mispair to prevent spontaneous G:C→C:G transversions. Nucleic Acids Res., 26, 4669–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maki H. and Sekiguchi,M. (1992) MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature, 355, 273–275. [DOI] [PubMed] [Google Scholar]
- 14.Tajiri T., Maki,H. and Sekiguchi,M. (1995) Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res., 336, 257–267. [DOI] [PubMed] [Google Scholar]
- 15.Hazra T.K., Izumi,T., Venkataraman,R., Kow,Y.W., Dizdaroglu,M. and Wallace,S.S. (2000) Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J. Biol. Chem., 275, 27762–27767. [DOI] [PubMed] [Google Scholar]
- 16.van der Kemp P.A., Thomas,D., Barbey,R., de Oliveila,R. and Boiteux,S. (1996) Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc. Natl Acad. Sci. USA, 93, 5197–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruner S.D., Nash,H.M., Lane,W.S. and Verdine,G.L. (1998) Repair of oxidatively damaged guanine in Saccharomyces cerevisiae by an alternative pathway. Curr. Biol., 8, 393–403. [DOI] [PubMed] [Google Scholar]
- 19.Hazra T.K., Izumi,T., Maidt,L., Floyd,R.A. and Mitra,S. (1998) The presence of two distinct 8-oxoguaine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Res., 26, 5116–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senturker S., van der Kemp,P.A., You,H.J., Doetsch,P.W., Dizdaroglu,M. and Boiteux,S. (1998) Substrate specificities of the Ntg1 and Ntg2 proteins of Saccharomyces cerevisiae for oxidized DNA bases are not identical. Nucleic Acids Res., 26, 5270–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeda S., Biswas,T., Roy,R., Izumi,T., Boldogh,I., Kurosky,A., Sarker,A.H., Seki,S. and Mitra,S. (1998) Purification and characterization of a human NTH1, a homolog of Escherichia coli endonuclease III. J. Biol. Chem., 273, 21585–21593. [DOI] [PubMed] [Google Scholar]
- 22.Aspinwall R., Rothwell,D.G., Roldan-Arjona,T., Anselmino,C., Ward,C.J., Cheadle,J.P., Sampson,J.R., Lindahl,T., Harris,P.C. and Hickson,I.D. (1997) Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl Acad. Sci. USA, 94, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q.-M., Miyabe,I., Matsumoto,Y., Kino,K., Sugiyama,H. and Yonei,S. (2000) Identification of repair enzymes for 5-formyluracil in DNA: Nth, Nei and MutM proteins of Escherichia coli. J. Biol. Chem., 275, 35471–35477. [DOI] [PubMed] [Google Scholar]
- 24.Cupples C.G. and Miller,J.H. (1989) A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitution. Proc. Natl Acad. Sci. USA, 86, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Zhang Q.-M., Sugiyama,H., Miyabe,I., Matsuda,S., Saito,I. and Yonei,S. (1997) Replication of DNA templates containing 5-formyluracil, a major oxidative lesion of thymine in DNA. Nucleic Acids Res., 25, 3969–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabow L.E. and Kow,Y.W. (1997) Mechanism of action of base release by Escherichia coli Fpg protein: role of lysine 155 in catalysis. Biochemistry, 36, 5084–5096. [DOI] [PubMed] [Google Scholar]
- 29.Nash H.M., Bruner,S.D., Schärer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 30.Bailly V. and Verly,W.G. (1987) Escherichia coli endonuclease III is not an endonuclease but a β-elimination catalyst. Biochem. J., 242, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Augeri L., Lee,Y.M., Barton,A.B. and Doestsch,P.W. (1997) Purification, characterization, gene cloning, and expression of Saccharomyces cerevisiae redoxyendonuclease, a homologue of Escherichia coli endonuclease III. Biochemistry, 36, 721–729. [DOI] [PubMed] [Google Scholar]
- 32.Roldan-Arjona T., Anselmino,C. and Lindahl,T. (1996) Molecular cloning and functional analysis of a Schizosaccharomyces pombe homologue of Escherichia coli endonuclease III. Nucleic Acids Res., 24, 3307–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strniste G. and Wallace,S.S. (1975) Endonucleolytic incision of X-irradiated deoxyribonucleic acid by extracts of Escherichia coli. Proc. Natl Acad. Sci. USA, 72, 1997–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radman M. (1976) An endonuclease from Escherichia coli that introduces single polynucleotide chain scissions in ultraviolet-irradiated DNA. J. Biol. Chem., 251, 1438–1445. [PubMed] [Google Scholar]
- 35.Hatahet Z., Kow,Y.W., Purmal,A.A., Cunningham,R.P. and Wallace,S.S. (1994) New substrates for old enzymes. 5-Hydroxy-2′-deoxycytidine and 5-hydroxy-2′-deoxyuridine are substrates for Escherichia coli endonuclease III and formamidopyrimidine DNA N-glycosylase, while 5-hydroxy-2′-deoxyuridine is a substrate for uracil DNA N-glycosylase. J. Biol. Chem., 269, 18814–18820. [PubMed] [Google Scholar]
- 36.David S.S. and Williams,S.D. (1998) Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev., 98, 1221–1261. [DOI] [PubMed] [Google Scholar]
- 37.Melamede R.J., Hatahet,Z., Kow,Y.W., Ide,H. and Wallace,S.S. (1994) Isolation and characterization of endonuclease VIII from Escherichia coli. Biochemistry, 33, 1255–1264. [DOI] [PubMed] [Google Scholar]
- 38.Jiang D., Hatahet,Z., Melamede,R.J., Kow,Y.W. and Wallace,S.S. (1997) Characterization of Escherichia coli endonuclease VIII. J. Biol. Chem., 272, 32230–32239. [DOI] [PubMed] [Google Scholar]
- 39.Blaisdell J.O., Hatahet,Z. and Wallace,S.S. (1999) A novel role for Escherichia coli endonuclease VIII in prevention of spontaneous G→T transversions. J. Bacteriol., 181, 6396–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 41.Karran P. and Bignami,M. (1999) Mismatch repair and cancer. In Smith,P.J. and Jones,C.J. (eds), DNA Recombination and Repair. Oxford University Press, Oxford, UK, pp. 66–98.
- 42.Culp S.J., Cho,B.P., Kadlubar,F.F. and Evans,F.E. (1989) Structural and conformational analyses of 8-hydroxy-2′-deoxyguanosine. Chem. Res. Toxicol., 2, 416–422. [DOI] [PubMed] [Google Scholar]
- 43.Bauche C. and Laval,J. (1999) Repair of oxidized bases in the extremely radiation-resistant bacterium Deinococcus radiodurans. J. Bacteriol., 181, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Senturker S., Bauche,C., Laval,J. and Dizdaroglu,M. (1999) Substrate specificity of Deinococcus radiodurans Fpg protein. Biochemistry, 38, 9435–9439. [DOI] [PubMed] [Google Scholar]