

Abstract
Synapse loss, rather than the hallmark amyloid-β (Aβ) plaques or tau filled neurofibrillary tangles (NFT), is considered the most predictive pathological feature associated with cognitive status in the Alzheimer disease (AD) brain. The role of Aβ in synapse loss is well established, but despite data linking tau to synaptic function, the role of tau in synapse loss remains largely undetermined. Here we test the hypothesis that human mutant P301L tau over-expression in a mouse model (rTg4510) will lead to age-dependent synaptic loss and dysfunction. Using array tomography and two methods of quantification (automated, threshold-based counting and a manual stereology based technique) we demonstrate that overall synapse density is maintained in the neuropil, implicating synapse loss commensurate with the cortical atrophy known to occur in this model. Multi-photon in-vivo imaging reveals close to 30% loss of apical dendritic spines of individual pyramidal neurons suggesting these cells may be particularly vulnerable to tau-induced degeneration. Post-mortem, we confirm the presence of tau in dendritic spines of rTg4510-YFP mouse brain by array tomography. These data implicate tau-induced loss of a subset of synapses that may be accompanied by compensatory increases in other synaptic subtypes thereby preserving overall synapse density. Biochemical fractionation of synaptosomes from rTg4510 brain demonstrates a significant decrease in expression of several synaptic proteins, suggesting a functional deficit of remaining synapses in the rTg4510 brain. Together these data show morphological and biochemical synaptic consequences in response to tau over-expression in the rTg4510 mouse model.
Keywords: Alzheimer Disease, tauopathy, hyperphosphorylated tau, array tomography
Introduction
Alzheimer disease (AD) brain is defined not only by the hallmark protein aggregates of plaques and neurofibrillary tangles (NFT) but also by marked synapse and neuronal loss. The extracellular plaques are deposits of amyloid-β (Aβ), the peptide largely considered the initiator of the disease (Hardy and Selkoe, 2002), and yet amyloid burden does not correlate well with synapse and neuronal loss or severity of AD (Arriagada et al., 1992; Giannakopoulos et al., 2003; Ingelsson et al., 2004). Although intracellular NFT composed primarily of aberrantly phosphorylated and misfolded tau protein correlate more closely than plaque load with neuronal loss and cognitive deficits of AD, synapse loss is the most predictive of cognitive status (Giannakopoulos et al., 2003; Gomez-Isla et al., 1997; Ingelsson et al., 2004; Selkoe, 2002; Terry et al., 1991). Soluble oligomeric forms of Aβ are strongly implicated in synapse loss early in the disease process (Koffie et al., 2012; Koffie et al., 2009; Selkoe, 2008) but the contribution of tau is less well understood. Here we use a reversible mouse model of tauopathy (rTg4510), in combination with array tomography, in-vivo multiphoton imaging and synaptosome biochemical analysis to test the hypothesis that over-expression of human mutant tau in a mouse model will lead to age-dependent loss of synapse density and function in the cortex.
Tau is a member of the microtubule associated protein family, residing primarily in axons, playing an intrinsic role in microtubule stabilization and neuronal transport. Disruptions to normal tau function via hyperphosphorylation, mislocalization, misfolding and fibrilization occur in AD, and are thought to impede transport and result in distal sites devoid of essential machinery (Coleman and Yao, 2003; Cuchillo-Ibanez et al., 2008; Dixit et al., 2008; Hollenbeck and Saxton, 2005; Morfini et al., 2009; Stamer et al., 2002; Thies and Mandelkow, 2007; Zempel et al., 2010), ultimately leading to axonal and dendritic ‘dying back’ and neuronal death (Coleman and Yao, 2003; Morfini et al., 2009; Yoshiyama et al., 2007). In addition, recent studies place tau both physiologically and pathologically in dendritic spines, suggesting that tau may have a more direct role in dendritic degeneration as well (Hoover et al., 2010; Ittner et al., 2010; Kremer et al., 2011; Tai et al., 2012; Yu et al., 2012).
Experimental results of whether changes to tau are related to synaptic deficits have been extremely diverse, with reports of pathological tau causing synaptic loss or dysfunction, maintenance and homeostasis and even compensatory increases in synaptic density and function. A complication in interpreting these results arises from differing tau models both in vitro and in vivo, in which isoforms of human wild type, mutant, pro- or anti-aggregant, fluorescent protein fused and even endogenous tau are used for drawing broad conclusions about repercussions of pathological changes in tau (see Table 1 for details).
Table 1. Simplified summary of selected literature on AD and tau-associated synaptic alterations.
Studies shaded in red found tau-associated synaptic loss or dysfunction, green indicates compensatory increases in synaptic numbers or function, and blue indicates mixed results or no change. AD = Alzheimer Disease, htau = human tau, 3R or 4R = 3 or 4 microtubule binding domains (MTBD), FL = full length, WT = wild-type, PHP = pseudophosphorylated AA = amino acids
| First Author & Year of Publication | Model | Form of tau | Argument |
|---|---|---|---|
| Alldred (2012) | htau mouse | all 6 isoforms of human non-mutant tau | LOSS & DYSFUNCTION |
| Amadoro (2010) | synaptosomes of human AD brain | - | COMPENSATION |
| Bittner (2010) | 3×TG (PS1/APP/tau)-YFP mouse | P301L human mutant tau | LOSS & COMPENSATION |
| Boekhoorn (2006) | Tau-P301L and 4R-wt-tau mouse | P301L mutant and full length non mutant human tau | COMPENSATION |
| Coleman (2003) | human AD brain | - | LOSS & DYSFUNCTION |
| Crimins (2011) | rTg4510 mouse | P301L human mutant tau | LOSS & COMPENSATION |
| David (2005) | P301L tau mouse | P301L human mutant tau | LOSS & DYSFUNCTION |
| DeKosky (1996) | human AD brain | - | LOSS & COMPENSATION |
| Dickstein (2010) | htau mouse | all 6 isoforms of human non-mutant tau | EQUAL/COMPENSATION |
| Dubey (2008) | primary neuronal culture | CFP tau | LOSS |
| Dumitriu (2010) | aging monkey prefrontal cortex | - | LOSS & COMPENSATION |
| Eckermann (2007) | ΔK280 and ΔK280/PP mice | ΔK280 = proaggregant full length human tau with FTDP17 deletion. ΔK280/PP = anti-aggregant tau with mutations I277P & I308P | LOSS |
| Gylys (2004) | synaptosomes of human AD brain | - | EQUAL/LOSS |
| Hall (2000) | lamprey central neurons | human tau filaments of shortest human tau (3R, 352AA) | LOSS |
| Hoover (2010) | rTgP301L and rTgWT mice | P301L mutant and non mutant human tau | DYSFUNCTION |
| Ingelsson (2004) | human AD brain | - | LOSS |
| Jaworski (2011) | YFP mice | AAVP301L human mutant tau and AAVnon-mutant human tau | LOSS |
| Kimura (2010) | P301L and WT tau mouse | P301L mutant and non mutant human tau | LOSS & COMPENSATION |
| Kremer (2011) | TauP301L and Tau4R mice | P301L mutant and non mutant human tau | LOSS & COMPENSATION |
| Lasagna-Reeves (2011) | WT mice | monomeric, oligomeric or fibrillar full length human tau | DYSFUNCTION |
| Leuba (2008) | human AD brain | - | COMPENSATION |
| Mocanu (2008) | ΔK280 and ΔK280/PP mice | ΔK280 = proaggregant truncated human tau with FTDP17 deletion. ΔK280/PP = anti-aggregant truncated tau with mutations I277P & I308P | LOSS |
| Polydoro (2009) | htau mouse | all 6 isoforms of human non-mutant tau | DYSFUNCTION |
| Rocher (2010) | rTg4510 mouse | P301L human mutant tau | LOSS |
| Scheff (1990) | human AD brain | - | LOSS & COMPENSATION |
| Shahani (2006) | WT mouse hippocampal slices | eGFPtau: 352wt, 352PHP, 441wt, 441PHP | EQUAL |
| Sydow (2011) | ΔK280 and ΔK280/PP mice | ΔK280 = proaggregant truncated human tau with FTDP17 deletion. ΔK280/PP = anti-aggregant truncated tau with mutations I277P & I308P | LOSS & DYSFUNCTION |
| Tackenberg (2009) | APPsdl TG mice | eGFPtau: 352wt, 352PHP, 352Ala, 441R406W, 441P301L | EQUAL |
| Terry (1991) | human AD brain | - | LOSS & COMPENSATION |
| Thies (2007) | primary neuronal culture | AAVhtau40-CFP (full length non-mutant human tau) and AAVhtau40/KXGA-CFP (ser to Ala in all four KXGS motifs) | LOSS |
| Yoshiyama (2007) | PS19 (P301S) mouse | P301S human mutant tau | LOSS & DYSFUNCTION |
| Yu (2012) | primary neuronal culture | endogenous and non-phosphorylatable tau | LOSS |
| Zempel (2012) | primary neuronal culture | endogenous tau | LOSS |
In vitro studies with changes to or over-expression of tau have shown substantial disruption of organelle transport, calcium changes and dendritic spine loss (Hall et al., 2000; Stoothoff et al., 2009; Thies and Mandelkow, 2007; Yu et al., 2012; Zempel et al., 2010). Transfection of lamprey central neurons with the shortest isoform of human tau led to filament formation associated with dendritic degeneration and synapse loss, resultant from trafficking impairments (Hall et al., 2000). Similar findings emerged from over-expression of the longest human tau isoform in mouse primary hippocampal neurons in which missorting of tau and transport deficits preceded synaptic degeneration (Stoothoff et al., 2009; Thies and Mandelkow, 2007). Even without introducing exogenous tau, abnormal phosphorylation and missorting contribute to disturbance of organelle transport, calcium levels and synapse loss in response to exogenous Aβ application (Yu et al., 2012; Zempel et al., 2010).
Several animal models, with transgenes for over-expression of human mutant tau known to lead to dementia such as P301L or P301S, introduction of mutant tau via adeno-associated virus or tau mutant mice expressing a pro-aggregating variant of tau have all established distinct spine loss and/or depletion of synaptic proteins concomitant with tau over-expression (Bittner et al., 2010; Dubey et al., 2008; Eckermann et al., 2007; Jaworski et al., 2011; Mocanu et al., 2008; Rocher et al., 2010; Sydow et al., 2011a; b). In other tau models, dysregulation of synaptic proteins has also been reported, with expression differentially influenced by tau (Alldred et al., 2012; David et al., 2005; Hoover et al., 2010; Lasagna-Reeves et al., 2011; Mocanu et al., 2008; Yoshiyama et al., 2007). Even with no evident change in structural components such as synaptic vesicle related proteins or post-synaptic density scaffolding protein PSD95, alterations in NMDA and AMPA receptor subunit representation may be evident, implying functional deficits since these are imperative to processes of neuronal function, synaptic transmission, learning and memory (Hoover et al., 2010; Sabatini et al., 2001; Yoshiyama et al., 2007). AMPA receptors appear particularly vulnerable to tau-induced deficits as subunits GluR1, 2 and 3 are reportedly diminished in several models (Hoover et al., 2010; Morris et al., 2011; Yoshiyama et al., 2007; Yu et al., 2012). This change in AMPA receptor content has been likened to long-term depression, a form of synaptic plasticity that can lead to regression of dendritic spines (Berridge, 2011; Crimins et al., 2011; D'Amelio et al., 2011; Huang and Mucke, 2012; Spires-Jones and Knafo, 2012; Yu and Lu, 2012; Yu et al., 2012). Contrary to these findings, however, others have reported no evidence of change in receptor composition, pre- and/or post-synaptic markers and have, therefore, postulated that synapse loss does not occur in conjunction with alterations to the same isoforms or mutations of tau that are associated with deficits in other studies (Hoover et al., 2010; Kimura et al., 2010; Kremer et al., 2011; Shahani et al., 2006; Tackenberg and Brandt, 2009).
Yet another series of studies have proposed not only that tau does not induce synapse loss, but rather that compensatory mechanisms in remaining neurons may be at play in tau models, leading to increases in synaptic proteins or synapses themselves, particularly at early stages (Amadoro et al., 2010; Bittner et al., 2010; Boekhoorn et al., 2006; Crimins et al., 2011; David et al., 2005; Dickstein et al., 2010; Kimura et al., 2010; Kremer et al., 2011). In human wild type or P301L tau over-expressing mice, formation of new synapses or changes in spine morphology, provide potential mechanisms for maintenance of normal network activity even at advanced stages of tauopathy (Crimins et al., 2011; Dickstein et al., 2010). Even in human AD brain, compensatory increases in synapse size and protein content have been suggested to counteract declines in synapse density (Amadoro et al., 2010; DeKosky et al., 1996; Gylys et al., 2004; Leuba et al., 2008), and there is clear evidence of neural system plasticity (Geddes and Cotman, 1986; Geddes et al., 1985; Hyman et al., 1987; Scheff et al., 1990; Scheff and Price, 2003).
In the present study we reconcile some of these previously disparate findings by addressing the effects of pathological changes to tau on synapses using two high resolution imaging techniques, array tomography and two-photon in-vivo microscopy, alongside biochemical fractionation experiments in rTg4510 mice. These mice over-express human mutant P301L tau and suffer progressive neuronal loss and cognitive deficits in conjunction with accumulation of NFT. We observe maintenance of total synapse density but loss of dendritic spines of cortical pyramidal neurons, indicating that synapse loss parallels cortical atrophy known to occur in this mouse model (Spires et al., 2006) and that spines of pyramidal neurons may be particularly vulnerable to tau-induced dendritic degeneration. Compensatory increases in other synapse subtypes may contribute to preservation of overall synapse density. We confirm the presence of tau in dendritic spines of rTg4510-YFP mice, post-mortem, indicating a potentially direct role of tau in dendritic spine collapse. However, presence of misfolded tau, detected by the conformational antibody Alz50 which persists in NFT, does not appear to impact linear synaptic densities, arguing against aggregates of tau as being locally toxic. Biochemical data indicate that remaining synapses are depleted of synaptic proteins, thereby revealing likely synaptic dysfunction. Taken together our data suggest a morphological and functional synaptic deficit in rTg4510 tau over-expressing mice.
Materials and Methods
Animals
The rTg4510 mouse line used for these experiments is a well-characterized model of tauopathy (de Calignon, 2010; Fox et al., 2011; Kopeikina et al., 2011; Santacruz et al., 2005; Spires et al., 2006). These bigenic mice have P301L human mutant four-repeat tau downstream of a tetracycline-operon-responsive element (TRE) on the responder transgene, and a tet-off open reading frame downstream of a Ca2+ calmodulin kinase II promotor within the activator transgene that lead to over-expression of human mutant tau in forebrain structures when both responder and activator are present. Littermate animals with either only the activator or responder transgene, which do not over-express tau were used as controls.
A second line of rTg4510-YFP mice was used for array tomography and in-vivo imaging experiments to allow for visualization of dendritic spines. To generate these mice, rTg4510 mice were crossed with transgenic mice expressing yellow fluorescent protein (YFP) driven by the Thy1 promotor (eYFP-H; Jackson Laboratories), yielding rTg4510 mice that over-express P301L human mutant tau in the forebrain with a subset of these neurons also expressing YFP for enhanced visualization.
Institutional guidelines and those of the National Institutes of Health were implemented for all animal housing and treatment protocols.
Array Tomography
Preparation of tissue, immunohistochemical and microscopy techniques, and image analysis methods related to array tomography have been described elsewhere (de Calignon et al., 2012; Koffie et al., 2012; Koffie et al., 2009; Kopeikina et al., 2011; Micheva et al., 2010; Micheva and Smith, 2007). Immediately following euthanasia by CO2 inhalation, small blocks of somatosensory cortex were immersion fixed for 3h at room temperature (RT) in 4% paraformaldehyde and 2.5% sucrose in phosphate-buffered saline solution (PBS), pH 7.4. A graded series of ethanols was used to dehydrate tissue into LRWhite resin (Electron Microscopy Sciences), at which point tissue was embedded in gelatin capsules and polymerized at 53C for 24h. Gelatin capsules were then removed and blocks cut with a Jumbo Histo Diamond Knife (Diatome) into ribbons of 7-30 ultra-thin 70nm consecutive sections, which were mounted on coverslips (Fisher Scientific, Pittsburgh PA: 12-544-E; No. 1.5; 0.16-0.19μm thick).
Immunostaining for analysis of synapse density was performed on tissue from rTg4510 and control mice at 5.5 and 8.5 months of age (n=4 per group, total 16 mice). A solution of 50mM glycine in Tris was used to wash ribbons, followed with a blocking solution of 0.05% Tween and 0.1% BSA in Tris. Primary antibodies (Table 2) mouse IgM anti-Alz50, mouse IgG anti-tubulin (Sigma), goat anti-PSD95 (Abcam), and rabbit anti-Synapsin I (Millipore) were diluted in block buffer at 1:100, applied to ribbons for 2h then rinsed with TBS. Fluorescent secondary antibodies AMCA- conjugated donkey anti-mouse IgM, donkey anti-goat Alexa-Fluor 488, donkey anti-rabbit Cy3 (Jackson ImmunoResearch) and chicken anti-mouse IgG Alexa-Fluor 647 (Invitrogen) were diluted at 1:500 in block buffer and incubated on ribbons for 30m. Prior to imaging, coverslips were then inverted onto a slide in mounting solution with or without nuclear stain. Areas of interest, containing nuclei or Alz50-positive cells within layers II/III of the somatosensory cortex, were imaged on 7-32 serial sections. Images of 1,024 × 1,024 pixels were collected with a Zeiss Axioplan LSM510 confocal/multiphoton microscope (63× numerical aperture Plan Apochromatic oil objective). Tile-scan images were collected on a Zeiss Axio Imager Z epifluorescence microscope equipped with AxioVision software and modified for automated acquisition of array tomography images.
Table 2. Antibodies used in array tomography and western blot analyses.
AD = Alzheimer Disease, AA = amino acids, WB = western blot, array = array tomography, IHC = Immunohistochemistry, NFT = neurofibrillary tangles, NT = neuropil threads
| Name | Immunogen/epitope | Manufacturer | Catalog # | Species | Mono/Polyclonal | Characterization |
|---|---|---|---|---|---|---|
| Alz50 | Human AD homogenate; AA2-10 & 312-342 (folded conformation) | Peter Davies | NA | mouse | monoclonal | WB: 64-68kD IHC: pretangles, NFT and NT (Wolozin et al., 1986; Hyman et al., 1988; Carmel et al., 1996; Jicha et al., 1997) |
| HT7 | Purified human tau AA159-163 | ThermoScientific | MN1000 | mouse | monoclonal | WB: 55-64kD IHC: human tau; NFT; NT (Eckermann et al., 2007; Kremer et al., 2011; de Calignon et al., 2012) |
| PHF1 | Paired helical filament proteins from AD brain pS396/pS404 of tau | Peter Davies | NA | mouse | monoclonal | WB: 64kD IHC: phosphorylated tau of NFT (Greenberg and Davies, 1990; Greenberg et al., 1992; Davies, 2000; de Calignon et al., 2012; Tai et al., in press) |
| GFP | Purified recombinant full length GFP made in e. coli | Abcam | ab6556 | rabbit | polyclonal | WB: 27kD IHC: GFP, YFP, CFP, RFP, eGFP |
| Tubulin | Acetylated tubulin from strongylocentrotus purpuratus (sea urchin) | Sigma | T6793 | mouse | monoclonal | WB: 55kD IHC: Neuronal cytoplasm and processes (Kopeikina et al., 2011) |
| β-actin | β-cytoplasmic actin N-terminal peptide | Sigma | A5316 | mouse | monoclonal | WB: 42kD (de Calignon et al., 2012) |
| PSD95 (array) | Synthetic peptide AA1-100 of mouse PSD95 | Abcam | ab12093 | goat | polyclonal | WB: 95kD IHC: Post-synapse (Koffie et al., 2009; Micheva et al., 2010; Koffie et al., 2012; Tai et al., in press) |
| PSD95 (WB) | Synthetic peptide of human PSD95 | Cell Signaling | 2507S | rabbit | polyclonal | WB: 95kD (Tai et al., in press) |
| Synapsin I | Synapsin I (mix of Ia & Ib) purified from bovine brain | Millipore | AB1543P | rabbit | polyclonal | WB: 77 & 80kD IHC: Pre-synapse (Koffie et al., 2009; Micheva et al., 2010; de Calignon et al., 2012; Koffie et al., 2012) |
| GluR1 | Recombinant GluR1 | Millipore | 05-855R | rat | monoclonal | WB: 106 & 200kD IHC: GluR1 subunit of AMPA receptors of post-synapse |
| GluR2 | Recombinant GluR2 AA175-430 | Millipore | MAB397 | mouse | monoclonal | WB: 102kD IHC: GluR2 subunit of AMPA receptors of post-synapse |
| NMDAR1 | His-tag AA834-938 rat NR1 | Millipore | 05-432 | mouse | monoclonal | WB: 130kD IHC: NR1 subunit of NMDA receptors of post-synapse |
| NMDAR2A | His-tag AA1265-1464 mouse NR2A | Upstate/Millipore | 07-632 | rabbit | polyclonal | WB: 170kD IHC: NR2 subunit of NMDA receptors of post-synapse |
To determine human tau localization within dendritic spines of rTg4510-YFP neurons, tissue of somatosensory cortex was similarly processed and stained for YFP with rabbit anti-GFP (Abcam), mouse HT7 for human tau (Thermo Scientific) or mouse PHF-1 (generous gift of Peter Davies, Albert Einstein College of Medicine) for pathologically phosphorylated tau, and counterstained with DAPI. Immunostainings in which secondaries were applied without primaries served as controls for all array tomography experiments.
Open source software from National Institutes of Health (ImageJ) was used for image viewing and analysis. Images from each ribbon were opened sequentially, converted to a stack and aligned with the MultiStackReg and StackReg plugins (courtesy of B. Busse at Stanford University and (Thevenaz et al., 1998)). Crop boxes (10.01μm × 10.01μm) were selected so as to exclude neuronal cell bodies or other obscuring features, realigned and re-cropped to exclude empty space created by realignment. For automated image analysis, crops of interest (synapsin and PSD95) were automatically thresholded with the MaxEntropy ImageJ option and run through an automated, threshold based detection program that counts puncta appearing in more than one consecutive section and reports dimensions of each (WaterShed program provided by B. Busse, S. Smith, and K. Micheva, Stanford University). Density was calculated based on the output of the watershed program and the volume sampled within the crop box. Linear spine density (number of spines per micrometer) along the length of a small subset of tubulin-only-filled or Alz50 (tau) positive dendrites (n = 14) was manually calculated from the synapsin and PSD95 watershed output files associated with a dendrite of interest. Two cortical blocks from the somatosensory cortex from each rTg4510 and control mouse were imaged, yielding close to 30,000 pre- and post-synaptic elements counted. The output of this automated analysis was then utilized in combination with a MATLAB script to determine co-localization density of synapsin and PSD95 puncta within 0.5μm of one another within each crop box.
In order to confirm the accuracy of the threshold based detection program, a modified physical disector analysis was also performed (Gundersen et al., 1988; West et al., 1988). In short, all puncta appearing in a single section were marked manually. New puncta appearing on the following section were then identified. Those new puncta confirmed to still be present in the subsequent (third) section were counted (see Figure 4) thereby combining traditional two serial section physical disector used in EM with the array tomography analysis principle of only counting puncta present in at least two sections as puncta present in only one section are too thin to be synapses and likely represent staining noise. Three such physical disectors were applied to each stack of images, ensuring that the same sections were not reused, in order to count at least 100 new puncta per animal.
Figure 4. Physical disector method.

Panels A-C show a cartoon diagram of the physical disector analysis method with 5 synapses identified in the first section (A). New puncta (green, pointed out with arrow) appearing in the next consecutive section (B) are located and if they persist into the third section (C), they are counted as ‘new’ synapses. Three consecutive 70nm sections (s1-s3) from somatosensory cortex of 5.5 month old control animals stained for synapsin (magenta) and PSD95 (green) are shown in D-F and G-I respectively. New puncta are identified in section 2 (B and E) and counted only if they are still present in the subsequent sections (F and I), shown by the arrows. Merged images of each section are shown in panels J-L. Scale bar = 2μm.
Tissue Processing and Immunohistochemistry
Mice were euthanized by CO2 inhalation (one 9m old rTg4510 and one 9m old rTg4510-YFP) and brains removed and drop fixed in 4% PFA in phosphate buffer (PBS) containing 15% glycerol cryoprotectant. Following two days in fixative, one hemisphere per animal was sectioned coronally on a freezing microtome to 50μm. Sections were washed with TBS, permeabilized with 0.5% Triton-X in TBS for 20m, blocked for one hour with 5% Normal goat serum (NGS) in TBS then immunostained with rabbit anti-GFP (Abcam) and mouse PHF1 (1:1000 with 1% NGS in TBS for both) with secondaries goat anti-rabbit Alexa-Fluor 488 and goat anti-mouse Cy3 (1:500 for both) then counterstained with DAPI. Images were collected on a Zeiss Axio Imager Z epifluorescence microscope equipped with AxioVision software and a 10× objective.
Cranial Window Implantation and Multiphoton in-vivo imaging
rTg4510-YFP and control mice (n = 4 per group; 9-10 months of age) received cranial windows for in-vivo imaging of dendritic spines (Bacskai et al., 2002; Klunk et al., 2002; Kuchibhotla et al., 2008; Spires-Jones et al., 2008). Mice were anaesthetized with isoflurane, secured in a stereotax and skin on the top of the head removed following sterilization and injection of a local anesthetic. Using a high-speed drill, a 6mm circular craniotomy was established, the brain washed with PBS and covered with an 8mm glass coverslip. The coverslip was secured with a dental cement-Krazy glue mixture and mice given three weeks recovery to account for any consequences of surgery.
For imaging, mice were again anesthetized with isoflurane and secured in a stereotax. A restraining ring of low-melting point wax was applied to the coverslip and filled with distilled water for the Olympus 20× 0.95N water immersion objective. The stereotax was then placed on the stage of an Olympus BX61WI upright microscope equipped with pre-chirp optics, fast AOM, Olympus Fluoview 1000MPE software, and a series of photomultiplier tube detectors (Hamamatsu, Ichinocho, Japan). A MaiTai titanium/sapphire laser (Spectra-Physics Fremont CA) generated two-photon fluorescence with 800nm excitation. Images were collected with z-axis resolution of 0.8μm, scanning speed of 8μs/pixel and zoom between 5 and 10, giving high magnification and high resolution of individual spines.
Spine density analysis was performed with Image J and NeuronStudio (Mt Sinai School of Medicine, Computational Neurobiology and Imaging Center). Dendrites of at least 20μm in length and a minimum of three spines along that length were chosen for analysis. Images were opened in NeuronStudio and run through a median blur filter. The dendritic segment and associated spines were selected semi-automatically and confirmed manually by comparison to the original image stack viewed in Image J. Spine densities were calculated as the number of spines per micrometer. At the end of in-vivo imaging experiments, rTg4510-YFP mice were also prepared for array tomography as described previously.
Western blotting
rTg4510 and control mice (n = 4 per group; 12 months of age) were euthanized with CO2, and the cortical tissue dissected, snap frozen in liquid nitrogen, and stored at -80 °C. After thawing, each hemi-cortex was gently ground in a Potter-Elvehjem homogenizer with 1.5 mL ice-cold buffer A (20 mM HEPES pH 7.5, 0.3 M DTT), supplemented with 2 mM DTT, protease inhibitors (Roche Complete tablet) and phosphatase inhibitors (Sigma cocktail 2 and 3). The homogenate was passed through two layers of 80μm nylon filters (Millipore) to remove tissue debris, and a 200μL aliquot was saved. The saved aliquot was mixed with 200μL water and 70μL 10% SDS, passed through a 27 gauge needle, and boiled for 5 min to prepare total extract. The rest of the homogenate was centrifuged at 500 ×g for 3 min to remove nuclei. This was followed by centrifugation at 12,000 ×g for 15 min to pellet synaptic terminals. The pellet was washed once with buffer A and centrifuged again, yielding the crude synaptosome pellet. Supernatant from the first 12,000 ×g centrifugation step was clarified by centrifugation at 100,000 ×g for 1h to obtain the cytosol fraction. Cytosolic extract was prepared by adding 1.5% SDS and boiling for 5 min. Synaptosome pellets were extracted with 0.5 mL buffer B (50 mM Tris pH 7.5, 1.5% SDS, 2 mM DTT) and boiled for 5 min. Protein concentration was determined by BCA assay (Pierce).
For standard SDS-PAGE (Invitrogen), protein lysates (∼20μg/lane) were boiled in sample buffer containing reducing agent and lithium dodecyl sulfate (Invitrogen), then resolved on NuPage Bis-Tris 4-12% gels and transferred to nitrocellulose membranes (Whatman). Membranes were blocked with Odyssey Block buffer (Li-Cor Biosciences) and incubated with a subset of the following primary antibodies (Table 2); β-actin (Sigma; 1:10,000), GluR1 (Millipore; 1:500), GluR2 (Chemicon; 1:500), NMDAR1 (Millipore; 1:500), NMDAR2A (Upstate/Millipore; 1:500), PSD95 (Cell Signaling; 1:5,000), and Synapsin 1 (Millipore; 1:5,000). Secondary antibodies anti-mouse or anti-rabbit IgG conjugated to IRDye 680 or 800 (Li-Cor Biosciences; 1:5,000 and 1:10,000 respectively) were used for detection on an infrared imaging system (Li-Cor Odyssey). Band intensities were measured by densitometry using ImageJ and normalized to β-actin. Data presented are an average of three experiments in which three animals per condition are represented.
Antibody Characterization
Antibody information is summarized in Table 2.
The antibodies to tau protein used in these experiments have been widely used and previously characterized. The monoclonal Alz50 mouse IgM antibody was a generous gift of Peter Davies at Albert Einstein College of Medicine, and was first prepared against homogenates of brain tissue from temporal cortex of human AD patients (Wolozin et al., 1986). The Alz50 epitope was later determined to be two discontinuous segments of tau protein (AA7-9 and AA312-342), representative of an intramolecular conformational change (Carmel et al., 1996; Jicha et al., 1997). In western blot, Alz50 recognizes a 64-68kD species highly enriched in the AD brain and largely undetected in control brain (Wolozin et al., 1986). Immunohistochemistry and array tomography with Alz50 in both mouse and human brain tissue identifies pre-tangles, a subpopulation of mature neurofibrillary tangles (NFT) and neuropil threads and demonstrates no staining in control mouse and minimal staining in control human brain (Hyman et al., 1988; Kopeikina et al., 2011; Wolozin et al., 1986). Monoclonal mouse IgG antibody PHF1 was also received as a gift from Peter Davies, and was first prepared against purified paired helical filaments of human AD brain (Davies, 2000; Greenberg and Davies, 1990; Greenberg et al., 1992). PHF1 is a phosphorylation dependent antibody with the epitope consisting of pSer396/pS404, which is considered a late stage phosphorylation in tau pathology. Western blots of mouse or human tissue with PHF1 reveal a band in the 64-68kD range and immunohistochemistry shows fibrillar pathology of both NFT and neuropil threads with little to no reactivity in control tissues (de Calignon et al., 2012; Fox et al., 2011; Polydoro et al., 2009; Tai et al., 2012). The third tau antibody used was human tau specific HT7 (Thermo Scientific), a mouse monoclonal antibody raised to purified human tau (AA159-163) that recognizes 55-64kD species of human tau in western blots (de Calignon et al., 2012; Eckermann et al., 2007; Kremer et al., 2011). In transgenic mouse tissue, HT7 recognizes human tau over-expression, including NFT and neuropil threads, and yields no staining in control animals (de Calignon et al., 2012).
Antibodies for synaptic proteins PSD95 (Abcam) and Synapsin I (Millipore) have been used for array tomography in several previous studies (Koffie et al., 2012; Koffie et al., 2009; Micheva et al., 2010). Polyclonal PSD95 was raised in goats to a synthetic peptide consisting of AA1-100 of mouse post-synaptic scaffolding protein post-synaptic density 95. Immunohistochemistry with PSD95 shows post-synaptic elements that in control tissue show 90% co-localization with pre-synaptic elements (Koffie et al., 2009). These pre-synaptic elements can be identified with rabbit polyclonal antibody for vesicle tethering protein Synapsin 1. This antibody was raised against a mix of Synapsin Ia and Ib purified from bovine brain. Western blot analyses show bands at 77 and 80kD while immunohistochemistry shows pre-synaptic elements (de Calignon et al., 2012; Koffie et al., 2012; Koffie et al., 2009; Micheva et al., 2010). A second PSD95 polyclonal antibody (Cell Signaling) was used for western blot experiments. This antibody was raised in rabbits against a synthetic peptide of human PSD95 and recognizes a 95kD band in homogenates of human and mouse brain and is enriched in synaptosome fractions (Tai et al., 2012). Monoclonal rat GluR1 (Millipore) was generated with recombinant GluR1 and recognizes this subunit of post-synaptic AMPA receptors in immunostains, and appears as monomers (106kD) and dimers (200kD) in western blot. Monoclonal mouse GluR2 raised against recombinant GluR2 (AA175-430) recognizes another subunit of AMPA receptors and has been shown to co-localize with other post-synaptic elements and to be in close proximity to pre-synaptic elements in array tomographic immunostains (Micheva et al., 2010). In western blot analyses, GluR2 reveals a band at 102kD. The NMDAR1 mouse monoclonal antibody (Millipore) identifies the NR1 subunit of post-synaptic NMDA receptors, which is seen on western blot at 130kD. This antibody was raised against a his-tagged peptide of residues 834-938 of rat NR1. Similarly, the NMDAR2A rabbit polyclonal antibody (Upstate/Millipore) was raised against a his-tagged mouse NR2A fragment (residues 1265-1464) and runs at 170kD in western blot. This antibody recognizes another subunit (NR2) of the post-synaptic NMDA receptor.
The immunogen for the rabbit polyclonal GFP antibody (Abcam) was highly purified recombinant GFP made in e. coli, resulting in an antibody reactive to all variants of Aequorea victoria GFP such as GFP, YFP, CFP, RFP and eGFP. In western blot this antibody detects a band at approximately 27kD, while in immunostains the antibody detects this family of fluorescent proteins. In our array tomography stains of YFP-4510 mice this antibody reveals YFP positive neurons in transgenic tissue, with no staining evident in control animals.
A mouse monoclonal β-actin antibody (Sigma) served as the loading control for western blot experiments. This antibody recognizes an N-terminal epitope of the β-isoform of the cytoskeletal protein actin due to its preparation from an immunogen derived from a slightly modified N-terminal peptide of β-actin conjugated to KLH. Western blot of mouse or human brain tissue reveals a band at 42kD (de Calignon et al., 2012; Tai et al., 2012).
Tubulin, another cytoskeletal element, was distinguished with a mouse monoclonal acetylated-tubulin antibody (Sigma). This antibody was raised to acetylated α-tubulin from the outer arm of sea urchin sperm axonemes, and recognizes an epitope on α-tubulin found in the majority of species due to homology. In western blot this antibody reveals a band at 50-55kD and in immunostain shows neuronal cell bodies and processes (Kopeikina et al., 2011).
Statistics
Synapse density measurements were recorded for each individual crop box for both synapsin and PSD95, then grouped by age and genotype. No statistically significant differences between animals of each group were identified by analysis of variance (ANOVA), allowing us to combine all crops for each genotype and age rather than using an average for each animal. Co-localization density of synapsin and PSD95 puncta within 0.5μm of one another was similarly recorded for each crop box. Each of these crop boxes then had three physical disectors applied, then averaged, for manual confirmation of synapse density, again grouped by age and genotype. Normality of data was assessed with a Shapiro-Wilks test. For data lacking normal distribution, Mann-Whitney U tests were applied, while one-way ANOVA or Student's t-test were applied for normally distributed data sets. Significance was determined as a p-value of less than 0.05. Non-normally distributed data are presented as box plots, which display the median value (line inside the box), upper quartile (top of the box), lower quartile (bottom of the box), 90th percentile (top whisker), 10th percentile (bottom whisker), with all values below the 10th and above the 90th percentile (potential outliers) shown as dots. Normally distributed data are shown in bar graphs as mean and standard deviation unless otherwise noted.
Results
Overall synapse density in the neuropil is maintained in rTg4510 mice
Array tomography (Figure 1) was used to quantify synaptic density in two age groups of rTg4510, prior to significant neuronal loss (5.5m) and following significant neuronal loss (8.5m), and control mice to determine whether tauopathy in this model is associated with age-dependent synaptic loss and whether this loss precedes or parallels the neuronal loss evident in this model (Spires et al., 2006). A hybrid of conventional confocal and electron microscopy techniques, array tomography allows rapid and automated quantification of structures too small to be properly identified or localized with traditional confocal microscopy methods. Ultra-thin sections of tissue, allowing for a z-axis resolution of approximately 70nm, were stained with tubulin, Alz50 (an antibody directed at a conformational state of tau also seen in NFT), and reliable synaptic markers synapsin I and PSD95 for identification of pre- and post- synaptic elements respectively and are shown in Figure 2 (Koffie et al., 2012; Koffie et al., 2009; Kopeikina et al., 2011; Micheva et al., 2010; Micheva and Smith, 2007; Spires-Jones and Knafo, 2012).
Figure 1. Array tomography method.

A tile-scan image, taken with a 10× objective, of a ribbon of 70nm sections (s1-25 in panel A) of rTg4510-YFP brain with YFP shown in green, PHF1 (neurofibrillary tangles) in magenta and DAPI (nuclei) in gray. The area outlined in (A) is enlarged in (B), demonstrating a higher-resolution view of a segment of sections 17 and 18. YFP and PHF1 are shown to co-localize in a subset of cells (white). The area outlined in (B) was imaged with the 63× objective to yield a higher magnification image shown in (C). The 10μm by 10μm crop box shown in panel (C) serves as an example of the crop boxes used for synapse density analyses and is shown at higher magnification in panel (D), with the arrow denoting the presence of a dendritic spine. Scale bars = 1000μm (A), 100μm (B), 10μm (C), 1μm (D).
Figure 2. Array tomograms of neuropil synapses in rTg4510 and control mice.

Array tomography images of synapse density by volume (A-P) based on synapsin I (magenta) and PSD95 (green) staining in layer II/III of somatosensory cortex of 5.5 month (A-H) and 8.5 month (I-P) rTg4510 (E-H and M-P) and control mice (A-D and I-L) . Synapsin and PSD95 images of single 70nm sections are shown in the first two columns (A, E, I, M and B, F, J, N respectively) then merged in the third (C, G, K, O). The final column demonstrates three-dimensional renderings of the volume sampled, consisting of 20-30 individual 70nm sections (D, H, L, P). Array tomography images for linear spine density analyses are shown in (Q-T) . Neuropil threads were identified as Alz50+, an antibody directed at a conformational state of tau also seen in NFT (Q) and co-stained with Synapsin (R) and PSD95 (S). Each panel represents a volume of 1.5μm in depth with the last panel (U) showing the zoomed image of the white boxed area in the merged image (T). The Alz50+ neurite of interest is shown outlined in white. Scale bars = 2μm.
At 5.5 months of age, the somatosensory cortex of rTg4510 mice has not yet suffered significant neuronal loss, though a few NFT have begun to accumulate (Spires et al., 2006). When applying an automated, threshold-based detection program that excludes puncta too small to be synapses, no significant changes were identified in synapsin I or PSD95 density between 5.5m rTg4510 and control mice (Figure 3A). This automated analysis method allows quantification of tens of thousands of synaptic puncta with relative ease compared to manual methods of counting synapses. Automated analysis also provides the opportunity for additional data to be extracted from the same raw files. Here we applied a Matlab script to quantify co-localization density of synapsin and PSD95 puncta within 0.5μm of one another (Figure 3E). No difference in co-localization density was detected with this method, suggesting that synaptic partners remain intact rather than orphaning pre- or post-synaptic elements. To confirm the validity of the automated analysis paradigm, we also applied a traditional stereology approach of the physical disector method (Figure 4) to the array tomography data (Gundersen et al., 1988; West et al., 1988). This labor-intensive method of quantification does not rely on selecting a threshold, but rather uses raw images and counts ‘new’ puncta that appear in sequential serial sections. We modified the physical disector method, which is traditionally carried out on electron micrographs where synapses are defined by ultrastructure instead of immunolabeling, to include a third serial section and only ‘new’ puncta which remain present in the third section were counted as synapses. This excludes puncta too thin to be synapses that are likely noise from immunostaining (non-specific secondary staining). This stereological, non-threshold based approach is more sensitive than the automated threshold-based detection and generates slightly higher absolute synaptic densities. With physical disector counts we confirm maintenance of synaptic density in rTg4510 cortex (Figure 3C) and with this more sensitive method we were in fact able to detect a small (∼14%) but statistically significant elevation of PSD95 in 5.5 month old rTg4510 mice when compared to controls (p < 0.05).
Figure 3. Synapse density in the neuropil is maintained in rTg4510 mice.

Automated threshold based quantification of synapse density obtained by array tomography for 5.5 month (A) and 8.5 month old (B) rTg4510 and control mice is shown in the box and whisker plots of the top row. No statistically significant change in synapse density is identified for either age group with this method. Manual stereology based physical disector quantification of synapse density is shown in the box and whisker plots of the second row (C and D) and confirm the findings with the automated analysis. This more sensitive analysis reveals slightly greater absolute values for synapse density altogether and a significant increase in PSD95 density in rTg4510 mice at 5.5 months of age that may represent compensatory responses (C). The last box and whisker plot (E) shows co-localization density of synapsin and PSD95 within 0.5μm of one another for 5.5 and 8.5 month old rTg4510 and control mice obtained following the automated assessment. No statistically significant differences in co-localization was detected. (*p < 0.5)
Substantial tauopathy is evident in the somatosensory cortex of rTg4510 mice at 8.5m of age, when significant accumulation of NFT, neuronal loss and cortical atrophy occur (Spires et al., 2006). Even in the face of marked neurodegeneration, synaptic density based on synapsin I and PSD95 staining remained stable in 8.5m old rTg4510 mice when compared to controls (Figure 3B and D), regardless of method of analysis (MaxEntropy or physical disector). Co-localization density following automated analysis also demonstrates a preservation of pairing of synapsin and PSD95 in the 8.5m rTg4510 mice as compared to controls (Figure 3E). These findings suggest that synapse density in the neuropil is maintained while the volume of the neuropil significantly decreases (Spires et al., 2006) resulting in a net loss of synapses (total number of synapses = synapse density * neuropil volume). These synapses are likely lost with equal preference for pre- and post-synaptic elements, regardless of the presence of aggregated misfolded tau, as co-localization and neuropil thread associated linear spine densities are maintained. This suggests that neuronal loss and neuropil remodeling proceed in tandem in this model. In addition, Mann Whitney U analysis of linear synapse density demonstrated no statistically significant difference between neuropil thread containing (Alz50+) versus tubulin only filled processes in 8.5m old rTg4510 brain, indicating that fibrillar tau in dendrites is not causing local loss of synapses in this model.
In-vivo imaging demonstrates loss of dendritic spine density in rTg4510 mice
By contrast to the results noted above using array tomography, Rocher et al (2010) found loss of apical dendritic spines in biocytin filled neurons in frontal cortex of 8.5m old rTg4510 mice. Since array tomography detects all synapses, we next examined dendritic spine density of apical dendrites of pyramidal neurons to determine whether these spines are particularly vulnerable to tau-induced toxicity. To enable visualization of dendritic spines in-vivo, cranial windows were implanted over the somatosensory cortex of 9-10 month old rTg4510-YFP mice, which over-express human mutant tau and also have a subset of neurons filled with yellow fluorescent protein (YFP), driven by a separate promoter (Thy1) from the human mutant tau. To ensure that introduction of YFP did not hinder accumulation of tau pathology, side-by-side comparison of hippocampus and cortex of 9m old rTg4510 and rTg4510-YFP was performed with immunostaining for YFP, PHF1 and DAPI as shown in Figure 5. Both transgenic lines demonstrate similar thinning of the CA1 subregion of the hippocampus and accumulation of tangles (PHF1+) in the cortex with significant co-labeling for YFP and PHF1. YFP is largely expressed in layer V pyramidal neurons in cortex, while tau is present in both layer V and layer II-III neurons (Figure 5). Dendritic spines were imaged in-vivo with multi-photon microscopy and quantified in rTg4510-YFP and control animals (Figure 6). A one-way analysis of variance (ANOVA) shows significant loss of dendritic spine density (close to 30%) in aged rTg4510-YFP mice as compared to control (F(1,60) =19.8; p < 0.0001). These data argue that while synaptic density may remain unchanged, synapses are distinctly lost along apical dendrites of vulnerable neurons.
Figure 5. rTg4510 and rTg4510-YFP show similar degeneration and tau pathology at 9 months of age.

Tile-scans of coronal sections of 9 month old rTg4510 (A, C) and rTg4510-YFP (B, D-F) cortex and hippocampus demonstrate similar loss of neurons in CA1 (asterisks) and tangle pathology based on DAPI (gray), PHF1 (magenta), and YFP (green) immunostaining in 50μm floating sections. Higher resolution images (C and D) demonstrate similar levels of tau pathology in the cortex of both transgenic mouse lines. In the rTg4510-YFP cortex, some co-localization of YFP and tau pathology occurs (white) in cell bodies (arrows, E) and neuropil threads (arrowhead, F). Scale bars = 1000μm (A and B), 100μm (C and D), 50μm (E), 10μm (F).
Figure 6. Decreased dendritic spine density in the intact living rTg4510-YFP brain.

In-vivo multiphoton images of linear dendritic spine density of YFP filled dendrites of littermate control (A) and rTg4510 (B) brains. Dendrites and associated spines were isolated from background for easier viewing. Scale bar = 2μm. Bar graph in C shows quantification of linear spine density and one-way ANOVA demonstrates significant loss of spine density in aged rTg4510 mice (p < 0.0001). Graph shows means and standard deviations.
Though tau is historically considered an axonal protein, recent studies indicate that tau can be present at the dendritic spine, either to play a physiological role or as a result of pathological missorting, and may contribute to degeneration of dendritic spines (Hoover et al., 2010; Ittner et al., 2010; Kremer et al., 2011; Tai et al., 2012; Yu et al., 2012). To confirm the presence of tau at dendritic spines, the rTg4510-YFP mice were processed for array tomography studies (Figure 7). Staining human tau and YFP showed localization of human tau (HT7) and hyperphosphorylated tau (PHF1) at a subset of dendritic spines on pyramidal neurons, indicating that the presence of tau at spines may, in fact, contribute to their collapse.
Figure 7. Human tau in dendritic spines of rTg4510-YFP mice.
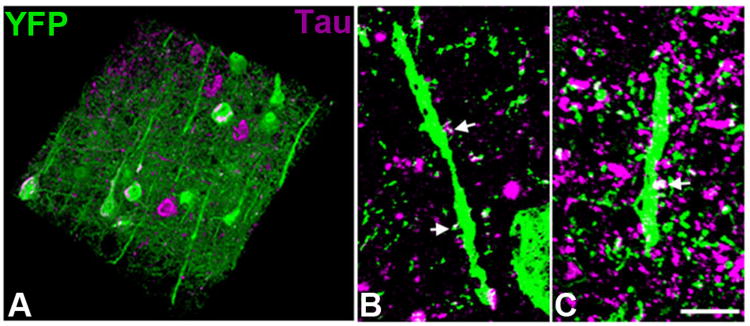
Post-mortem analysis of array tomography stacks (A) stained with phosphorylated tau (PHF1) shows neurofibrillary tangles and neuropil threads (tau aggregation in neurites) in a subset of both YFP positive and YFP negative neurons (three-dimensional reconstruction of 100 serial sections of 100nm thickness 143μm × 143μm × 10μm volume). Staining with both PHF1 (B) and total human tau (HT7; C) antibodies shows tau staining in a subset of dendritic spines (arrows, maximum intensity projections of three 100nm sections). Scale bar = 5μm.
Biochemical analyses suggest loss of function in synapses of rTg4510
Several studies have suggested that alterations to tau, such as hyperphosphorylation, mislocalization or over-expression, induce toxicity by diminishing functionality of synapses rather than causing their overt loss (Hoover et al., 2010; Polydoro et al., 2009). To assess the levels of synaptic proteins in remaining synapses of the rTg4510 mouse brain at an advanced age (12m), biochemical fractionation of the brains was carried out to isolate synaptosomes (Tai et al., 2012), which were probed by western blot for functional components of the dendritic spine (Figure 8). NMDA and AMPA receptors, generally anchored in the post-synaptic density (PSD95), are thought to subserve the processes underlying learning and memory through their presence and function. Loss of functional subunits (NMDAR1, NMDAR2A, GluR1, GluR2) would indicate a deficit or alteration in synaptic function. In 12 month old rTg4510 mice, levels of PSD95, Synapsin I, NMDAR1 and GluR1 are all significantly decreased (p < 0.05 for all) while NMDAR2A and GluR2 expression do not demonstrate a deficit. These data suggest a preferential change to the constitution of pre- and post-synaptic elements, along with NMDA and AMPA receptors and imply a potential loss of function in remaining synapses of rTg4510 mice. These data support electrophysiological findings in rTg4510 and other tau models, which demonstrate synaptic functional impairments due to tau over-expression (Polydoro et al., 2009; Rocher et al., 2010; Sydow et al., 2011b).
Figure 8. Loss of functional components of remaining synapses in rTg4510 brain.
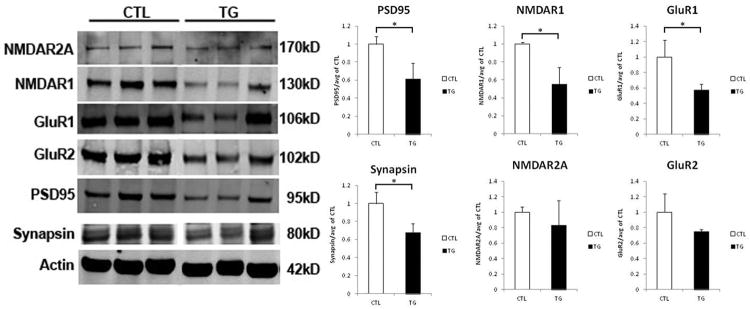
Synaptosomes were isolated from the brains of aged rTg4510 and littermate control mice and probed by Western blot (shown on the left) for functional components of the synapse. Quantification of the average of three trials is shown in the bar graphs to the right. PSD95, Synapsin I, NDMAR1, and GluR1 levels are significantly decreased in remaining synapses of rTg4510 mouse brain when compared to littermate controls, while NMDAR2A and GluR2 levels are maintained. (*p < 0.05). Graphs show means and standard deviations.
Discussion
Degeneration of synapses is a strong correlate of cognitive decline in Alzheimer Disease (AD) brain, even more closely related to severity and progression of disease than the hallmark neurofibrillary tangles (NFT), extracellular amyloid plaques, and neuronal loss (Arriagada et al., 1992; Coleman and Yao, 2003; DeKosky et al., 1996; Giannakopoulos et al., 2003; Gomez-Isla et al., 1997; Terry et al., 1991). Though the amyloid cascade hypothesis posits that disease pathogenesis is initiated by Aβ with downstream consequences executed by the necessary presence of tau, it remains unclear how Aβ and or tau may cause synaptic decay. The synapse has however, been proposed as a point of convergence for both Aβ and tau toxicity (Ittner et al., 2010; Roberson et al., 2011). Previous studies linking tau and synapses have had complicated results, with some studies indicating distinct dendritic spine loss or dysfunction (Bittner et al., 2010; Crimins et al., 2011; Eckermann et al., 2007; Mocanu et al., 2008; Polydoro et al., 2009; Rocher et al., 2010; Sydow et al., 2011b; Yoshiyama et al., 2007), others indicating no significant alteration in synapse density or function (Hoover et al., 2010; Kimura et al., 2010; Shahani et al., 2006; Tackenberg and Brandt, 2009) and a last set of studies suggesting compensatory increases in synapse density or function (Amadoro et al., 2010; Boekhoorn et al., 2006; Crimins et al., 2011; David et al., 2005; Dickstein et al., 2010; Kimura et al., 2010; Kremer et al., 2011). Here we used two high-resolution imaging techniques to resolve some of these conflicting data in the rTg4510 model. We find preservation of overall synapse density in the neuropil even in the context of significant dendritic spine loss along apical dendrites of cortical pyramidal neurons, suggesting these neurons may be particularly vulnerable to tau-induced dendritic spine collapse, and furthermore indicating a compensatory response of other populations of synapses.
Assessment of synaptic structural and functional components in remaining synaptosomes from aged rTg4510 mice revealed reductions in synapsin I and PSD95 along with components of NMDA and AMPA receptors, NMDAR1 and GluR1, while two other constituents of these receptors (NMDAR2A and GluR2) remained stable. These synaptic components are critical to neuronal function and dysregulation of individual elements suggests likely coincident synaptic dysfunction. Taken together, our data argue that synapse loss and dysfunction occur in rTg4510 mice.
Since distinguishing individual synapses is beyond the limits of routine light microscopy methods we applied array tomography, a recently developed high-resolution immunohistochemical technique for localization and quantification of pre-synaptic and post-synaptic elements (Micheva et al., 2010; Micheva and Smith, 2007). We have previously used this technique to show synapse loss associated with oligomeric Aβ in both mouse models and human AD brain (Koffie et al., 2012; Koffie et al., 2009). In this study, we use array tomography to probe the role of tau in synaptic degeneration in rTg4510 mice. Somatosensory cortex of mice at an age prior to significant neuronal loss (5.5 months) and following significant neuronal loss (8.5 months) was prepared for array tomography and stained with reliable synaptic markers Synapsin I and PSD95 (Micheva et al., 2010). Quantification with a threshold based automated program and manual physical disector stereology to confirm the validity of our automated analysis technique showed little evidence of change in synaptic density across both age groups of rTg4510 mice when compared to littermate controls. The comparison of automated and manual analysis of array tomography stacks indicates that automated, threshold-based analysis, which is much less time and labor intensive, is adequate for comparisons of groups if the difference between groups is large enough (more than 10%), as we observed previously in examining synapse loss near plaques (Koffie et al., 2012; Koffie et al., 2009). For more subtle differences, manual stereology or new non-threshold based automated image analysis methods may be more appropriate. Another advantage, however, of automated analysis is the ability to extrapolate additional data from the same set of raw images such as analysis of colocalization or proximity of different channels.
The maintenance of synaptic density in the neuropil along with the known cortical atrophy in this model by 8.5 months of age (Spires et al., 2006) indicates loss of synapses in parallel with cortical atrophy. An approximately 30% deficit in linear dendritic spine density was observed using in vivo imaging of the aged rTg4510-YFP brain in comparison to control animals. It should be noted that these rTg4510-YFP mice have an additional YFP transgene driven by the Thy1 promoter alongside the CaMKII derived over-expression of mutant tau. Both promoters drive expression in pyramidal neurons of the forebrain, thereby generating high likelihood of double-expression, though this is, of course, not a guarantee. Furthermore, the use of separate promoters suggests expression of both transgenes without interference and yields no evidence in delay of degenerative processes as a result of YFP introduction. Our findings in the rTg4510-YFP mice are consistent with ∼30% dendritic spine loss observed in virus filled neurons in 9m rTg4510 mice (data not shown), and data from Rocher et al (2010), in which a 30-40% deficit of dendritic spines was identified in biocytin filled neurons of 8.5m old rTg4510 mice and electron microscopy data from Mocanu et al (2008) demonstrating a ∼30% loss of spine-synapses in tau transgenic mice expressing a pro-aggregant form of tau. The similarity across data sets indicates that YFP expression in rTg4510 mice does not interfere with the timeline of degenerative events.
Both the maintenance of synapse density as measured by array tomography and loss of linear spine density by in-vivo imaging in the context of cortical atrophy demonstrate that tau over-expression results in synapse degeneration. Furthermore, pyramidal neurons may be particularly vulnerable to such decay, potentially resulting in compensatory responses from remaining neurons, with increases in other synapses, to augment network function. In support of this idea, Crimins et al 2011 found an increase in filopodia (which are difficult to detect with in vivo imaging resolution) and electrophysiological evidence of synaptic compensation in aged rTg4510 mice. In addition, neuronal cultures exposed to Aβ treatment have also demonstrated similar increases in filopodia (Wu et al., 2012), suggesting a potential common mechanism of synaptic response to both Aβ and tau. Such filopodia have been shown to have variable number of post-synaptic densities, ranging from zero to multiples (Crimins et al., 2011; Petrak et al., 2005) and may therefore be associated with delocalization of PSD95 and destabilization of dendritic spines. A recent study on mechanisms of Aβ and tau toxicity to synapses and dendritic spines in neuronal cultures observed a shift of PSD95 from the synapse to the dendritic shaft (Yu et al., 2012). Changes like these have also been seen in the aging non-human primate brain (Dumitriu et al., 2010) and could be exacerbated in an age-related disease such as AD and suggest a second synaptic subtype that may be increased by compensatory responses in remaining neurons of rTg4510 brain.
Redistribution or mislocalization of PSD95, which serves as a scaffold for essential post-synaptic elements such as NMDA and AMPA receptors, may have negative ramifications for their localization or function as well (Chakroborty and Stutzmann, 2011; D'Amelio et al., 2011; Hoover et al., 2010; Huang and Mucke, 2012). Our data indicate a significant loss of PSD95, synapsin, and subunits NMDAR1 and GluR1 of NMDA and AMPA receptors respectively, while NMDAR2A and GluR2 levels are maintained, in synaptosomes of aged rTg4510 mice. These data are consistent with findings in neuronal cultures over-expressing P301L tau, in which deficits were detected in NMDAR1, GluR1 but also in GluR2/3 levels (Hoover et al., 2010). Human AD brain also exhibits preferential loss of NMDAR1 subunits while NMDAR2A levels remain stable (Bezprozvanny and Mattson, 2008), which we also observe in the rTg4510 mouse brain. These data argue that pathological changes in tau may influence synaptic make-up and function in human AD brain as well as in tauopathy models. One of the mechanisms responsible for removal of synaptic receptors, known as long term depression, is thought to be more efficient in disease models (D'Amelio et al., 2011) and may lead to an increase in silent synapses or dendritic spine collapse (Hoover et al., 2010). Recent studies that place tau both physiologically and pathologically within the dendritic spine rather than solely in the axonal compartment indicate that tau may play a more direct role than previously believed in regulating the presence and function of post-synaptic receptors (Hoover et al., 2010; Ittner et al., 2010; Tai et al., 2012). Here we demonstrate by array tomography that tau is present at dendritic spines and may therefore be implicated in the process of receptor subunit loss, thereby contributing to synaptic dysfunction and degradation. However, the presence of misfolded aggregates of tau as detected by the Alz50 antibody does not exacerbate local linear synapse density loss, suggesting that aggregates themselves may not enhance spine degradation.
Taken together our data indicate dendritic spine loss and alterations in receptor constitution in aged rTg4510 mice with concomitant maintenance of synapse density, suggesting the possibility that compensatory responses or homeostatic mechanisms may counteract loss of dendritic spines.
Acknowledgments
This work was supported by National Institute of Health Grants AG08487, T32AG000277, AG026249, R00AG33670 and the Alzheimer's Association Zenith Award.
Footnotes
Conflict of interest statement: None
Role of authors: All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: KJK, TLSJ, BTH. Acquisition of data: RP, KJK, HCT, MP. Analysis and interpretation of data: KJK, EY, MP, TLSJ, BTH. Drafting of manuscript: KJK. Critical revision of manuscript for important intellectual content: GAC, RP, MP, TLSJ, BTH. Statistical analysis: KJK. Obtained funding: TLSJ, BTH. Administrative, technical and material support: HCT, GAC, RP, MP. Study supervision: TLSJ, BTH.
Literature cited
- Alldred MJ, Duff KE, Ginsberg SD. Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol Dis. 2012;45(2):751–762. doi: 10.1016/j.nbd.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadoro G, Corsetti V, Stringaro A, Colone M, D'Aguanno S, Meli G, Ciotti M, Sancesario G, Cattaneo A, Bussani R, Mercanti D, Calissano P. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis. 2010;21(2):445–470. doi: 10.3233/JAD-2010-100120. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42(3 Pt 1):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22(18):7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling and Alzheimer's disease. Neurochem Res. 2011;36(7):1149–1156. doi: 10.1007/s11064-010-0371-4. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31(9):454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Ochs SM, Hoffmann N, Mitteregger G, Kretzschmar H, LaFerla FM, Herms J. Multiple events lead to dendritic spine loss in triple transgenic Alzheimer's disease mice. PLoS One. 2010;5(11):e15477. doi: 10.1371/journal.pone.0015477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, Ramakers GJ, de Vos K, Krugers H, Tomiyama T, Mori H, Joels M, van Leuven F, Lucassen PJ. Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci. 2006;26(13):3514–3523. doi: 10.1523/JNEUROSCI.5425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271(51):32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Stutzmann GE. Early calcium dysregulation in Alzheimer's disease: setting the stage for synaptic dysfunction. Sci China Life Sci. 2011;54(8):752–762. doi: 10.1007/s11427-011-4205-7. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24(8):1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Crimins JL, Rocher AB, Peters A, Shultz P, Lewis J, Luebke JI. Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol. 2011;122(5):551–564. doi: 10.1007/s00401-011-0877-0. [DOI] [PubMed] [Google Scholar]
- Cuchillo-Ibanez I, Seereeram A, Byers HL, Leung KY, Ward MA, Anderton BH, Hanger DP. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008;22(9):3186–3195. doi: 10.1096/fj.08-109181. [DOI] [PubMed] [Google Scholar]
- D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011;14(1):69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Drose S, Brandt U, Muller WE, Eckert A, Gotz J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. 2005;280(25):23802–23814. doi: 10.1074/jbc.M500356200. [DOI] [PubMed] [Google Scholar]
- Davies P. Characterization and use of monoclonal antibodies to tau and paired helical filament tau. Methods Mol Med. 2000;32:361–373. doi: 10.1385/1-59259-195-7:361. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73(4):685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, F LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010 doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5(4):417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- Dickstein DL, Brautigam H, Stockton SD, Jr, Schmeidler J, Hof PR. Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct Funct. 2010;214(2-3):161–179. doi: 10.1007/s00429-010-0245-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319(5866):1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey M, Chaudhury P, Kabiru H, Shea TB. Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: neurofilaments attenuate tau-mediated neurite instability. Cell Motil Cytoskeleton. 2008;65(2):89–99. doi: 10.1002/cm.20243. [DOI] [PubMed] [Google Scholar]
- Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, Rapp PR, Morrison JH. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30(22):7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schonig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007;282(43):31755–31765. doi: 10.1074/jbc.M705282200. [DOI] [PubMed] [Google Scholar]
- Fox LM, William CM, Adamowicz DH, Pitstick R, Carlson GA, Spires-Jones TL, Hyman BT. Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J Neuropathol Exp Neurol. 2011;70(7):588–595. doi: 10.1097/NEN.0b013e318220a658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JW, Cotman CW. Plasticity in hippocampal excitatory amino acid receptors in Alzheimer's disease. Neurosci Res. 1986;3(6):672–678. doi: 10.1016/0168-0102(86)90062-3. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC. Plasticity of hippocampal circuitry in Alzheimer's disease. Science. 1985;230(4730):1179–1181. doi: 10.1126/science.4071042. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60(9):1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41(1):17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87(15):5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267(1):564–569. [PubMed] [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, et al. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96(10):857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165(5):1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall GF, Chu B, Lee G, Yao J. Human tau filaments induce microtubule and synapse loss in an in vivo model of neurofibrillary degenerative disease. J Cell Sci. 2000;113(Pt 8):1373–1387. doi: 10.1242/jcs.113.8.1373. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118(Pt 23):5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Kromer LJ, Van Hoesen GW. Reinnervation of the hippocampal perforant pathway zone in Alzheimer's disease. Ann Neurol. 1987;21(3):259–267. doi: 10.1002/ana.410210307. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Wolozin BL, Davies P, Kromer LJ, Damasio AR. Alz-50 antibody recognizes Alzheimer-related neuronal changes. Ann Neurol. 1988;23(4):371–379. doi: 10.1002/ana.410230410. [DOI] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62(6):925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142(3):387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jaworski T, Lechat B, Demedts D, Gielis L, Devijver H, Borghgraef P, Duimel H, Verheyen F, Kugler S, Van Leuven F. Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol. 2011;179(4):2001–2015. doi: 10.1016/j.ajpath.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48(2):128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Kimura T, Fukuda T, Sahara N, Yamashita S, Murayama M, Mizoroki T, Yoshiike Y, Lee B, Sotiropoulos I, Maeda S, Takashima A. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synaptic loss. J Biol Chem. 2010;285(49):38692–38699. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61(9):797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, Holtzman DM, Hyman BT, Spires-Jones TL. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012 doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106(10):4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol. 2011;179(4):2071–2082. doi: 10.1016/j.ajpath.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer A, Maurin H, Demedts D, Devijver H, Borghgraef P, Van Leuven F. Early improved and late defective cognition is reflected by dendritic spines in Tau.P301L mice. J Neurosci. 2011;31(49):18036–18047. doi: 10.1523/JNEUROSCI.4859-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59(2):214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba G, Walzer C, Vernay A, Carnal B, Kraftsik R, Piotton F, Marin P, Bouras C, Savioz A. Postsynaptic density protein PSD-95 expression in Alzheimer's disease and okadaic acid induced neuritic retraction. Neurobiol Dis. 2008;30(3):408–419. doi: 10.1016/j.nbd.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68(4):639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55(1):25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schonig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28(3):737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29(41):12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70(3):410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrak LJ, Harris KM, Kirov SA. Synaptogenesis on mature hippocampal dendrites occurs via filopodia and immature spines during blocked synaptic transmission. J Comp Neurol. 2005;484(2):183–190. doi: 10.1002/cne.20468. [DOI] [PubMed] [Google Scholar]
- Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29(34):10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-{beta}/Fyn-Induced Synaptic, Network, and Cognitive Impairments Depend on Tau Levels in Multiple Mouse Models of Alzheimer's Disease. J Neurosci. 2011;31(2):700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, Luebke JI. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2010;223(2):385–393. doi: 10.1016/j.expneurol.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2001;11(3):349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990;11(1):29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24(8):1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192(1):106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani N, Subramaniam S, Wolf T, Tackenberg C, Brandt R. Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of Alzheimer's disease-relevant tau constructs in organotypic hippocampal slices. J Neurosci. 2006;26(22):6103–6114. doi: 10.1523/JNEUROSCI.4245-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168(5):1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T, Knafo S. Spines, plasticity, and cognition in Alzheimer's model mice. Neural Plast. 2012;2012:319836. doi: 10.1155/2012/319836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28(4):862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156(6):1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff W, Jones PB, Spires-Jones TL, Joyner D, Chhabra E, Bercury K, Fan Z, Xie H, Bacskai B, Edd J, Irimia D, Hyman BT. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J Neurochem. 2009;111(2):417–427. doi: 10.1111/j.1471-4159.2009.06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM. Reversibility of Tau-related cognitive defects in a regulatable FTD mouse model. J Mol Neurosci. 2011a;45(3):432–437. doi: 10.1007/s12031-011-9604-5. [DOI] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011b;31(7):2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009;29(46):14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. Am J Pathol. 2012 doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thevenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7(1):27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27(11):2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG. Estimating the number of granule cells in the dentate gyrus with the disector. Brain Res. 1988;448(1):167–172. doi: 10.1016/0006-8993(88)91114-6. [DOI] [PubMed] [Google Scholar]
- Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232(4750):648–650. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ, Hyman BT. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-beta revealed by a novel fluorescence resonance energy transfer assay. J Neurosci. 2012;32(15):5298–5309. doi: 10.1523/JNEUROSCI.0227-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Yu W, Lu B. Synapses and dendritic spines as pathogenic targets in Alzheimer's disease. Neural Plast. 2012;2012:247150. doi: 10.1155/2012/247150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Polepalli J, Wagh D, Rajadas J, Malenka R, Lu B. A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Abeta on synapses and dendritic spines. Hum Mol Genet. 2012;21(6):1384–1390. doi: 10.1093/hmg/ddr576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30(36):11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]