

Abstract
Objective
To investigate the pathogenesis and transmission of elephant endotheliotropic herpesvirus (EEHV1) by analyzing various elephant fluid samples with a novel EEHV1-specific real-time PCR assay.
Animals
5 apparently healthy captive Asian elephants (Elephas maximus) from the same herd.
Procedures
A real-time PCR assay was developed that specifically detects EEHV1. The assay was used to evaluate paired whole blood and trunk-wash samples obtained from the 5 elephants during a 15-week period. Deoxyribonucleic acid sequencing and viral gene subtyping analysis were performed on trunk-wash DNA preparations that had positive results for EEHV1. Viral gene subtypes were compared with those associated with past fatal cases of herpesvirus-associated disease within the herd.
Results
The PCR assay detected viral DNA to a level of 1,200 copies/mL of whole blood. It was used to detect EEHV1 in trunk secretions of 3 of the 5 elephants surveyed during the 15-week period. Viral gene subtyping analysis identified 2 distinct elephant herpesviruses, 1 of which was identical to the virus associated with a previous fatal case of herpesvirus-associated disease within the herd.
Conclusions and Clinical Relevance
EEHV1 was shed in the trunk secretions of healthy Asian elephants. Trunk secretions may provide a mode of transmission for this virus. Results of this study may be useful for the diagnosis, treatment, and management of EEHV1-associated disease and the overall management of captive elephant populations.
Elephant endotheliotropic herpesviruses can cause acute hemorrhagic disease in endangered Asian and African (Loxodonta africana) elephants, resulting in considerable illness, reproductive loss, and death in captive elephant populations.1–8 This herpesvirus-associated disease primarily affects juvenile Asian elephants and results in rapid-onset endotheliolytic disease with a mortality rate of 85% in elephants that have positive results for the disease as indicated by semiquantitative conventional PCR-assay blood tests.9 After the original report of EEHV in 1999,2 > 60 cases of systemic herpesvirus-associated hemorrhagic disease have been identified worldwide, with most disease developing in captive-born juvenile Asian elephants.5,9
The clinical course of herpesvirus-associated disease has a rapid progression over 1 to 7 days.1–3,5,10 It begins with the onset of lethargy, anorexia, edema, and cyanosis of the tongue and often culminates in death with pathological findings of disseminated hemorrhagic lesions characterized by intranuclear herpesvirus inclusions in capillary endothelial cells.2,3,5,8,10,11 World-wide, there have been 8 confirmed cases of elephants surviving detectable EEHV infection.2,11 All but 2 of these elephants were clinically ill with herpesvirus-associated disease.a Those elephants that survived EEHV-associated disease were treated with the guanine-analog antiviral drug famciclovir.2,11 Therefore, it is presumed that early intervention with antiviral medication is beneficial for elephants with herpesvirus-associated disease.2,11 Both EEHV1 and EEHV2 carry the putative genes for thymidine kinase and protein kinase,12 but it remains unclear whether these proteins possess the rel-evant activities that confer therapeutic benefit from the antiviral medications.
Since the first 2 EEHV species, EEHV1 and EEHV2, were described nearly 10 years ago, 3 additional species have been identified,5 and all 5 EEHVs are classified as members of the Proboscivirus genus within the subfamily Betaherpesvirinae.13 Through DNA sequence analysis, EEHV1 has been shown to have 2 distinct genotypes, EEHV1A and EEHV1B, which together have accounted for 26 of the 31 (84%) cases of herpesvirus-associated disease in North America in which the EEHV species has been identified.2,6,9,14 In addition to the illness associated with EEHV1, EEHV2 has been associated with 2 elephant deaths, and the recently identified EEHV3 and EEHV4 (formerly EEHV3B) have each been associated with 1 case of fatal hemorrhagic disease in Asian elephants.2,5 In addition to the probosciviruses, 5 distinct elephant gammaherpesviruses have been identified but have not been associated with considerable illness or death in infected elephants.14,15
Although substantial advances have been made in the identification of herpesviruses that infect elephants, little progress has been made toward elucidating the potential mechanisms of virus transmission.15,16 Herpesviruses infect nearly every animal species examined to date.17 Following primary infection, herpesviruses usually establish a latent subclinical infection in host cells (usually neurons, lymphocytes, or monocytes), with intermittent periods of reactivation and shedding of new virus particles in a range of host fluid samples.17 It is through these virus-containing fluids that herpesviruses are transmitted from one host to the next.17 Currently available PCR tests have been used to detect EEHV1 DNA in cutaneous papillomas and vestibular lesions of otherwise healthy African elephants.2,18 In addition, EEHV2 and EEHV3 DNA have been detected in pulmonary lymphoid nodules of healthy culled African elephants.2 Attempts to detect the EEHV1 strains associated with fatal hemorrhagic disease in the fluids or tissues of healthy Asian elephants have been unsuccessful except in 1 recent situation.a The presumed apathogenic EGHVs have been detected in samples of conjunctival and vaginal secretions obtained from healthy Asian and African elephants15 and routine samples of blood from Asian elephants.a Semiquantitative EEHV-specific PCR assays are available to confirm EEHV in clinically ill elephants.2,5 However, these tests have been unable to detect EEHV DNA present in fluid samples other than blood and are not widely applicable for the routine monitoring of elephant herds for the detection of preclinical viremia and early intervention with antiviral medications and other supportive care.
Extensive genetic analysis has revealed there are 2 major distinctive genotypes among EEHV1 strains, referred to as EEHV1A and EEHV1B.2,6,12,14 However, the relationship between the 2 is that of complex chimeras, with several genes having large differences of 15% to 40% at the predicted encoded protein level (U39 [glycoprotein B], U46 [glycoprotein N], U48 [glycoprotein H], and open reading frame E [thymidine kinase]), but with many others having much smaller differences of 1% to 2% (U57 [major capsid protein], U66 [terminase], and U43 [helicase-primase complex protein]) and some genes having essentially no differences at all (U41 [MDBP]).14 Within the 2 original small diagnostic PCR loci from the DNA polymerase and terminase genes, both loci usually but not always display a common pattern of 15 single nucleotide polymorphisms over a 500-bp region when comparing the EEHV1A and 1B subtypes. Two additional EEHV1 gene loci that provide further useful strain discrimination by falling into 3 and even 5 distinct subtypes are U71 (gM; A, B and C subtypes) and U51 (vGPCR; A, B, C, D, and E).14
A rapid, sensitive, and specific real-time qPCR assay is needed for detecting the EEHV subtypes most commonly associated with herpesvirus-associated disease in captive Asian elephants in North America. Such an assay would be useful for the detection of EEHV1 viral DNA in blood samples for the purposes of screening clinically ill animals and monitoring susceptible elephants for early viremia. The ideal assay would have the potential to be applied to multiple clinical samples with the intent of identifying elephants latently infected with EEHV1 subtypes by detecting viral DNA present in various fluid samples. The purpose of the study reported here was to establish an EEHV herd-monitoring program for the detection of preclinical viremia and the detection of EEHV1 DNA in routine trunk-wash samples from healthy Asian elephants obtained over a 15-week period. We sought to use gene subtyping on the viral DNA in positive trunk-wash samples to establish an epidemiological link with a previous lethal case of EEHV1A-associated disease in the monitored herd.
Materials and Methods
Animals
Five healthy captive Asian elephants (elephants 1 to 5) from June 22, 2009, to September 28, 2009, were used for the study. Sample collection was performed in accordance with recommended guidelines for research involving animals, and the sample collection protocol was approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Archival DNA samples including NAP case numbers (NAP#) 14, 17, 19, 22, 23, 26, 27, 29, 31, 32, 33, and 34, as well as the first archive elephant and second archive elephant, represent previous cases of elephant herpesvirus infection for which gene subtyping data had been previously determined.4-6,9,14 These samples were used to validate the specificity of our real-time qPCR assay and to perform gene subtyping on novel isolates of elephant herpesvirus DNA.
Sample collection and processing
For the study, beginning June 22, 2009, and ending September 28, 2009, routine blood samples and trunk washes were obtained from each elephant. To do so, 2 mL of blood was collected from an auricular vein into EDTA anticoagulant tubes. Samples were collected 1 to 3 times weekly and stored at 4°C until DNA processing, which was always within 24 hours after collection. Generally, 200 μL was processed for DNA, and the surplus was stored in 500-μL aliquots at −80°C. Trunk-wash samples containing elephant nasal secretions were collected by means of the standard recommended technique for collection of samples for Mycobacterium tuberculosis testing by the USDA. Briefly, 50 mL of sterile saline (0.9% NaCl) solution was poured into the nares of each elephant, and the proboscis was elevated for 20 to 30 seconds. The elephant was then instructed to blow the instilled saline solution into a fresh plastic 1-gallon freezer bag. The resulting effluent was transferred to a sterile 50-mL conical tube and then chilled on ice until processed. Trunk-wash samples were centrifuged at 1,500 × g for 10 minutes, the supernatant was discarded, and the cell pellet was stored at −80°C until the DNA was processed.
DNA extraction
Whole blood samples were processed by use of a commercial kit,b in accordance with the manufacturer’s recommended protocol for 200 μL of whole blood. The DNA was eluted in 110 μL of buffer.c A different commercial kitd was used to isolate larger quantities of Elephas maximus DNA for generating qPCR standards. Concentrations of DNA were calculated by use of a spectrophotometer.e
Nasal secretions obtained via trunk washes were processed with a tissue kitf by use of a modified protocol for purification of genomic DNA from 50 to 100 mg of fresh or frozen tissues. Briefly, cell pellets were digested overnight at 55°C in 3 mL of cell lysis buffer and proteinase K. Samples were then treated with ribonuclease A for 3 minutes, 1 mL of protein precipitation solution was added, and the mixture was incubated at 4°C and then centrifuged for 10 minutes at 2,000 × g. The supernatant was transferred to a new 15-mL conical tube, incubated on ice for 5 minutes, and then centrifuged again for 10 minutes at 2,000 × g. The supernatant was poured into a tube containing 3 mL of isopropanol and mixed by inversion 50 times, then centrifuged for 3 minutes at 2,000 × g. The DNA pellets were washed with 3 mL of 70% ethanol and then centrifuged at 2,000 × g. The DNA pellets were dried for 15 to 20 minutes, and 100 to 150 μL of kit DNA hydration solution was added. Samples were incubated at 65°C for 1 hour. Afterward, samples were removed from heat and stored at room temperature (approx 22°C) overnight. The DNA concentration was determined with a spectrophotometer.g Extraction of DNA from elephant tissue samples was achieved by use of commercial kits,f,h in accordance with manufacturer recommendations. Control samples obtained from 10 captive elephants at other institutions were processed as described elsewhere.5
DNA constructs
The DNA sequence for EEHV1A MDBP was amplified from DNA isolated from NAP#32 heart tissue by use of the following primers: forward, CGATGATACCCGATCCCTAGTC; and reverse, CGGACCACATACGGGATTCT. Oligonucleotide sequences were based on the published EEHV1B MDBP DNA sequence6,12 (GenBank accession No. AF322977) and unpublished EEHV1A MDBP DNA sequence.a The product was purified with a purification kiti and subcloned into a cloning vectorj to produce the plasmid pJT255. A second recombinant plasmid, pJT256, was constructed by PCR amplification of the E maximus IFN-γ gene and cloning into the cloning vector.j The following primers were used to amplify IFN-γ for construction of pJT256: forward, GCGTGAAGACCCTTGAGGAA; and reverse, TCATTTACCGGAGTTTGCATCA. The cloned inserts for pJT255 and pJT256 were verified by sequencing.k Plasmids pJT255 and pJT256 were linearized with Not1 and then purified.i Linearized plasmid DNA concentrations were quantified with a spectrophotometer,g and copy numbers were calculated with the following formula:
Tenfold serial dilutions of the linearized plasmid from 2 × 106 to 0.2 copies/μL were made in 20 ng of E maximus DNA/μL for pJT255 or 50 ng of yeast transfer RNA/μL for pJT256. For each point on the standard curve, 5 μL of standard curve solution was used for each real-time qPCR reaction, which was performed by use of EEHV1 primers and hydrolysis probe in master mix.l
Real-time qPCR assay
The primers and probes used for quantitative real-time qPCR were designed with the assistance of computer software.m Primer oligonucleotidesn and custom probeso were synthesized. Real-time qPCR experiments were performed by use of a real-time PCR thermocycler,p and data were analyzed with associated software.q Primers and probes for EEHV1 and IFN-γ were as follows: EEHV1 MDBP forward primer, CGATGATACCCGATCCCTAGTC; EEHV1 MDBP reverse primer, GGCGCCGAAGCTTAGATG; EEHV1 MDBP hydrolysis probe, 6FAM-CAGCACACCGCAAAACCAAAAAATCTTAAA-MGB-NFQ; IFN-γ forward primer, GCGTGAAGACCCTTGAGGAA; IFN-γ reverse primer, TCATTTACCGGAGTTTGCATCA; and IFN-γ hydrolysis probe, VIC-TTCTTCAATAGCAGCTCC-MGB-NFQ. The qPCR reactions were performed with a 20-μL total reaction volume resulting from 10 μL of master mix,l,r,s a 900nM final concentration of each primer, and a 250nM final concentration of the hydrolysis probe, when used. Amplification of archival EEHV DNA samples was performed by use of a master mix.l
Routine EEHV1 blood-screening tests were performed by use of SYBR green chemistry, and each blood sample was screened for EEHV1 with 200 ng of genomic DNA and IFN-γ with 50 ng of genomic DNA. The IFN-γ serves as an internal amplification control sample to ensure that a quality elephant genomic DNA preparation is being screened and that the PCR master mix used for the assay is adequate. The PCR cycling conditions for real-time qPCR assays with SYBR green detection were as follows: 1 cycle consisting of 95°C for 10 minutes, then 40 cycles of 95°C for 15 seconds followed by 60°C for 1 minute. A melting curve analysis was subsequently performed.
The EEHV1 trunk-wash screening assays were performed with 200 ng of genomic DNA in a multiplex qPCR reaction for EEHV1 and IFN-γ; DNA prepared from trunk-wash samples was amplified by use of a master mix.s Trunk-wash screening assays were performed by use of the presence-absence format of the thermocycler-associated software.q A confidence value of 99.9% was applied to all samples screened. Cycling conditions for real-time qPCR assays with hydrolysis probe detection were as follows: 1 cycle consisting of 50°C for 2 minutes followed by 95°C for 10 minutes, then 40 cycles consisting of 95°C for 15 seconds followed by 60°C for 1 minute.
Amplification and PCR sequencing of trunk-wash DNA samples
Eight DNA samples prepared as described above were first subjected to whole genome amplification by use of a commercial kit.t Two microliters of trunk-wash DNA preparation was used in a 20-μL total amplification reaction volume. This initial step, which was presumed to have the principle effect of diluting PCR inhibitors while maintaining approximately the same viral DNA concentration, proved to be critical for obtaining efficient PCR assay results with the trunk-wash samples.
Two microliters of amplified DNA was then used for each of 3 first-round PCR reactions, along with outside primer pairs specific for 3 selected EEHV1 gene loci representing subsegments of the U38 (POL), U71 (gM), and U51 (vGPCR) genes. The PCR primers used to amplify the EEHV1 POL locus were as follows: first round (530 bp), LGH7445 (GATTTTGCGAG[C/T]CTGTA[C/T]CC) and LGH7446 (CACGCTGTCAGTATCTCCGTA); second round A (500 bp), LGH7446 and LGH7447 (CCCAGTATCATTCAAGCATAC); second round B (500 bp), LGH7445 and LGH7448 (CTGTCTACAGGGCA[A/G]TCAAC); and third round (480 bp), LGH7447 and LGH7448. The PCR primers used to amplify the EEHV1 gM locus were as follows: first round (750 bp), LGH6749 (CTATGGGATCCGAACTTTC) and LGH6750 (CTTTCTAAGGGGGTTTGTTGC); second round A (730 bp), LGH6749 and LGH6752 (CTACATGCCCATGCAGATAGG); second round B (700 bp), LGH6750 and LGH6751 (GAAGTCCTGCTAGCCCC[C/T]TAC); and third round (710 bp), LGH6751 and LGH6752. The PCR primers used to amplify the EEHV1 vGPCR locus were as follows: first round (930 bp), LGH7506 (GATTGTGAACGCTGTATGTC) and LGH4963 (GACTTTCTTCGTCGTAGCCCTCGTCTT); second round A (880 bp), LGH7506 and LGH5200 (CGTGATACGCTTCCAAACATACA); second round B (700 bp), LGH7470 (GGTGGTACTGTATGATGTGC) and LGH4963; and third round (550 bp), LGH7470 and LGH5200.
For multiple-round PCR reactions, 2 μL of a 20-μL PCR reaction was used in subsequent PCR reactions. All PCR amplification reactions involved the following conditions: 1 cycle consisting of 95°C for 2 minutes; then 45 cycles consisting of 95°C for 40 seconds, 50°C for 45 seconds, and 73°C for 1 minute; then 1 cycle consisting of 73°C for 5 minutes. All PCR product bands were purified as isolated agarose gel bands by use of an extraction kit.u The DNA sequencing was carried out by direct cycle sequencing on both strands with a cycle sequencing kitv and analyzed on a DNA sequencer.w Resulting DNA sequences totaling 500 bp for POL, 700 bp for gM, and 840 bp for vGPCR were analyzed and edited.x The assembled sequences were then compared with previous sequencing data for the same 3 gene loci from 22 samples of EEHV from North American and European elephants with acute EEHV-associated hemorrhagic disease by use of a software program.9,y
GenBank accession numbers
For consistency in future publications, elephants 2, 3 and 4 have been designated NAP#36, NAP#37, and NAP#38, respectively. Data files for all sequence data regarding the gene subtyping analysis were deposited in GenBank under the following accession numbers: NAP#32 EEHV1A, U38/POL, 491 bp, GU350751; NAP#14 EEHV1B, U38/POL, 491 bp, GU350752; NAP#36 EEHV1A, U38/POL, 491 bp, GU350754; NAP#37 EEHV1A, U38/POL, 491 bp, GU350755; NAP#38 EEHV1A, U38/POL, 491 bp, GU350756; NAP#32 EEHV1A, U51/vGPCR, 876 bp, GU350760; NAP#14 EEHV1B, U51/vGPCR, 885 bp, GU350759; NAP#36 EEHV1A, U51/vGPCR, 876 bp, GU350761; NAP#37 EEHV1A, U51/vGPCR, 876 bp, GU350762; NAP#38 EEHV1A, U51/vGPCR, 888 bp, GU350763; NAP#32 EEHV1A, U71/gM, 648 bp, GU350766; NAP#14 EEHV1B, U71/gM, 637 bp, GU350767; NAP#36 EEHV1A, U71/gM, 648 bp, GU350769; NAP#37 EEHV1A, U71/gM, 648 bp, GU350770; and NAP#38 EEHV1A, U71/gM, 642 bp, GU350771.
Results
To detect the viral genomes of EEHV1A and EEHV1B, we identified a gene sequence encoding the MDBP that was 99% identical at the nucleotide level between the 2 virus subtypes. The qPCR primers and probe sequences used for the EEHV1 assay were 100% conserved between EEHV1A and EEHV1B (Figure 1). Through use of standard curves containing known amounts of EEHV1A MDBP DNA (Figure 2), the limit of detection was established as 10 copies/reaction with 100% sensitivity in curves containing 5 replicates/dilution point for both SYBR green detection (CT = 33.0) and hydrolysis probe detection methods (CT = 35.5). The assay was capable of detecting 1 copy of target DNA/reaction; however, the sensitivity of the assay decreased to approximately 75% (data not shown). Both assays yielded statistical slopes, y-intercepts, coefficients of determination, and PCR efficiencies within acceptable limits. Considering a mean genomic DNA yield of 5 μg of DNA from 200 μL of whole blood and that 200 ng of DNA/screening qPCR was used, the limit of detection (with 100% sensitivity) in blood samples was established as 1,200 VGCs/mL of whole blood.
Figure 1.
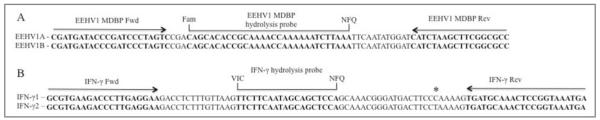
Aligned nucleotide sequences of EEHV1A and EEHV1B (A) and Elephas maximus IFN-γ (B) with locations of qPCR primers and hydrolysis probes for a real-time qPCR assay. The IFN-γ 1 sequence was obtained from GenBank accession number EU000432, and the IFN-γ 2 sequence was obtained from GenBank accesion number EF203241. The location of an SNP within the known E maximus IFN-γ nucleotide sequences is indicated with an asterisk. Fam = Carboxyfluorescein. Fwd = Forward. NFQ = Nonfluorescent quencher. Rev = Reverse. VIC = Proprietary fluorophore.z
Figure 2.
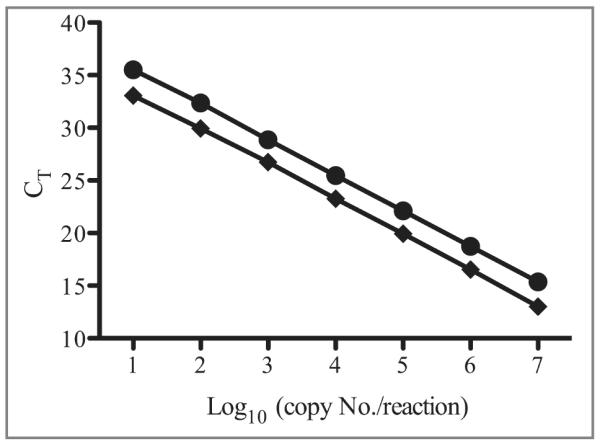
Standard curves for 2 types of real-time qPCR assays of EEHV1 MDBP plasmid diluted in E maximus DNA. Each standard curve is representative of 3 independent experiments. The assay performed with SYBR green detection (diamonds) yielded the following reaction parameters: regression equation, y = −3.338x + 36.57; R2 = 0.999; assay efficiency, 99.4%. The assay performed with hydrolysis probe (circles) yielded the following reaction parameters: y = −3.376x + 38.99; R2 = 0.999; assay efficiency, 97.8%.
Specificity assay
To determine the specificity of the real-time qPCR assay for EEHV1A and EEHV1B, we obtained archived DNA samples derived from whole blood or tissues from elephants that had been diagnosed with EEHV and in which the viral strain had been identified. The DNA amounts from archived DNA samples used for the specificity assay were the same amounts as those used in the EEHV PCR clinical test that was used to originally diagnose each affected elephant. In samples from all cases of EEHV1A and EEHV1B assayed, including a case involving an EEHV1A/B chimeric genome, the test provided a positive result with the SYBR green and hydrolysis probe detection systems. The EEHV1 hydrolysis probe assay did not detect any viral DNA from samples taken from cases involving EEHV3 and EEHV4. Although the hydrolysis probe assay provided a clear distinction between positive and negative (CT = undetermined) samples, the SYBR green detection system required further analysis of a melting curve to determine a negative result. Because the mean negative CT for non-EEHV1A or 1B samples was 33.64, which was within the detectable range for EEHV1, an analysis of the melting curves for the non-EEHV1A or 1B samples was required to determine whether the amplicons could be nonspecific PCR products. Analysis of the melting curve revealed that the amplicons generated did not denature at the expected melting temperature for the target amplicon; therefore, they were judged nonspecific PCR products (data not shown).
Real-time qPCR assay
The real-time qPCR assay also provided quantitative results for each sample tested. The quantitation from SYBR green (data not shown) and hydrolysis probe (Table 1) detection systems correlated well, with a range of disparity between the calculated values from the same sample of 1 to 2.4 times, based on the number of VGCs per nanogram of test DNA (data not shown). The range of VGCs per milliliter of blood ranged from 1 × 104 to 2.6 × 107, with elephants that died of EEHV1 having higher blood viral loads than those that were subclinically infected or clinically ill from the disease. Our calculated limit of detection in blood samples (1,200 VGCs/mL) was lower than the viral load calculated from all the elephant blood samples screened, which included subclinically infected, clinically ill, and deceased elephants.
Table 1.
Results of an EEHV1 real-time qPCR assay designed to specifically detect EEHV1A and EEHV1B in various samples from elephants with NAP infection for which archived samples were available.
| Elephant (source) | EEHV strain |
SYBR green CT* |
Hydrolysis probe CT† |
Test result | No. of VGCs‡ | VGCs/mL of blood§ | Clinical result |
|---|---|---|---|---|---|---|---|
| First archive elephant (heart) |
1A | 28.26 | 36.61∥ | Positive | 18 | NA | Death |
| First archive elephant (spleen) |
1A | 24.90 | 28.94∥ | Positive | 3 × 103 | NA | Death |
| NAP#26 (heart) | 1A | 18.81 | 22.92 | Positive | 1.8 × 105 | NA | Death |
| NAP#29 (heart) | 1A | 24.31 | 31.28∥ | Positive | 654 | NA | Death |
| Second archive elephant (blood) |
1A | 30.91 | 35.40 | Positive | 41 | 1 × 104 | Clinically ill |
| NAP#31 (blood) | 1A | 21.11 | 23.72 | Positive | 1 × 105 | 2.6 × 107 | Death |
| NAP#19 (blood) | 1A/B | 23.95 | 27.92 | Positive | 1.3 × 104 | 3.4 × 106 | Death |
| NAP#33 (blood) | 1B | 23.19 | 28.72 | Positive | 8 × 103 | 2 × 106 | Clinically ill |
| NAP#34 (blood) | 1B | 27.24 | 32.45 | Positive | 647 | 1.6 × 105 | Subclinically infected |
| NAP#27 (kidney) | 3 | 33.32 | U | Negative | NA | NA | Death |
| NAP#22 (blood) | 4 | 33.28 | U | Negative | NA | NA | Death |
SYBR green reaction parameters: regression equation, y = −3.324x + 37.181; R2 = 0.999; assay efficiency, 99.9%.
Hydrolysis probe reaction parameters: regression equation, y = −3.421x + 40.911; R2 = 0.999; assay efficiency, 96.0%.
Double-stranded copies of target DNA in individual test reactions based on a qPCR assay with hydrolysis probes; equivalent to VGCs.
Viral genome copy per milliliter of blood calculated on the basis of known amount of genomic DNA yield from 200 μL of whole blood used in clinical assays.
Because of a limited sample supply, samples used in hydrolysis probe assays were diluted 1:10 in double-distilled water relative to SYBR green.
NA = Not applicable. U = Undetectable (samples did not yield a reaction that crossed the threshold signaling a productive PCR resulting in an undetermined CT; CT for nontemplate control reactions was undetermined for all hydrolysis probe assays and was ≥ 35 for SYBR green assays).
EEHV screening protocol
Using the screening assay, we developed a protocol to screen for the presence of EEHV1 DNA in trunk-wash samples collected from a herd of healthy Asian elephants (Table 2). During the screening period, we were able to detect the presence of EEHV1 at a frequency of 31% for the entire herd, with individual elephants yielding positive trunk-wash samples with various frequencies (Table 3). The elephants that had positive trunk washes most often were pregnant elephants (elephants 2 and 3). Elephant 2 had positive trunk-wash samples consistently from June 22 through August 24, 2009, and then yeilded no positive samples between August 26 and September 28. Elephant 3 had positive trunk-wash samples throughout the screening period. Elephant 4 yielded only 2 positive trunk-wash samples during the entire screening period. No viral DNA was detected in samples from elephant 1 or 5. Importantly, all EEHV1 qPCR assays performed on whole blood obtained from these elephants at the same time as the trunk washes failed to detect EEHV1 viremia when SYBR green chemistry was used (data not shown).
Table 2.
Results of detection of EEHV1 DNA in trunk-wash samples obtained from 5 captive Asian elephants of the same herd, from June through August 2009.
| Date of test (mo/d) |
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Elephant No. |
6/22 | 6/29 | 7/6 | 7/13 | 7/20 | 7/27 | 8/3 | 8/10 | 8/14 | 8/17 | 8/19 | 8/21 | 8/24 | 8/26 | 8/28 | 9/3 | 9/8 | 9/10 | 9/14 | 9/21 | 9/28 |
| 1 | − | − | − | NR | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 2 | + | + | + | + | + | +* | +† | +† | +† | + | + | + | + | NR | − | − | − | − | − | − | − |
| 3 | − | +† | + | + | +† | +† | − | NR | NT | + | NT | NT | + | + | − | − | + | + | + | + | + |
| 4 | − | − | − | +† | − | − | − | − | NT | + | NT | NT | − | NT | − | NT | − | NT | − | − | − |
| 5 | − | − | − | − | − | − | − | − | NT | − | NT | NT | − | NT | − | NT | − | NT | − | − | − |
Positive result obtained with SYBR green methods (CT = 33.9).
Gene subtyping analysis was available on selected positive samples (see tables 4).
− = Test result indicated EEHV1 was absent, with 99.9% confidence. + = Test result indicated EEHV1 was present, with 99.9% confidence in all wells tested in triplicate. NR = No result from assay because of lack of amplification of internal positive control sample (IFN-γ). NT = No test performed on this date.
Test triplicates with a mean CT ≥ 36.2 were deemed negative. Mean positive test triplicate CT values ranged from 26.6 to 36.2.
Table 3.
Frequency of detection of EEHV1A or EEHV1B DNA in trunk-wash samples obtained from 5 captive Asian elephants of the same herd.
| Elephant No. |
Age (y) | Origin | Sex | Total No. of samples assayed |
No. (%) of positive samples |
|---|---|---|---|---|---|
| 1 | 4 | CB | M | 20 | 0 |
| 2 | 28 | WB | F (pregnant) | 20 | 12 (60) |
| 3 | 19 | CB | F (pregnant) | 17 | 13 (76) |
| 4 | 40 | WB | F | 15 | 2 (13) |
| 5 | 44 | WB | M | 15 | 0 |
| Herd total | NA | NA | NA | 87 | 27 (31) |
CB = Captive born. F = Female. M = Male. NA = Not applicable. WB = Wild born.
DNA sequencing
Through viral DNA sequence analysis and EEHV1 gene subtyping from select positive samples, 2 distinct EEHV1 genomes were identified as being shed from the elephants within the study herd (Table 4). One of the viral genomes was identical to that associated with a previous case of EEHV1A-associated death within this elephant herd, whereas the other genome was unique when compared with all previously identified EEHV genomes. The 3 loci examined for gene subtyping were U38 (POL), U51 (vGPCR), and U71 (gM). Three separate trunk-wash DNA samples from which qPCR-positive results were obtained (elephants 2 and 3), taken from different screening dates (Table 2), were analyzed for EEHV gene subtypes. All 3 trunk-wash DNA samples from elephant 2, but not those from elephant 3 or 4, were positive after the first-round PCR assay (Figure 3). For all samples, 2 μL of the first-round 20-μL PCR assay was then further incubated in a second-round PCR assay with 2 separate pairs of seminested primers for each locus. All 3 samples from elephant 3 and 1 of the 2 from elephant 4 also then yielded correct-sized products from all 3 loci, but only the POL locus was positive for all 8 samples. A blood sample containing a low concentration of EEHV1B DNA from case NAP#34 was also amplified after the second-round PCR assay, whereas a negative control sample of Asian elephant DNA failed to be amplified. Finally, a third-round, fully nested PCR assay was required to produce correct-sized PCR bands for gM and vGPCR for the additionally purified 7/13 DNA sample from elephant 4 (data not shown).
Table 4.
Results of EEHV1 gene subtyping in trunk-wash samples from 3 captive Asian elephants (Nos. 2, 3, and 4) and archived samples from other elephants with fatal EEHV1 infection.
| Gene subtype |
|||
|---|---|---|---|
| Elephant No. | POL* | vGPCR | gM |
| 2 | A | D2 | A |
| 3 | A | D2 | A |
| NAP#32† | A | D2 | A |
| NAP#23† | A | A1 | A |
| NAP#17† | A | D2 | D2 |
| 4 | B | A/B | B |
| NAP#33 | B | E/B | B |
| NAP#34 | B | E/B | B |
| NAP#19 | B | B | A |
| NAP#14 | B | B | BΔ |
Locus (0.55 kilobase) discriminates EEHV1A from EEHV1B subtypes.
Previous EEHV-associated fatalities, within the screened herd, for which gene subtyping analysis is available. Letters denoting viral gene subtypes have been arbitrarily assigned to denote the unique nucleotide sequence of each gene subtype.
Figure 3.

Photographs of electrophoretic gels for results of PCR amplification of EEHV1 loci for gene subtyping analysis (vGPCR and POL subtypes) in trunk-wash DNA preparations from study elephants (Nos. 2 through 4) and archived samples from other elephants from the same herd. Lane assignments (numbers below each gel) and dates (mo/d) samples were obtained were as follows: 1 (8/14), elephant 2; 2 (7/20), elephant 3; 3 (6/29), elephant 3; 4 (7/13), elephant 4; 5 (7/13), elephant 4; 6 (7/27), elephant 3; 7 (8/3), elephant 2; 8 (8/10), elephant 2; 9, elephant NAP#34 (archived); and 10, uninfected elephant. Unnumbered lanes indicate molecular-weight markers with size (base pairs) indicated to the left of the gel. The DNA samples analyzed in lanes 4 and 5 represent independent DNA preparations from the same trunk wash obtained on July 13, 2009, from elephant 4.
Comparison of identified DNA sequences with those of different samples of EEHV from North American and European elephants with acute EEHV-associated hemorrhagic disease revealed all 3 trunk-wash DNA samples from elephant 2 and all 3 from elephant 3 were identical, and both had a 100% match with the previous data for a single EEHV1A sample (EEHV-associated hemorrhagic disease case NAP#32) from the same facility. However, sequences differed significantly from those of the other 17 known EEHV1A strains. In contrast, the 2 DNA samples from elephant 4 were fully matched with each other but represented a novel EEHV1B strain, distinct from the other 3 known examples of EEHV1B strains (NAP#15, NAP#19, and NAP#33/34).
Discussion
In the present study, a real-time qPCR assay was developed to detect the presence of pathogenic EEHVs in trunk-wash samples obtained from healthy Asian elephants. This is the first report of pathogenic EEHVs being reproducibly detected in fluid samples obtained from healthy elephants. To our knowledge, this study represented the first attempt to screen elephant trunk-wash samples for elephant herpesviruses.
Within individual elephants, detection of EEHV1 DNA in trunk-wash samples varied over the screening period (Table 1). Detection frequency for EEHV1 among individual elephants varied from 0% to 76%. A variation in the frequency with which people reactivate and shed virus is a common finding in studies19,20 of several human herpesviruses. The overall frequency of detection within the elephant herd in our study was 31%. The elephants that were found to shed the most often were pregnant females; however, there were insufficient data to determine whether pregnant elephants were more likely to shed EEHV1 when compared to nonpregnant elephants. Analysis of a larger number of elephants, including pregnant and nonpregnant females, will be required to evaluate such associations. It would also be interesting to evaluate elephants 2 and 3 once they have given birth. The available data suggested that EEHVs, previously associated only with lethal infection of Asian elephants, are capable of producing subclinical, persistent infections with periods of reactivation and shedding of the virus in healthy adult Asian elephants.
Our data also provided the first epidemiological link connecting the death of an elephant from herpesvirus-associated disease with the shedding of EEHV by herd mates. By use of extensive DNA sequence data collected from historical cases of EEHV from the study herd, we were able to compare viruses shed by elephants through nasal secretions with those from past cases of EEHV infection within the herd. Among the 3 elephants shedding EEHV, 2 (elephants 2 and 3) were shedding an identical EEHV1A virus, and elephant 4 was shedding a unique EEHV1B virus (Table 4). Comparison with viral DNA from past cases of elephant herpesvirus-associated disease within this herd of elephants revealed that the viruses shed by elephants 2 and 3 were identical to the DNA isolated from tissue samples from EEHV1A case NAP#32, an elephant calf that died of EEHV1A while all study elephants were present in the herd. No viruses from previous cases of EEHV infection, either within this herd or throughout North America or Europe, are known to match the virus shed by elephant 4.
Several scenarios are possible for the course of events that led to the infection of elephants 2, 3, and NAP#32. One scenario is that elephant 2 was the source elephant and, upon entry into the herd, infected elephants NAP#32 and 3. Another possibility is that elephant 3 was the source elephant and infected elephants NAP#32 and 2 upon its entry into the herd. We cannot rule out the possibility that elephant NAP#32 was the source of the infection and developed viral reactivation that overwhelmed the immune system. Finally, all 3 elephants may have acquired the infection from a common source that has not yet been determined. In the aforementioned scenarios, a horizontal mode of disease transmission through nasal secretions is possible. The factors that could produce a lethal infection in elephant NAP#32 yet a subclinical, persistent infection in elephant 3 remain unclear. Multiple host factors are likely to play a role in EEHV pathogenicity, including age at exposure to a novel virus, acquisition of passively transferred protective maternal antibodies during gestation or nursing, previous exposure to or latent infection with EGHVs or other EEHV subtypes, and the preexisting presence of immunocompromising disease states. A definitive epidemiological link specifically to elephant 2 or 3 cannot be made for elephant NAP#32. Analysis of other elephant herds with a history of EEHV-associated disease and active screening of herds with susceptible elephants prior to disease transmission would be needed for making such connections. Overall, these data provided evidence supporting the hypothesis that trunk secretions may be a mode of transmission of pathogenic EEHVs and that gene subtyping analysis of viral DNA in trunk-wash secretions obtained from herds with a history of EEHV-associated disease may be useful in establishing epidemiological connections among elephants that shed viruses and those with lethal herpesvirus-associated disease.
The real-time qPCR test developed in the present study was useful for detection of EEHV1 DNA in trunk-wash secretions; however, another study goal was the testing of elephant blood samples for the presence of pathogenic EEHVs. Such an assay has 2 potential, primary uses as a blood-screening assay: to screen clinically ill elephants for the presence of pathogenic EEHV viremia and to routinely monitor healthy elephants for early EEHV1 viremia. Early detection of viremia would make possible early intervention with antiviral medication to reduce morbidity and mortality rates associated with elephant herpesvirus infection. Our data suggested the assay developed yielded a positive result in all the historical cases of EEHV1A and EEHV1B disease evaluated. In addition, the assay appeared to have the potential to detect viremia in elephants prior to the onset of considerable illness. Such evidence was accrued through knowledge regarding the health status of particular elephants when the historical samples were obtained, quantitation of the viral genomes in these samples, and establishment of a limit of detection of 1,200 VGCs/mL of blood. In 3 elephants that were subclinically infected or had clinical signs of EEHV infection but survived the disease, the EEHV blood titer ranged from 1 × 104 VGCs/mL to 1 × 106 VGCs/mL. This viral load is at a minimum 1 log10 higher than our estimated lower limit of detection. Whereas it would have been ideal to evaluate samples obtained from other subclinically or clinically infected but not moribund elephants, such samples are in limited supply.
An important limitation about the reported method of EEHV1 detection is that it does not indicate whether the virus detected is infectious. In addition, it is unknown whether the concentration of virus in trunk washes would be a sufficient infectious dose if the secretions were indeed infectious. Many unsuccessful attempts have been made to cultivate elephant herpesviruses in vitro, thus limiting our ability to assay the infectivity of the virus identified via qPCR assay. Clearly, cultivation of pathogenic EEHVs in vitro not only would provide a valuable tool to further address the possibility that nasal secretions or other fluids provide a medium for transmission of infectious virus, but also would provide an invaluable reagent for the development of a vaccine.
Finally, the presence of PCR inhibitors was consistently detected in our trunk-wash DNA preparations, which potentially limits the sensitivity of the trunk-wash assays. Because of this, we anticipate that the sensitivity of the assay will be less in trunk-wash DNA preparations than that calculated for blood DNA preparations. The presence of PCR inhibitors in various body fluids21,22 is not unusual, and use of the IFN-γ internal amplification control in our study allowed us to identify the inhibition. The presence of PCR inhibitors was suspected when qPCR assays using 200 ng of DNA purified from trunk washes did not yield a positive IFN-γ reaction. The impact of the PCR inhibitors was reduced at least partially by the use of commercial PCR master mix. For example, in an IFN-γ reaction, a CT obtained via use of 200 ng of DNA purified from blood is, on average, 6 cycles less than that obtained from 200 ng of trunk-wash DNA (data not shown). This indicates the trunk-wash DNA preparations contained < 100% E maximus DNA or there were still PCR inhibitors present in the reactions. Until the DNA purification protocol and qPCR assay conditions can be further optimized to remove inhibitors, accurate quantitative analysis of viral DNA in trunk-wash preparations is not possible, and such quantitation will likely be useful only to indicate patterns in virus secretion or large changes (orders of magnitude) in secretion.
Acknowledgments
Supported by Houston Zoo Incorporated, Elephant Managers Association, and National Institutes of Health training grant T32-AI-07471.
Abbreviations
- CT
Cycle threshold
- EEHV
Elephant endotheliotropic herpesvirus
- EGHV
Elephant gammaherpesvirus
- gM
Glycoprotein M
- IFN-γ
Interferon γ
- MDBP
Major DNA-binding protein
- NAP
North American proboscivirus
- POL
DNA polymerase
- qPCR
Quantitative PCR
- VGC
Viral genome copy
- vGPCR
Viral G-protein coupled receptor
Footnotes
Hayward G, Viral Oncology Program, Johns Hopkins University, Baltimore, Md: Unpublished data, 2009.
QIAamp Blood Mini DNA Kit, QIAGEN Inc, Hilden, Germany.
Buffer AE, QIAGEN Inc, Hilden, Germany.
QIAamp DNA Blood Midi/Maxi Kit, QIAGEN Inc, Hilden, Germany.
Ultraspec 2000, GE Healthcare Life Sciences, Piscataway, NJ.
Gentra Puregene Tissue Kit, QIAGEN Inc, Hilden, Germany.
Nanodrop ND-1000 UV spectrophotometer, NanoDrop Technologies, Wilmington, Del.
Generation Capture Column Kit, QIAGEN Inc, Hilden, Germany.
QIAquick PCR Purification Kit, QIAGEN Inc, Hilden, Germany.
pCR-Blunt Cloning Vector, Invitrogen/Life Technologies Inc, Carlsbad, Calif.
SeqWright DNA Sequencing, Houston, Tex.
TaqMan Universal PCR Mastermix, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
Primer Express, version 3.1, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
Integrated DNA Technologies Inc, Coralville, Iowa.
Custom TaqMan probes, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
StepOne thermocycler, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
StepOne, version 2.1, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
POWER SYBR Green PCR Mastermix, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
TaqMan Environmental PCR Mix, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
GenomiPhi HY DNA Amplification Kit, GE Healthcare Life Sciences, Piscataway, NJ.
Qiagen II Gel Extraction Kit, QIAGEN Inc, Hilden, Germany.
ABI PRISM DigDye Terminator cycle sequencing kit, version 3.1, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
ABI-310 DNA sequencer, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
ABI Assembly-Align implemented in MacVector 7.0, Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
ClustalW, version 1.3, Bioinformatics Center, Institute for Chemical Research, Kyoto University, Kyoto 611-0011, Japan. Available at: align.genome.jp. Accessed Apr 27, 2010.
Applied Biosystems/Life Technologies Inc, Carlsbad, Calif.
References
- 1.Ossent P, Guscetti F, Metzler AE, et al. Acute and fatal herpesvirus infection in a young Asian elephant (Elephas maximus) Vet Pathol. 1990;27:131–133. doi: 10.1177/030098589002700212. [DOI] [PubMed] [Google Scholar]
- 2.Richman LK, Montali RJ, Garber RL, et al. Novel endotheliotropic herpesviruses fatal for Asian and African elephants. Science. 1999;283:1171–1176. doi: 10.1126/science.283.5405.1171. [DOI] [PubMed] [Google Scholar]
- 3.Richman LK, Montali RJ, Cambre RC, et al. Clinical and pathological findings of a newly recognized disease of elephants caused by endotheliotropic herpesviruses. J Wildl Dis. 2000;36:1–12. doi: 10.7589/0090-3558-36.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Fickel J, Richman LK, Montali R, et al. A variant of the endotheliotropic herpesvirus in Asian elephants (Elephas maximus) in European zoos. Vet Microbiol. 2001;82:103–109. doi: 10.1016/s0378-1135(01)00363-7. [DOI] [PubMed] [Google Scholar]
- 5.Garner MM, Helmick K, Ochsenreiter J, et al. Clinico-pathologic features of fatal disease attributed to new variants of endotheliotropic herpesviruses in two Asian elephants (Elephas maximus) Vet Pathol. 2009;46:97–104. doi: 10.1354/vp.46-1-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehlers B, Burkhardt S, Goltz M, et al. Genetic and ultrastructural characterization of a European isolate of the fatal endotheliotropic elephant herpesvirus. J Gen Virol. 2001;82:475–482. doi: 10.1099/0022-1317-82-3-475. [DOI] [PubMed] [Google Scholar]
- 7.Reid CE, Hildebrandt TB, Marx N, et al. Endotheliotropic elephant herpes virus (EEHV) infection. The first PCR-confirmed fatal case in Asia. Vet Q. 2006;28:61–64. doi: 10.1080/01652176.2006.9695209. [DOI] [PubMed] [Google Scholar]
- 8.Metzler AE, Ossent P, Guscetti F, et al. Serological evidence of herpesvirus infection in captive Asian elephants (Elephas maximus) J Wildl Dis. 1990;26:41–49. doi: 10.7589/0090-3558-26.1.41. [DOI] [PubMed] [Google Scholar]
- 9.Zong JC, Latimer E, Heaggans S, et al. Analysis of species and strain differences among elephant endotheliotropic herpesviruses by gene subtyping: absence of epidemiological connections among 20 cases of EEHV1 associated with acute hemorrhagic disease in captive Asian elephants. Proceedings; 2010 Int Elephant Conserv Res Symp; 2010. in press. [Google Scholar]
- 10.Richman LK, Montali RJ, Hayward GS. Review of a newly recognized disease of elephants caused by endotheliotropic herpesviruses. Zoo Biol. 2000;19:383–392. doi: 10.7589/0090-3558-36.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Schmitt DL, Hardy DA, Montali RJ, et al. Use of famciclovir for the treatment of endotheliotrophic herpesvirus infections in Asian elephants (Elephas maximus) J Zoo Wildl Med. 2000;31:518–522. doi: 10.1638/1042-7260(2000)031[0518:UOFFTT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 12.Ehlers B, Dural G, Marschall M, et al. Endotheliotropic elephant herpesvirus, the first betaherpesvirus with a thymidine kinase gene. J Gen Virol. 2006;87:2781–2789. doi: 10.1099/vir.0.81977-0. [DOI] [PubMed] [Google Scholar]
- 13.Davison AJ, Eberle R, Ehlers B, et al. The order Herpesvirales. Arch Virol. 2009;154:171–177. doi: 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zong JC, Latimer E, Heaggans S, et al. Pathogenesis and molecular epidemiology of fatal elephant endotheliotropic disease associated with the expanding Proboscivirus genus of the Beta-herpesvirinae. Proceedings; 2007 Int Elephant Conserv Res Symp.2008. pp. 23–35. [Google Scholar]
- 15.Wellehan JF, Johnson AJ, Childress AL, et al. Six novel gamma-herpesviruses of Afrotheria provide insight into the early divergence of the Gammaherpesvirinae. Vet Microbiol. 2008;127:249–257. doi: 10.1016/j.vetmic.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 16.Fickel J, Lieckfeldt D, Richman LK, et al. Comparison of glycoprotein B (gB) variants of the elephant endotheliotropic herpesvirus (EEHV) isolated from Asian elephants (Elephas maximus) Vet Microbiol. 2003;91:11–21. doi: 10.1016/s0378-1135(02)00264-x. [DOI] [PubMed] [Google Scholar]
- 17.Pellett PE, Roizman B. The family Herpesviridae: a brief introduction. In: Knipe DM, Howley PM, editors. Fields’ virology. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2479–2499. [Google Scholar]
- 18.Jacobson ER, Sundberg JP, Gaskin JM, et al. Cutaneous papillomas associated with a herpesvirus-like infection in a herd of captive African elephants. J Am Vet Med Assoc. 1986;189:1075–1078. [PubMed] [Google Scholar]
- 19.Ling PD, Lednicky JA, Keitel WA, et al. The dynamics of herpesvirus and polyomavirus reactivation and shedding in healthy adults: a 14-month longitudinal study. J Infect Dis. 2003;187:1571–1580. doi: 10.1086/374739. [DOI] [PubMed] [Google Scholar]
- 20.Hadinoto V, Shapiro M, Sun CC, et al. [Accessed Jun 14, 2009];The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009 5:e1000496. doi: 10.1371/journal.ppat.1000496. [serial online] Available at: www.plospathogens.org/article/info:doi%2F10.1371%2Fjournal.ppat.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratcliff RM, Chang G, Kok T, et al. Molecular diagnosis of medical viruses. Curr Issues Mol Biol. 2007;9:87–102. [PubMed] [Google Scholar]
- 22.Ochert AS, Boulter AW, Birnbaum W, et al. Inhibitory effect of salivary fluids on PCR: potency and removal. PCR Methods Appl. 1994;3:365–368. doi: 10.1101/gr.3.6.365. [DOI] [PubMed] [Google Scholar]