

Abstract
[VO(Sal-L-tryp)(H2O)] 1 (where sal-L-tryp = N-salicylidene-L-tryptophanate) was used as a precursor to produce the novel complexes, [VO(Sal-L-tryp)(MeATSC)].1.5C2H5OH 2 (where MeATSC = 9-Anthraldehyde-N(4)-methylthiosemicarbazone), [VO(Sal-L-tryp)(N-Ethhymethohcarbthio)].H2O 3 (where N-Ethhymethohcarbthio = (E)-N-ethyl-2-(4-hydroxy-3-methoxybenzylidene)hydrazinecarbothioamide), and [VO(Sal-L-tryp)(acetylethTSC)].C2H5OH 4 (where acetylethTSC = (E)-N-ethyl-2-(1-(thiazol-2-yl)ethylidene)hydrazinecarbothioamide), by reaction with the respective thiosemicarbazone. The chemical and structural properties of these ligands and complexes were characterised by elemental analysis, ESI MS, FT-IR, UV-visible, ESR, 1H and 13C NMR spectroscopy, and X-ray crystallography. DMSO and DMSO-d6 solutions of compounds 1-4 were oxidised in air to produce vanadium(V) species which were verified by ESI MS and 51V NMR spectroscopy. Anti-cancer properties of compounds 2-4 were examined with three colon cancer cell lines, HTC-116, Caco-2, and HT-29, and also with non-cancerous colonic myofibroblasts, CCD18-Co. Compounds 2-3 exhibited less inhibitory effects in the CCD-18Co cells, indicating a possible cytotoxic selectivity towards colon cancer cells. In general, those compounds which exhibited anti-proliferative activity on cancer cells, but did not affect non-cancerous cells, may have a potential in chemotherapy.
Keywords: vanadium(IV), vanadium(V), 51V NMR, ESR spectroscopy, thiosemicarbazone, colorectal cancer
Introduction
The limited efficacy of current treatments for advanced colon cancer serves an impetus for a concerted effort to identify chemo-preventive agents for treatment. This process has always involved metal complexes. Cisplatin is widely used for the treatment of many cancers[1] despite its high toxicity, undesirable side effects, and problems with drug resistance in primary and metastatic cancers.[2] These limitations have spurred a growing interest in novel non-platinum metal complexes that can show anti-cancer properties.[3] Ruthenium has been reported to possess several favourable properties suited to rational anti-cancer drug design[4] and ruthenium complexes of various types are actively studied as metallodrugs as they are believed to have low toxicity and good selectivity for tumours.[5] Recently, we reported the effect of ruthenium(II) complexes with new chelating thiosemicarbazones on growth inhibition of MCF-7 and MDA-MB-231 (breast adenocarcinoma) as well as HCT 116 and HT-29 (colorectal carcinoma) cell lines.[6] Thiosemicarbazones and their metal complexes are used in many applications ranging from pharmacology to nuclear medicine.[7]
Here we present our efforts to expand our study to the non-ruthenium systems. There have been few systematic studies of the use of vanadium compounds as potential anti-cancer agents, for example, there was a report of vanadium(V) complexes with salicylaldehyde semicarbazone derivatives featuring in vitro anti-tumour activity towards kidney tumour cells (TK-10).[8] Also, several vanadium(IV) compounds are known to exhibit anti-cancer activities.[9] Previously, bis(4,7-dimethyl-1,10-phenanthroline)sulfatooxovanadium(IV) (metvan) was identified as the most promising multi-targeted anti-cancer compound with apoptosis-inducing activity.[9c]
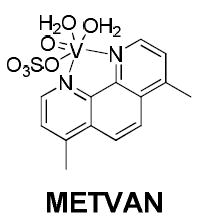
Interestingly, in this study, metvan was found to be highly effective against cisplatin-resistant ovarian and testicular cancer cell lines.[9c] Given the paucity of data regarding the use of vanadium(IV) complexes with thiosemicarbazones as ligands, as potential anti-cancer agents, we now describe in this report the synthesis and chemical characterisation of a series of such complexes and the preliminary results of a biological study against several colorectal cancer cell lines in order to evaluate their potential as chemotherapeutic candidates.
Chemistry and pharmacology
9-Anthraldehyde-N(4)-methylthiosemicarbazone (MeATSC) was synthesised as by Beckford et al.;[6] while two novel thiosemithiocarbazones (E)-N-ethyl-2-(4-hydroxy-3-methoxybenzylidene)hydrazinecarbothioamide (N-Ethhymethohcarbthio) and (E)-N-ethyl-2-(1-(thiazol-2-yl)ethylidene)hydrazinecarbothioamide (acetylethTSC) were also synthesised (Scheme S1, Supporting Information). 2-(2-Hydroxybenzylamino)-3-(1H-indol-3-yl)propanoic acid (the reduced Schiff base) was prepared using a known procedure[10] involving salicylaldehyde, amino acids, and NaBH4, but with L-tryptophan (Scheme S2, Supporting Information). [VO(Sal-L-tryp)(H2O)] 1 (where sal-L-tryp = N-salicylidene-L-tryptophanate) was prepared as by Pessoa et al.[11] [VO(Sal-L-tryp)(MeATSC)].1.5C2H5OH 2, [VO(Sal-L-tryp)(N-Ethhymethohcarbthio)].H2O 3, and [VO(Sal-L-tryp)(acetylethTSC)].C2H5OH 4 were synthesised by reacting [VO(Sal-L-tryp)(H2O)] 1 and the respective thiosemicarbazones (Scheme 1). Compounds 2–4 were isolated in yields ranging from 57 to 78%.
Scheme 1.
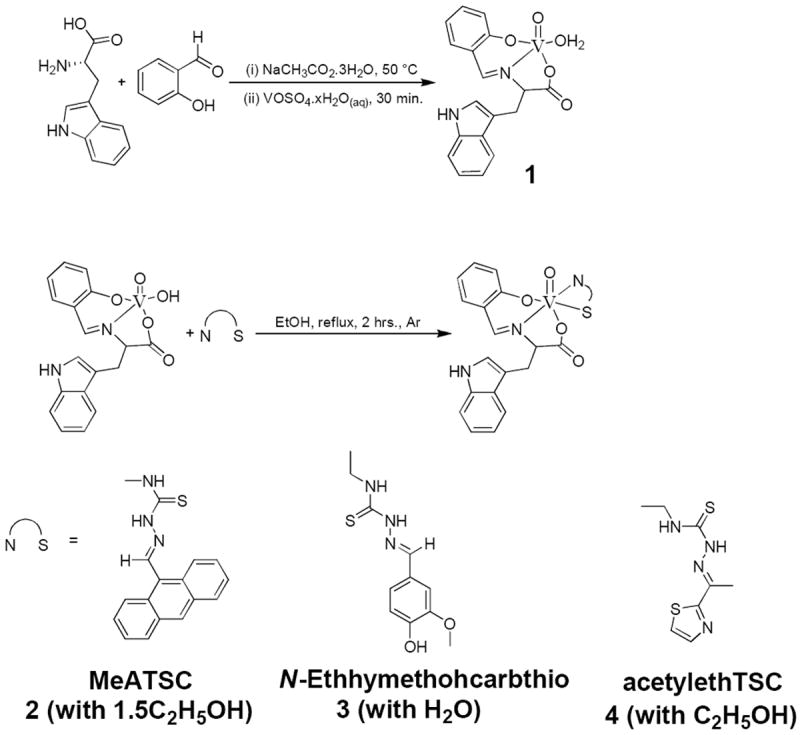
Synthesis of compounds 1–4.
In the present study, we investigated the cytotoxic effects of our complexes against three colon cancer cell lines, HTC-116, Caco-2, and HT-29, along with a comparative anti-proliferative study on non-cancerous colonic myofibroblasts, CCD-18Co using the standard MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfenyl)-2H-tetrazolium, inner salt]-dye reduction assay for cell viability.
Results and Discussion
All compounds were characterised by elemental analysis. The structural components of the ligands and complexes were confirmed by FT IR spectroscopy, 1H and 13C NMR spectroscopy, mass spectroscopy, and X-ray crystallography, where appropriate.
Elemental and Mass spectroscopic analyses
Elemental analyses were carried out on N-Ethhymethohcarbthio and compounds 1–4. The percentage of N found in compound 4 was 10.91% versus the calculated value of 12.98%. The elemental analysis data for the percentage of N is not fully consistent with the calculated value, but other spectroscopic methods have confirmed the identity of compound 4. Due to the fact that the discrepancy is only in the nitrogen, it is more likely due to an error in the analysis process, or the loss of ethanol as solvate during the analytical procedure. However, ESI MS analysis proved the existence of the complex [VO(Sal-L-tryp)(acetylethTSC)] without any ethanol as a solvate.
ESI mass spectra were acquired for compounds 2–4 and the products obtained from oxidised DMSO solutions of compounds 1 and 3 (Figures S1–S9, and Schemes S3–S5 for the respective spectra and proposed fragmentation patterns, Supporting Information). The results thus obtained are in agreement with metal/ligand ratio 1:1 in the investigated complexes. Table 1 shows the ESI MS data acquired for the respective ligands and complexes (see Figures S1–S9, Supporting Information). Overall, the suggested formulae were further confirmed by mass spectral fragmentation analysis.
Table 1.
Mass spectroscopic data of ligands and compounds.
| (A) Reduced Schiff base, K[Sal-L-tryp), and compounds 1-4. | ||||
|---|---|---|---|---|
| Species | Exact Mass [g mol-1] | Species formed in the spectrometer | m/z | Relative Intensity |
| Reduced Schiff base # | 310.13 | [M + H]+ | 311.08 | 100.00 |
| [M − CHO3 + H]+ | 242.42 | 13.04 | ||
| [2M + H]+ | 620.36 | 12.77 | ||
| [3M + H]+ | 930.39 | 5.02 | ||
| [4M + H]+ | 1240.92 | 3.92 | ||
| K[(Sal-L-tryp)] # | 346.07 | [M + H]+ | 344.05 | 39.65 |
| [M − OH − K + H]+ | 300.03 | 100.00 | ||
| [M − OH − O − K + H]+ | 205.20 | 50.43 | ||
| [M − COO − K + H]+ | 242.62 | 64.65 | ||
| [M − COO − OH − K + H]+ | 262.12 | 1.51 | ||
| [M − C7H6O − K + H]+ | 281.14 | 46.08 | ||
| [VO(Sal-L-tryp)(MeATSC)] # | 666.14 | [M + H]+ | 666.17 | 100.00 |
| [M − C2H5N2S + H]+ | 577.08 | 33.10 | ||
| [M − C17H17N3 + H]+ | 405.14 | 31.86 | ||
| [MeATSC + H]+ | 292.14 | 54.60 | ||
| [VO(Sal-L-tryp)(N-Ethhymethohcarbthio)] † | 626.13 | [M − H]- | 624.87 | 100.00 |
| [M − C11H10N2O − H]- | 431.90 | 64.75 | ||
| [M − C11H15N3O2S - H]- | 407.85 | 20.99 | ||
| [[C11H15N3O2S − C8H7N] − H]- | 287.88 | 12.70 | ||
| [N-Ethhymethohcarbthio − H]- | 252.00 | 25.74 | ||
| [VO(Sal-L-tryp)(acetylethTSC)] # | 601.09 | [M + H]+ | 600.25 | 2.71 |
| [M − C3H7NS + H]+ | 513.57 | 100.00 | ||
| [M − C7H5NO + H]+ | 482.38 | 37.11 | ||
| [M − C17H15N2O + H]+ | 336.06 | 4.13 | ||
| (B) Oxidised products isolated from compounds 1 and 3. | ||||
| Species | Exact Mass [g mol-1] | Species formed in the spectrometer | m/z | Relative Intensity |
| Proposed [VO(Sal-L-tryp)(DMSO)(OH)] a, # intermediate | 468.06 | [M + H]+ | 468.60 | 1.67 |
| [M − OH − DMSO − C8H7N + H]+ | 242.41 | 100.00 | ||
| [M − OH + H]+ | 451.05 | 48.25 | ||
| [[V2O3(Sal-L-tryp)2] + H]+ | 763.68 | 30.06 | ||
| [[V2O3(Sal-L-tryp)2(H2O)] + H]+ | 776.77 | 49.36 | ||
| [[V2O3(Sal-L-tryp)2(DMSO)2] + H]+ | 918.28 | 3.13 | ||
| Proposed [VO(Sal-L-tryp)(DMSO)(OH)] b, # intermediate | 468.06 | [M + H]+ | 470.01 | 0.50 |
| [N-Ethhymethohcarbthio + H]+ | 252.26 | 100.00 | ||
| [M − OH + H]+ | 451.96 | 2.97 | ||
| [M − OH + N-Ethhymethohcarbthio + H]+ | 702.75 | 2.15 | ||
| [[V2O3(Sal-L-tryp)2 + H]+ | 763.22 | 0.74 | ||
| [[V2O3(Sal-L-tryp)2(H2O)] + H]+ | 775.40 | 1.08 | ||
| [[V2O3(Sal-L-tryp)2(DMSO)2] + H]+ | 916.44 | 0.75 | ||
positive mode
negative mode
proposed vanadium(V) intermediate obtained from compound 1
proposed vanadium(V) intermediate obtained from compound 3
Oxidovanadium(IV) complexes that contain N-salicylidene-L-aminoacidatos ligands can be oxidised to form vanadium(V) compounds.[12] It is important to determine whether our vanadium(IV) complexes can be oxidised to form vanadium(V) species when dissolved in DMSO. Compounds 1 and 3 were dissolved in DMSO, and the resulting solutions were allowed to evaporate to leave a solid. ESI mass spectral studies were carried out on the remaining solids. In our study, we have found out that compounds 1 and 3 were oxidised to form vanadium(V) species (see Table 1). Vanadium(V) species with coordinated DMSO were detected from ESI MS studies. Scheme 2 shows a proposed mechanism for the formation of the oxidised compounds in DMSO.
Scheme 2.
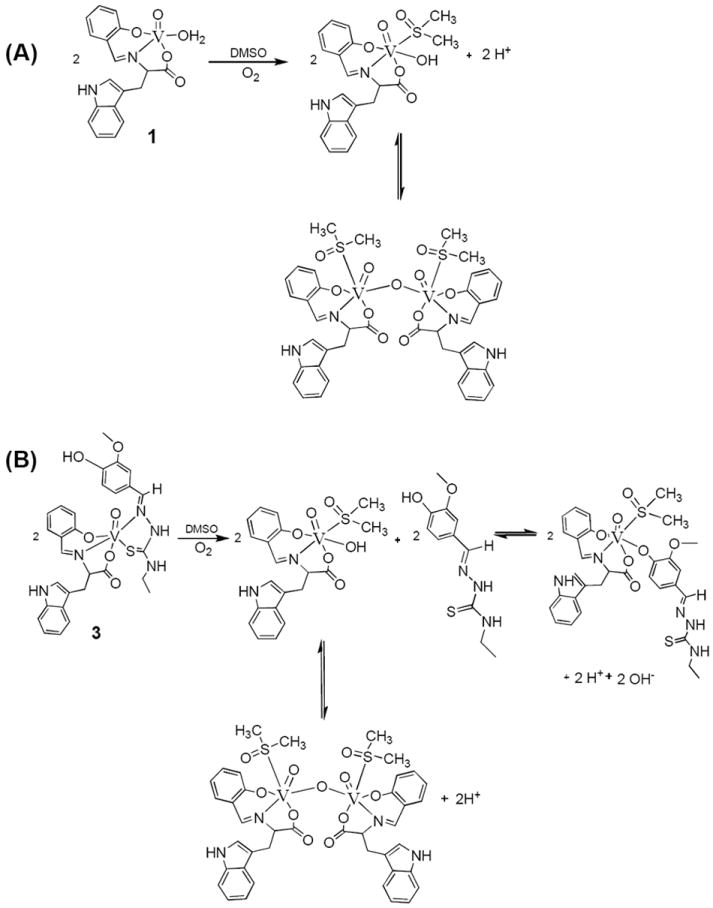
Proposed mechanism for the formation of the oxidised compounds in DMSO.
X-ray crystallographic studies on acetylethTSC
A single crystal of acetylethTSC was grown by slow evaporation of a methanol solution. The molecular structure of acetylethTSC, together with the atomic numbering scheme is shown in Figure 1, while Table 2 shows its crystal data and structure refinement (See Tables S1-S5, Supporting Information, for selected bond lengths and angles).
Figure 1.
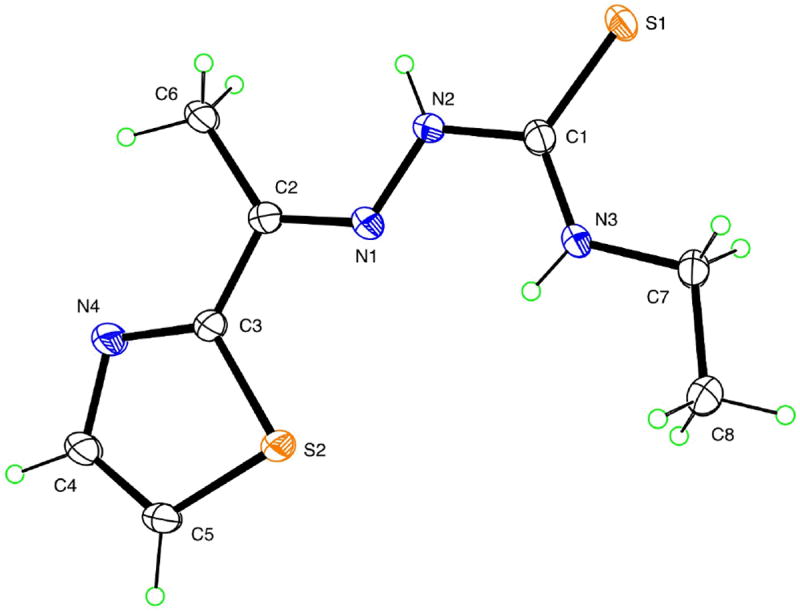
A thermal ellipsoid plot (50% probability envelopes) for acetylethTSC.
Table 2.
Crystal data and structure refinement for acetylethTSC.
| Identification code | acetylethTSC |
|---|---|
|
| |
| Empirical formula | C8H12N4S2 |
|
| |
| Formula weight | 228.34 |
|
| |
| Temperature | 90.0(5) K |
|
| |
| Wavelength | 1.54178 Å |
|
| |
| Crystal system, space group | Monoclinic, C2/c |
|
| |
| Unit cell dimensions | a = 10.4549(10) Å, α = 90° |
| b = 10.8970(10) Å, β = 91.544(5)° | |
| c = 18.7694(15) Å, γ = 90° | |
| Volume 2137.6(3) Å3 | |
|
| |
| Z, Calculated density | 8, 1.419 Mg/m3 |
|
| |
| Absorption coefficient | 4.251 mm-1 |
|
| |
| F(000) | 960 |
|
| |
| Crystal size | 0.18 × 0.17 × 0.09 mm |
|
| |
| Theta range for data collection | 5.8 to 68.1° |
|
| |
| Limiting indices | -12<=h<=12, -12<=k<=13, -22<=l<=22 |
|
| |
| Reflections collected / unique | 8017 / 1901 [Rint = 0.026] |
|
| |
| Completeness to theta = 66.6° | 99.1% |
|
| |
| Absorption correction | Semi-empirical from equivalents |
|
| |
| Max. and min. transmission | 0.701 and 0.515 |
|
| |
| Refinement method | Full-matrix least-squares on F2 |
|
| |
| Data / restraints / parameters | 1901 / 0 / 136 |
|
| |
| Goodness-of-fit on F2 | 1.065 |
|
| |
| Final R indices [I>2σ(I)] | R1 = 0.0276, wR2 = 0.0691 |
|
| |
| R indices (all data) | R1 = 0.0312, wR2 = 0.0715 |
|
| |
| Extinction coefficient | 0.00011(3) |
|
| |
| Largest diff. peak and hole | 0.257 and -0.262 e Å -3 |
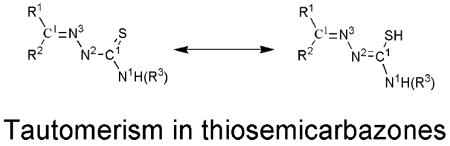
Thiosemicarbazones are known to exhibit thione-thiol tautomerism.[6] The Ortep diagram shows that acetylethTSC exists as a thione in the solid state. The C(1)-S(1) distance (1.6814 Å) is similar to that found in a methyl analogue of 2-acetyl-2-thiazoline thiosemicarbazone which is in the thione form as a solid.[13] AcetylethTSC adopts a syn conformation about the N1–N2 bond in a similar fashion as those thiosemicarbazones that were formed from 2-acetylthiazole as reported by Venkatraman et al.[14] The N2-H group donates an intermolecular hydrogen bond to thione S1, while forming centrosymmetric hydrogen-bonded dimers with N…S distance 3.4651(15) Å. Thiazole N atom N4 is not involved in hydrogen bonding as shown in Figure 2.
Figure 2.
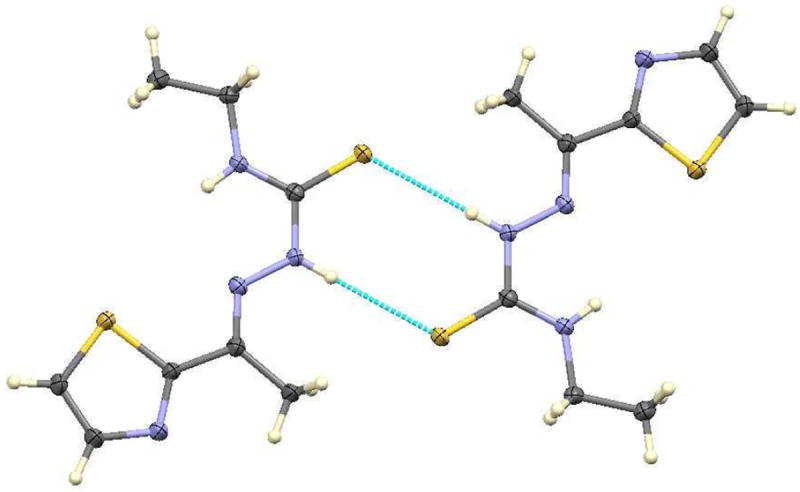
Hydrogen bonding in acetylethTSC.
FT IR Spectroscopy
Infrared spectra were acquired for the ligands and complexes (see Figures S10-S18, Supporting Information). Thiosemicarbazones exhibit characteristic bands corresponding to various functional groups in different energy regions. The most significant infrared bands in the region 4000–500 cm-1 are found between 3100–3500 (assigned to NH stretching vibrations); 1580–1630 (assigned to C=N + NH); and 1100–1300 and 820–900 (assigned to the C=S moiety). Thiosemicarbazones can coordinate as either a neutral (thione) or as a mono-anionic (thiolate) ligand.[15] The ligands (see Figures S10-S12, Supporting Information) in our case all seem to be in the thione form in the solid state [16]. This can be inferred primarily from the absence of a ν(S-H) absorption in the region 2600–2500 cm-1. There are two bands in the ν(N-H) region and the presence of the peak due to the hydrazinic hydrogen confirms the thione formulation. The thiolate form was not evident in our compounds (see tables 3 and 4 for the respective stretching frequencies for the ligands and complexes) in the solid state.
Table 3.
FT IR spectroscopic data for the ligands.
| Compound | Indolic ν(NH) [cm-1] | −N1H2 ν(NH) [cm-1] | N2H ν(NH) [cm-1] | ν(N-N) [cm-1] | ν(C-O) [cm-1] | νs (COO) [cm-1] | νas (COO) [cm-1] | TSC ν(C=N) [cm-1] | Sal-L-tryp ν(C=N) [cm-1] | ν(C=S) [cm-1] | ν(V-S) [cm-1] | ν(OH) [cm-1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MeATSC | - | 3399 (m) | 3201 (w) | 1075 (m) | - | - | - | 1621 (m) | - | 1255 (m), 841 (m) | - | - |
| N-Ethhymethohcarbthio | - | 3304 (m) | 3300 (w) | 1155 (m) | - | - | - | 1606 (m) | - | 1267 (m), 831 (m) | - | 3130 (br) |
| AcetylethTSC | - | 3164 (m) | 3054 (w) | 1059 (m) | - | - | - | 1543 (m) | - | 1296 (m), 813 (m) | - | - |
| K[(Sal-L-tryp)] | 3407 (m) | - | - | - | 1194 (m) | 1521 (m) | 1604 (m) | - | 1570 (m) | - | - | 3169 (br) |
| Reduced Schiff base ligand | 3392 (m) | - | - | - | 1208 (m) | 1494 (m) | 1592 (m) | - | - | - | - | 3055 (br) |
Table 4.
FT IR spectroscopic data for the complexes.
| A) Compound 1-4 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Indolic ν(NH) [cm-1] | −N1H2 ν(NH) [cm-1] | N2H ν(NH) [cm-1] | ν(N-N) [cm-1] | ν(C-O) [cm-1] | νs(COO) [cm-1] | νas(COO) [cm-1] | TSC ν(C=N) [cm-1] | Sal-L-tryp ν(C=N) [cm-1] | ν(C=S) [cm-1] | ν(V-S) [cm-1] | ν(V=O) [cm-1] | ν(OH) /cm-1 |
| 1 | 3478 (m) | - | - | - | 1540 (m) | 1490 (m) | 1597 (s) | - | 1600 (m) | - | - | 997 (s) | 3014 (br) |
| 2 | 3200 (m) | 3340 (m), | 2976 (w) | 1148 (m) | 1544 (m) | 1491 (m) | 1600 (s) | 1624 (m) | 1590 (m) | 1225 (m), 829 (m) | 455 (m) | 980 (s) | - |
| 3 | 3210 (m) | 3306 (m) | 2970 (w) | 1154 (m) | 1540 (m) | 1480 (m) | 1600 (s) | 1620 (m) | 1580 (m) | 1270 (m), 802 (m) | 454 (m) | 976 (s) | 3010 (br) |
| 4 | 3229 (m) | 3310 (m) | 2976 (w) | 1149 (m) | 1545 (m) | 1480 (m) | 1600 (s) | 1624 (m) | 1580 (m) | 1287 (m), 819 (m) | 455 (m) | 982 (s) | - |
| B) Oxidised products isolated from compounds 1 and 3 | |||||||||
| Compound | Indolic ν(NH) [cm-1] | ν(C-O) [cm-1] | νs(COO) [cm-1] | νas(COO) [cm-1] | TSC ν(C=N) [cm-1] | Sal-L-tryp ν(C=N) [cm-1] | ν(V=O) [cm-1] | ν(OH) [cm-1] | ν(S=O) [cm-1] |
| A | 3185 (m) | 1551 (m) | 1497 (m) | 1618 (s) | - | 1581 (m) | 949 (s) | 3194 (br) | 1164 (m), 1213 (m) |
| B | 3201 (m) | 1550 (m) | 1497 (m) | 1600 (s) | 1666 (m) | 1590 (m) | 946 (s) | 3193 (br) | 1133 (m), 1157 (m) |
A-oxidised product obtained from compound 1
B-oxidised product obtained from compound 3
Infrared spectra of all the complexes (see Figures S15-S18, Supporting Information) show a band in the ν(N-H) region which may be attributed to the hydrazinic nitrogen thus suggesting that the ligands are coordinated as the thione form. It must be noted that neither the hydrazinic nor the terminal (aminic) hydrogen has shifted significantly upon coordination of the ligand. The ligands all show a medium intensity band in the 1620–1605 cm-1 region, and this is assigned as the C=N (imine) linkage. On coordination of the ligand, this band shifts significantly for the acetylethTSC ligand (1607 to 1621 cm-1 in compound 4) and for the N-Ethhymethohcarbthio ligand (1605 to 1616 cm-1 in compound 3) ligands. The stretching frequency of the MeATSC ligand changed slightly on coordination to compound 1. The shift in this band is indicative of the iminic nitrogen being involved in the ligation.[7b, 17] The involvement of the thiocarbonyl group in binding can be similarly inferred from the fact that the peaks due to frequency changes to lower wave numbers (6 cm-1 for compound 2, 11 cm-1 for compound 3, and 2 cm-1 for compound 4) on coordination of the respective ligands to compound 1. The magnitude of the shifts suggest that the ligands coordinate as the neutral, bidentate (through the iminic nitrogen and the thiocarbonyl sulphur) form in all the complexes.
A ν(V-S) stretching frequency is observed in the region 470 – 400 cm-1 for all complexes. The absence of a similar band in the spectra of the free thiosemicarbazone ligands, confirms coordination via the thiocarbonyl sulphur atom. IR spectra of the reduced Schiff base and K[Sal-L-tryp)] (see Figures S13 and S14, Supporting Information) are used to assign the important stretching frequencies due to the coordination of the Schiff base moiety. A phenolate ν(C-O) stretching frequency in the region 1545-1540 cm-1 is observed for compounds 1–4. A broad ν(OH) stretching frequency is observed at 3014, 3010, and 3256 cm-1 for compounds 1 and 3, respectively. The absence of phenol’s ν(OH) stretching frequencies in compounds 2 and 4 confirms coordination of the ligand to the vanadium(IV) metal centre via the phenolate anion.[18]
All complexes exhibited ν(V=O) stretching frequencies in the region 997–980 cm-1, which is the typical range for oxidovanadium(IV) complexes.[19] Upon coordination of the respective thiosemicarbazone ligands to compound 1, a lowering of the V=O stretching frequency occurred, for example, a change of 17 cm-1 in compound 2, 21 cm-1 in compound 3, and 15 cm-1 in compound 4.
NMR spectral studies
1H and 13C NMR spectroscopy
1H and 13C NMR spectra were acquired for N-Ethhymethohcarbthio ligand, acetylethTSC, the reduced Schiff base, K[Sal-L-tryp)], and the oxidized products derived from compounds 1 and 3 (Figures S19-S26, Supporting Information). Figure 3 shows the 1H NMR spectra for N-Ethhymethohcarbthio ligand and acetylethTSC.
Figure 3.
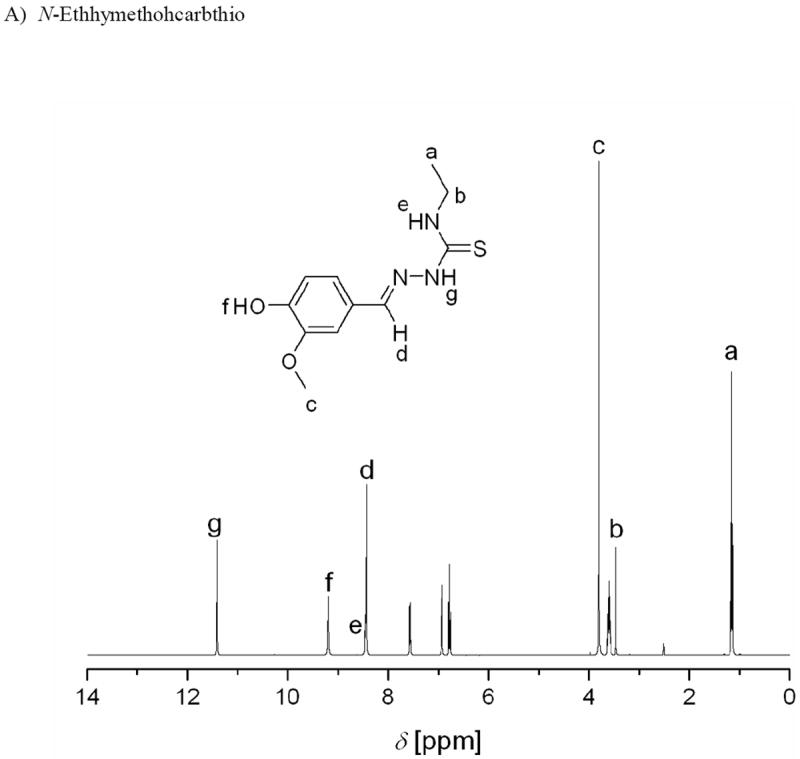
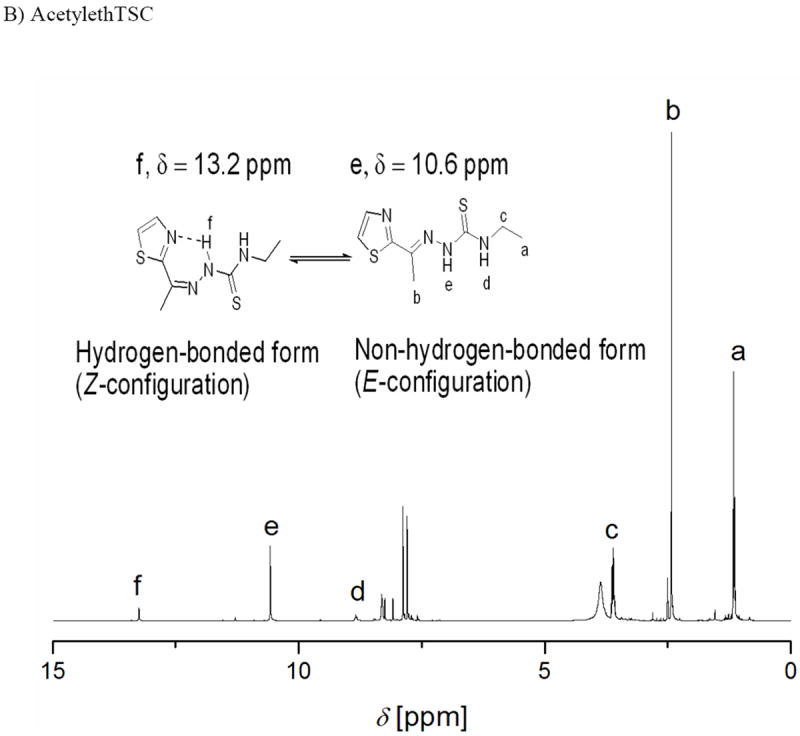
1H NMR spectra of N-ethhymethohcarbthio and acetylethTSC in DMSO-d6.
N-Ethhymethohcarbthio and acetylethTSC
The presence of singlets at 11.42 ppm and 10.59 ppm in the 1H NMR spectra of N-Ethhymethohcarbthio and acetylethTSC, respectively, are labelled as g in figure 3A and as e in figure 3B, respectively.[20] It is common for this signal to be used as a diagnostic test for the identification of E and Z isomers.[6] According to Afrasiabi,[21] the chemical shift δ = 13–15 ppm proves that the ligand is in the E form, whereas δ = 9–12 ppm proves that the ligand is in the Z form. Based on this analysis and coupled with the lack of a resonance signal at ca. δ = 4.0 ppm which is attributed to a −SH proton resonance, it can be inferred that both ligands exist as the Z isomer. An X-ray crystallographic study on acetylethTSC confirms the existence of the thione moiety in the “free” ligand. We also believe that the chemical shift of 13.2 ppm for acetylethTSC (see figure 3B) is due to equilibrium in solution between the hydrogen-bonded (Z-conformation) and the non-hydrogen-bonded (E-conformation) forms of acetylethTSC.
Oxidised products isolated from Compounds 1 and 3
It is important to determine the species that are formed when compounds 1 and 3 are oxidised in DMSO. In order to determine such species, 1H NMR spectra were acquired on the isolated oxidised products. All 1H NMR spectra exhibited broad signals (see Figures S21 and S22, Supporting Information). This phenomenon is attributed to the presence of some residual paramagnetic vanadium(IV) species which could be due to vanadium(IV) species which are in equilibrium with the vanadium(V) species that are formed upon aerial oxidation in DMSO.
The 1H NMR spectrum of the oxidised product isolated from compound 1 exhibited resonance signals that are due to aromatic protons in the region δ = 7.06 to 7.52 ppm. The 1H NMR spectra of the oxidised products isolated from compounds 1 and 3 exhibited resonance signals in the region δ = 7.82 to 8.09 ppm, and δ = 7.06 to 10.43 ppm, respectively. These resonance signals are assigned to the proton of the azomethine group and the −NH proton of the indole ring of the L-tryptophan moiety, respectively. The resonance signal corresponding to the N(2)-H proton of the N-Ethhymethohcarbthio ligand in the oxidised product isolated from compound 3 are overlapped with the resonance signal assigned to −NH proton of the indole ring of the L-tryptophan moiety.
Chemical shifts are observed at δ = 1.17 ppm and δ = 3.79 ppm, respectively, for the oxidised product isolated from compound 3. These signals are assigned to the methylene and methyl protons of the “free” N-Ethhymethohcarbthio ligand, respectively. Aromatic protons are also observed as multiplets in the range δ = 6.87 to 6.96 ppm, and δ = 7.00 to 7.43 ppm.
ESI mass spectroscopy was used to prove the presence of DMSO-containing vanadium(V)complexes, but 1H NMR spectroscopy has proven that chemical shifts for DMSO occur at δ = 2.7 ppm and δ = 3.0-3.6 ppm, thus proving the existence of O-bonded and S-bonded DMSO, respectively.[22] The absence of a peak at δ = 2.7 ppm implies the absence of O-bonded DMSO.[22] The resonance due to the presence of an S-bonded DMSO produces singlets at δ = 3.36 ppm and δ = 3.34 ppm for the oxidised products isolated from compounds 1 and 3, respectively.
1H and 51V NMR studies on oxidised solutions of compounds 1-4 in DMSO-d6
It is interesting to note that over a 24 hour period, DMSO-d6 solutions of compounds 1–4 were found to be oxidised, with the eventual formation of vanadium(V) species. In such a case, 51V NMR spectroscopy was used to definitively confirm the presence of vanadium(V) species. Figure 4 shows the 51V NMR spectra for DMSO-d6 solutions of compounds 1–4 (10 mM) at room temperature; while table 5 shows the chemical shifts and percentages of species for each vanadium(V) species. It is interesting that two species are formed on oxidation of compounds 1, 2, and 4, while three species are formed for compound 3. It is possible that a mononuclear species is formed in the range −480.5 to −467.7 ppm, based on the chemical shifts obtained for 51V NMR spectra of vanadate(V) esters of monoionised and diionised aromatic 1,2-diols as reported by Baruah et al.[23] For example, [VO(gsal)(HL1)], [VO(asal)(HL1)], and [VO(vsal)(HL1)] (where gsal, asal, or vsal = diionised salicylaldimine of L-glycine, L-alanine or L-valine; and HL1 = catecholate) have chemical shifts of −460, −457, and −455 ppm, respectively, when DMSO-d6 was used as solvent.
Figure 4.
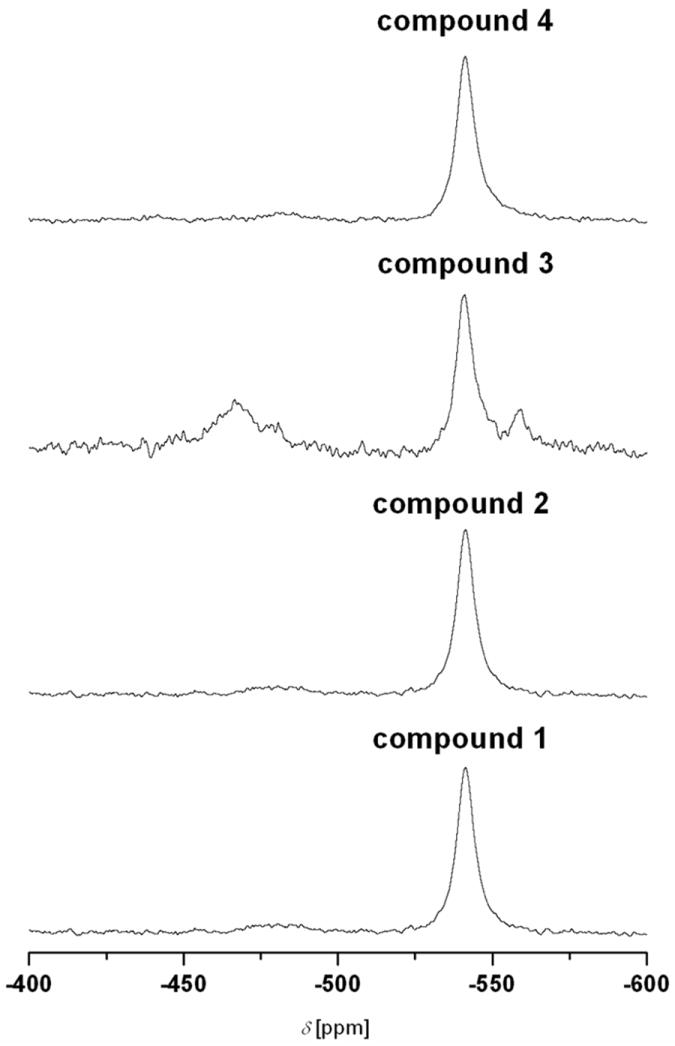
51V NMR spectra of compounds 1–4 after being oxidised in DMSO-d6 for 24 hours.
Table 5.
Chemical shifts and percentages of species of DMSO-d6 solutions for compounds 1–4 after being oxidised for 24 hours.
| Compound | δ [ppm] (%) | δ [ppm] (%) | δ [ppm] (%) |
|---|---|---|---|
| 1 | −480.5 (7.1) | −541.4 (92.9) | |
| 2 | −481.9 (9.2) | −542.2 (90.8) | |
| 3 | −467.7 (41.7) | −541.0 (47.9) | −559.3(10.4) |
| 4 | −481.9 (6.9) | −541.2 (93.1) |
It is interesting that Mauyra et al.[24] reported the synthesis of new oxidovanadium(V) complexes, [VOL(hq)] by the reaction of [VO(acac)2] with ligands LH2 (where LH2 is the dibasic tridentate ONO Mannich base [(S)-H2glysal, (S)-H2alasal, (S)-H2leusal and (S)-H2ileusal; S represents the S-enantiomer] obtained by the reduction of the Schiff bases of salicylaldehyde (sal) and the amino acids: glycine (gly), D,L-alanine (ala), leucine (leu) and isoleucine (ileu), respectively) in the presence of 8-hydroxyquinoline (Hhq). Spectral studies were used to confirm an octahedral structure for the complexes.[24] The complexes exhibited a single 51V NMR signals (with DMSO-d6 as solvent) in the range −464.6 to −468.0 ppm due to the existence of a single isomeric species in solution.[24] Due to the similar chemical shifts of the species in the range −480.46 to −467.69 ppm, we can conclude that a mononuclear vanadium(V) species is formed when the respective vanadium(IV) complexes are oxidised in DMSO-d6. The reason why the vanadium(V) species (which is formed from compound 3) has a chemical shift of −467.7 ppm, is yet to be determined.
Mauyra et al.[24] carried out a time dependent 51V NMR spectroscopic study (in DMF-d7 and CD3OD) in order to investigate the possible isomerisation and/or further reaction in solution. Freshly prepared solutions of [VO(S-alasal)(hq)] showed a signal at −466.7 ppm. As time elapsed, a second, broader signal at −516.3 ppm was formed, which had about the same integral intensity as the −466.7 ppm signal after three days, and about twice its intensity after one week. The authors concluded that the change in chemical shift was too large to represent diastereomers based on pairs of enantiomers, and therefore, assigned the new signal to a [VO2] or [(VO)2μ-O] species, i.e., the oxygenated product after a slow loss of 8-hydroxyquinoline. They also concluded that as generation of a [(VO)2μ-O] species usually dominates during oxygenation[24] in the absence of a base, the signal at −516.3 ppm is most probably due to the [(VO)2μ-O] species. In our case, the chemical shift of approximately −541 ppm accounts for the major species formed for compounds 1–4. We believe that this is the [V2O3(Sal-L-tryp)2(DMSO)2]. This species was also detected by ESI MS (m/z = 918.3, [[V2O3(Sal-L-tryp)2(DMSO)2] + H]+) from oxidised DMSO solutions of compounds 1 and 3. A similar complex with N-salicylidene-L-alaninate (sal-L-ala) as ligand, viz., [V2O3(sal-L-ala)2].2CH2Cl2, was also reported by Nakajima et al.[25]
We believe that [VO(Sal-L-tryp)(DMSO-d6)(OH)] is formed as an intermediate when compounds 2 and 4 are oxidised in DMSO-d6, all within experimental errors in the determination of the percentage species. We believe that the species that has a chemical shift at -559.3 ppm is mononuclear, and are proposing its structure as shown in Scheme 1. We propose its formula as [VO(sal-L-tryp)(DMSO)(N-Ethhymethohcarbthio)]. Based on 51V NMR spectroscopy, we propose the existence of equilibrium between mononuclear and binuclear vanadium(V) species (see Scheme 1).
Based on 1H NMR spectroscopic studies (See Figures S27-S30, Supporting Information), it appears as if the thiosemicarbazone ligands are now “free” when compounds 2-4 are oxidised in DMSO-d6 and DMSO. The 1H NMR chemical shifts of the “free” ligands are within the values shown in figures 3, and those chemical shifts obtained for MeATSC as reported by Beckford et al.[6] ESI MS have also proven the existence of a “free” thiosemicarbazone ligand for a mixture that resulted from the oxidation of compound 3 in DMSO. In this study, a m/z ratio of 252.3 accounts for the presence of the “free” ligand, N-Ethhymethohcarbthio.
UV-visible and fluorescence spectroscopic studies of compound 1-4
UV-visible spectra were acquired for compounds 1-4 in DMSO (see Figures S31-S34, Supporting Information for the data). Assignments of the absorption bands are shown in Table 6. Charge transfer bands in the region 266-292 nm which can be assigned to π→π* transitions associated with the thioamide moiety are observed for compounds 2–4.[18a, 26] These occurred at 266 nm and 292 nm (shoulder) for compound 2, 270 nm and 288 nm (both as shoulders) for compound 3, and 276 nm for compound 4, respectively. Charge transfer bands assigned to n→π* transitions of aromatic rings are observed in the region 316-352 nm.[18a, 19a, 26-27] These are observed as a shoulder at 316 nm for compound 2, at 330 nm for compound 3, and 352 nm for compound 4, respectively.
Table 6.
UV-visible spectroscopic data for compounds 1–4.
| Compound | λ [nm] | ε [M-1cm-1] | Assignments |
|---|---|---|---|
|
| |||
| 1 | 558 | 44 | LMCT (p → d transition) |
| 758 | 27 | d → d transition | |
|
| |||
| 2 | 266 | 74835 | π → π* (thioamide moiety) |
| 292 (sh) | 17954 | π → π* (thioamide moiety) | |
| 316 (sh) | 10046 | n → π* (aromatic rings) | |
| 388 | 15013 | π → π* (azomethine chromophore) | |
| 550 | 54 | LMCT (p → d transition) | |
| 592 (sh) | 45 | d → d transition | |
| 756 | 28 | d → d transition | |
|
| |||
| 3 | 270 (sh) | 10984 | π → π* (thioamide moiety) |
| 288 (sh) | 13874 | π → π* (thioamide moiety) | |
| 330 | 24837 | n → π* (aromatic rings) | |
| 390 (sh) | 18250 | π → π* (azomethine chromophore) | |
| 620 | 852 | d → d transition | |
|
| |||
| 4 | 276 | 21969 | π → π* (thioamide moiety) |
| 352 | 14227 | n → π* (aromatic rings) | |
| 558 | 80 | LMCT (p → d transition) | |
| 590 (sh) | 70 | d → d transition | |
| 756 | 38 | d → d transition | |
In the UV region, oxidovanadium(IV) complexes derived from salicylaldehyde generally possess a low-energy band around 375 nm which can be attributed to a π→π* transition originating mainly from the azomethine chromophore.[11] This occurs at 388 nm for compound 2, as a shoulder at 390 nm for compound 3, and hidden under the n→π* transitions at 352 nm for compound 4. A band in the region 550-558 nm is assigned to LMCT transitions of the type p→d where p and d represent phenolato oxygen’s lone pair and vanadium 3d orbitals, respectively.[28] This occurs at 558 nm for compound 1, 550 nm for compound 2, and 558 nm for compound 4. A similar band is absent for compound 3 since it is believed to be hidden under the band at 614 nm which is assigned as d→d transitions.
Bands assigned to d-d transitions were also observed in the region 756-758 nm for compounds 1, 2 and 4.[19b] This occurred at 758 nm for compound 1 and 756 nm for compounds 2 and 4. In the electronic spectra of compounds 2 and 4, bands at 592 nm for compound 2 and 590 nm for compound 4 are also assigned to d→d transitions, appearing as shoulders of the much stronger LMCT band in the region 550-558 nm.[19b]
Fluorescence spectra were acquired for compounds 2–4 in DMSO (See Figures S35-S37, Supporting Information). The excitation wavelengths for compounds 2, 3, and 4, were 400, 330, and 340 nm, respectively. Compound 2 which contains an anthracene unit in MeATSC produced the highest intensity in its fluorescence spectra; while compounds 3 and 4 produced the lower intensities.
ESR spectroscopy
Compounds 1–4 were also characterised by low-temperature ESR spectroscopy, where at 10 K, each complex exhibits a powder type axial g = 2 ESR signal (S = ½) with a minor rhombic distortion. The observed hyperfine structure shows sixteen line ESR signals with partial overlapping on three lines. Figure 5 shows two representative ESR spectra along with simulations for compounds 1 and 4. These spectra are characteristic of slowly tumbling macromolecular complexes consistent with vanadyl (VO2+)-containing complexes, indicating that the freshly prepared vanadium metal centre has a +4 oxidation state for the two complexes. The anisotropic sixteen line powder ESR spectra are due to the hyperfine interaction between the electron spin and the nuclear spin of 51V(IV) (I = 7/2, 100% natural abundance).
Figure 5.
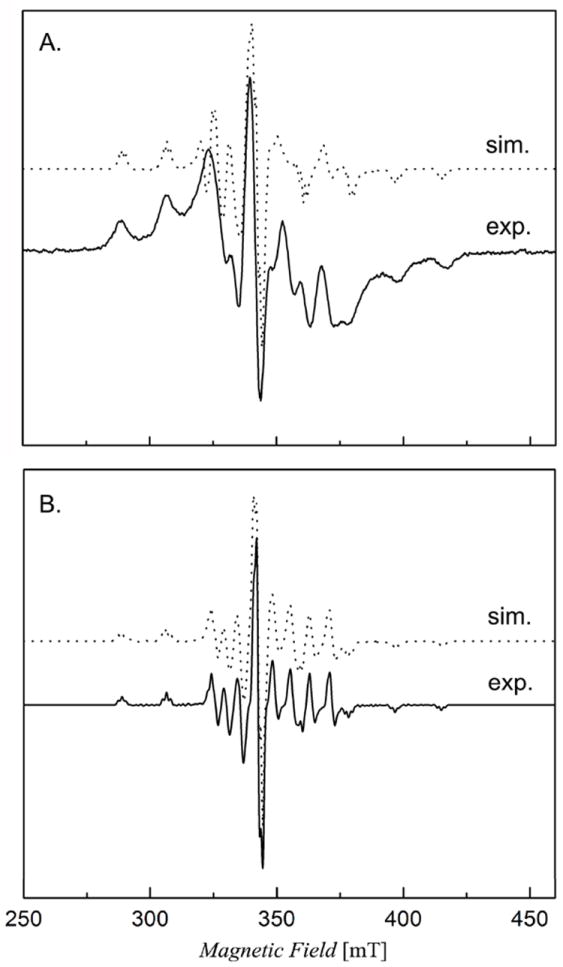
X-band continuous-wave ESR spectra of [VO(Sal-L-tryp) (H2O)] [compound 1 (A), 10 mM in DMSO] and [VO(Sal-L-tryp)(acetylethTSC)].C2H5OH [compound 4 (B), 10 mM in DMSO] overlaid with spectral simulations on top of the original data. Instrument conditions were as follows: temperature = 10 K, modulation frequency = 100 kHz, modulation amplitude = 0.3 G, microwave frequency = 9.64 GHz, and microwave power = 0.004 mW.
The powder ESR spectra of compounds 1–4 were simulated to determine the magnitudes of the principal 51V hyperfine coupling constants (Axx, Axx, and Azz) and g values using the ESR simulation program DOUBLET.EXE.[29] The ESR parameters obtained from spectral simulations are shown in Table 7. The large A values and broader linewidth are presumably due to the sulphur ligation and hexacoordination geometry. The vanadyl cation, VO2+, is known to form complexes that are pentacoordinate or square bipyramidal with a short V=O bond.[30] However, hexacoordinate vanadyl-containing complexes with mixed N/O/S ligands are thus poorly characterised so far. The ESR spectra of the complexes described here may be useful in future comparison studies of VO2+ in complexes and biological systems. Nevertheless, these ESR results are in agreement with those of UV-visible and FT IR spectral measurements, and thus prove that each synthesised compound has a vanadium(IV) metal centre prior to air oxidation of the DMSO solution. On another note, we plan to carry out future studies involving DFT calculations in order to elucidate the dependence of calculated 51V Az values on the orientation and geometry of coordinated thiosemicarbazone and water ligands in compounds 1–4. Such a study involving calculated and experimental Az values for compounds 1–4 would complement the excellent piece of data for VIVO complexes as reported by Garriba and co-workers.[31]
Table 7.
ESR parameters of compounds 1–4.a
| Compound | gx | gy | gz | Ax | Ay | Az |
|---|---|---|---|---|---|---|
| 1 | 1.958 | 1.988 | 1.953 | 80 | 68 | 180 |
| 2 | 1.98 | 1.98 | 1.968 | 64 | 65 | 178 |
| 3 | 2 | 1.995 | 1.965 | 63 | 63 | 175 |
| 4 | 1.98 | 1.98 | 1.968 | 64 | 65 | 178 |
ESR parameters were obtained from spectral simulations as described in the text.
Pharmacology
In vitro cytotoxicity
The objective of this research is to evaluate the anti-proliferative activity of compounds 2–4 against colon cancer cell lines (HTC-116, Caco-2 and HT-29), and to compare the anti-proliferative activity on one non-cancerous colon cell line (CCD-18Co). Compounds 2–4, cisplatin, and etoposide were evaluated for their cytotoxicity on three different cancer colon cell lines (HCT-116, Caco-2 and HT-29) by means of a colorimetric assay (MTS assay) which measures mitochondrial dehydrogenase activity as an indication of cell viability. The effects of the compounds on the viability of these cells were evaluated after continuous incubation (24, 48, and 72 hours).
As such, the growth inhibition effects of compounds were investigated in non-cancerous colonic myofibroblasts CCD-18Co cells and compared to cancer colon cell lines. The results demonstrated the greatest difference in the inhibition of cell proliferation between cancer lines and the non-cancerous CCD-18Co cells. Compounds 2–4 have been shown to time-dependently decrease cell viability of HCT-116, Caco-2 and more potently in HT-29 cells (Table 8). Compound 4 was the most active of the three tested; while compounds 2 and 3 showed similar activities in both cancer cell lines. Figure 6 shows a plot of percentage cell viability versus concentration of compound 4 against the HT-29 cancer cell line; while table 8 shows the IC50 values for inhibition of cell proliferation for compounds 2–4, with IC50 values generally around 100 μM concentration at 72 hours in compounds 2 and 3. None of the compounds had better efficacy against any of the cancer cell lines when compared to the standard, etoposide. It is very important to note that compound 4 has a better efficacy against the HT-29 cancer cell line when compared to the standard, cisplatin (Table 8).
Table 8.
Anti-proliferative effects of compounds 2–4, cisplatin, and etoposide on different cells lines after 24, 48, and 72 hours treatment. Data are expressed as IC50 (μM). IC50 is defined as the concentration required to achieve 50% inhibition over control cells (0.5% DMSO); IC50 values are shown as mean ± S.D. from three independent experiments.
| Species | HT-29 | Caco-2 | ||||
|---|---|---|---|---|---|---|
| 24 hours | 48 hours | 72 hours | 24 hours | 48 hours | 72 hours | |
|
| ||||||
| IC50 [μM] | IC50 [μM] | IC50 [μM] | IC50 [μM] | IC50 [μM] | IC50 [μM] | |
| 2 | 277.1±10.2 | 239.6±7.9 | 100.3±5.1 | 349.1±19.5 | 277.1±16.3 | 147.1±10.8 |
|
| ||||||
| 3 | 244.2±8.8 | 169.6±15.1 | 87.9±0.5 | 367.0±21.5 | 281.6±14.4 | 152.5±12.7 |
|
| ||||||
| 4 | 128.6±10.7 | 82.4±9.6 | 47.8±5.5 | 242.1±25.1 | 166.2±15.3 | 85.4±14.0 |
|
| ||||||
| Cisplatin | 84.7±1.9 | 80.6±1.6 | 69.1±3.2 | 32.0±1.6 | 22.8±1.5 | 17.9±1.8 |
|
| ||||||
| Etoposide | 19.2±0.8 | 17.3±1.9 | 15.8±2.4 | 47.6±0.8 | 46.2±1.9 | 40.9±1.5 |
|
| ||||||
| Species | HCT-116 | CCD-18Co | ||||
| 24 hours | 48 hours | 72 hours | 24 hours | 48 hours | 72 hours | |
|
| ||||||
| IC50 [μM] | IC50 [μM] | IC50 [μM] | IC50 [μM] | IC50 [μM] | IC50 [μM] | |
|
| ||||||
| 2 | 203.4±7.7 | 181.3±9.2 | 115.0±6.5 | 495.6±23.5 | 339.2±16.7 | 208.0±11.9 |
|
| ||||||
| 3 | 227.4±6.8 | 192.7±11.6 | 110.3±10.1 | 490.6±27.6 | 329.5±15.4 | 203.6±12.5 |
|
| ||||||
| 4 | 161.3±4.6 | 134.9±3.9 | 89.5±14.5 | 382.7±21.9 | 246.2±11.4 | 152.2±12.0 |
|
| ||||||
| Cisplatin | 53.8±2.1 | 49.7±1.3 | 41.0±2.7 | 83.0±1.1 | 72.0±1.5 | 64.1±1.6 |
|
| ||||||
| Etoposide | 40.8±0.5 | 36.2±2.0 | 38.4±1.2 | 82.5±3.1 | 79.2±1.2 | 73.2±1.9 |
Figure 6.
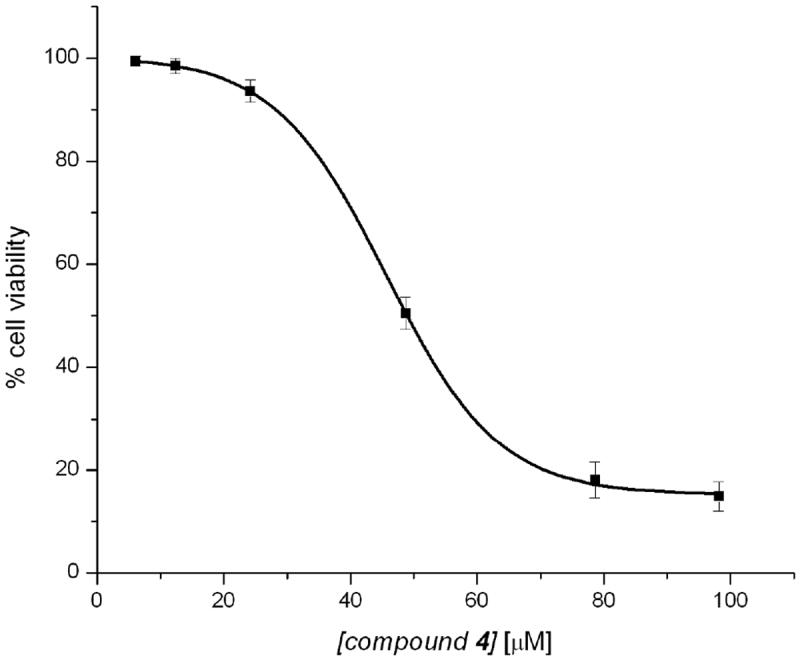
MTT cell proliferation assay of compound 4 against the HT-29 cancer cell line.
Compounds 2–4 exhibited less inhibitory effects in human normal CCD-18Co cells, indicating a possible cytotoxic selectivity towards colon cancer cells (Table 8). IC50 values in CCD-18Co were around 3-fold and 2-fold than HT-29 and Caco-2 and HCT-116, respectively. However, cytotoxic selectivity towards cancer colon cells of cisplatin (as a positive control) was slightly lower, except on Caco-2 cells (Table 8). These results are also in good agreement with those of a previous study where it was shown that cisplatin inhibited cell proliferation by about 70% in both cell lines (cancer and normal colon cells).[32] In general, compounds 2–4 which have been proven to be growth suppressors of cancer cells, but do not affect non-cancerous cells, may have a potential in chemotherapy.
Although metal complexes are being investigated as probes and therapeutics, there are a relatively few studies on their mechanism of uptake. It has been found that metal complexes that are lipophilic cations may passively diffuse across the plasma membrane in response to the membrane potential.[33] A ruthenium(II) complex containing the lipophilic 4,7-diphenyl-1,10-phenanthroline ligand has been shown to enter the membrane by passive diffusion in a membrane-potential dependent manner.[33] It is believed that the excellent efficacy of the ruthenium(II) complexes with thiosemicarbazones as reported in a study by Beckford and co-workers,[6] is likely due to the fact that those complexes are lipophilic cations. As a point of speculation, this may explain why cations such as [(bpy)2Ru(TSC)]2+ and [(phen)2Ru(TSC)]2+ (where TSC = 9-anthraldehydethiosemicarbazones),[6] have better efficacies against HT-29 and HCT-116 cell lines versus our neutral vanadium(IV) complexes. Based on 1H NMR spectroscopy and ESI MS studies, where the “free” ligand was observed, we believe that the “free” ligand is likely causing the cell death as apposed to the vanadium species in the media. In order to prove this belief, in the near future, we will have to carry out in vitro studies with the “free” ligands. A systematic study will have to be carried out with the ruthenium(II) complexes and compounds 2–4, all under the same conditions. Apart from such a study, in the future, a more detailed emphasis will be placed on the methods used to examine cellular accumulations to identify the mechanism(s) of uptake, and to monitor possible efflux of compounds 2–4, and their analogues.
Conclusions
Two novel thiosemicarbazone and three novel vanadium(IV) complexes with mixed-ligands were successfully synthesised and characterised. Anti-cancer properties of compounds 2–4 were examined with three colon cancer cell lines, HTC-116, Caco-2, and HT-29, along with a comparative anti-proliferative study on non-cancerous colonic myofibroblasts, CCD-18Co. Compounds 2 and 3 exhibited a less inhibitory effects in human non-cancerous CCD-18Co cells, indicating a possible cytotoxic selectivity towards colon cancer cells. In general, those compounds which exhibited anti-proliferative activity on cancer cells, but did not affect non-cancerous colorectal cells, may have a potential in chemotherapy.
Experimental Section
Materials and methods
Analytical or reagent grade chemicals were used throughout this study. All the chemicals including solvents were obtained from Sigma-Aldrich (St. Louis, MO, USA) or other commercial vendors, and used as received. Microanalyses (C, H, N) were performed by Desert Analytics, Tucson, U.S.A. and Columbia Analytical Services 3860 S. Palo Verde Road Suite 303 Tucson, AZ 85714, U.S.A. 1H and 13C NMR spectra were acquired in DMSO-d6 on a JEOL ECX-300, a Varian 300 MHz, Bruker 400 MHz, or a Varian 500 MHz spectrometer operating at room temperature. The residual 1H and 13C present in DMSO-d6 (2.49 and 39.7 ppm, respectively) were used as internal references. 51V NMR spectra were acquired on a Varian 500 MHz spectrometer with DMSO-d6 as solvent and VOCl3 as an external reference as described for vanadium(V) compounds.[34]
ESR spectra were acquired at either 10 or 20 K on a Bruker ER200D spectrometer using a 4116DM resonator. Sample temperature was maintained with a temperature controller (ITC503S), an ESR910 liquid helium cryostat and LLT650/13 liquid helium transfer tube (Oxford Instruments, Concord, MA, U.S.A.) spectrometer. Instrument conditions were as follows: modulation frequency = 100 kHz, modulation amplitude = 0.3 G, microwave frequency = 9.64 GHz, and microwave power = 0.004 mW.
FT IR spectra were acquired in the range 4000-400 cm-1 using the ATR accessory (with a diamond crystal) on a Nicolet 6700 FTIR spectrophotometer. Electronic spectra were acquired using quartz cuvettes on a HP8452 diode array spectrophotometer using DMSO as the solvent. Fluorescence spectra were acquired on a Cary Eclipse fluorescence spectrophotometer (Varian Inc.) using a slit width of 10 nm. ESI MS data was acquired on an HP Agilent 1956b single-quadrupole mass spectrometer. Samples were dissolved in an acetic acid/methanol mixture; then the solution was introduced by direct injection using a syringe pump with a flow rate of 100 μL s-1, while sweeping the cone voltage from 0 to 200 V at a rate of 10 V min-1. All m/z ratios and percentages presented as ESI MS data were determined by using the MestReNova software.
X-ray crystallography
Crystallographic data were collected at T = 90 K on Bruker Kappa Apex-II CCD diffractometer equipped with CuKα radiation and an Oxford Cryosystems Cryostream cooler. The structure of acetylethTSC was solved using SHELXS,[35] and refined using the SHELX97[36] software packages. All H atoms were visible in difference maps. Coordinates of the NH hydrogen atoms were refined individually, while those on C were placed in idealized positions, with torsional parameters refined for the methyl groups. Crystal data: C8H12N4S2, FW = 228.34, monoclinic, space group = C2/c, a =10.4549(10), b = 10.8970(10), c = 18.7694(15) Å, β = 91.544(5)°, V = 2137.6(3) Å3, Z = 8, T = 90.0(5)K, μ = 4.25 mm-1, dcalc = 1.419 g cm−3, colourless parallelepiped, dimensions = 0.18 × 0.17 × 0.09 mm, reflections collected = 8017, unique reflections = 1901, observed reflections (I > 2σ(I)) = 1738, Rint = 0.026, no. of parameters = 136, R1 = 0.028, wR2 = 0.069, GOF(F2) = 1.065, CCDC No. = 812348.
Synthesis of ligands
9-Anthraldehydethiosemicarbazone (ATSC), 9-anthraldehyde-N(4)-methylthiosemicarbazone (MeATSC) was prepared according to the procedure as by Beckford et al.[6]
Synthesis of N-Ethhymethohcarbthio
o-Vanillin (3.00 g, 19.7 mmol) and 4-ethyl-3-thiosemicarbazide (2.35 g, 19.7 mmol) were placed in a 250 ml round bottom flask, followed by absolute ethanol (100 ml). Approximately 10 drops of glacial acetic acid were added to the off-white suspension, and the reaction mixture was refluxed for three hours. The reaction mixture was then cooled to room temperature and filtered through a sintered glass crucible. The white residue was washed with ethanol (3 × 15 ml), followed by ether (3 ×10 ml) and allowed to air dry. Yield = 4.97 g (99%). The purity was checked by elemental analysis. Calc. for C10H13N3O2S: C, 52.15; H, 5.97; N, 16.59. Found: C, 52.53; H, 6.16; N, 17.02; Infrared spectrum (ν/cm-1): FT IR (ν/cm-1): 3304 (m) (-N1H), 3300 (m) (-N2H), 3130 (br) (OH), 1155 (m) (N-N), 1606 (m) (TSC (C=N)), (1267) (m) and (831) (m) (C=S). 1H NMR (400 MHz; DMSO-d6); δH = 1.15 (t, Jt = 7.10 Hz, 3H, -CH3), 3.46 (s, 1H, -CH3-OAr), 3.60 (q, 2H, CH2), 3.80 (s, 1H, (azomethine -CH=N)), 6.79 (t, Jt = 7.95 Hz, CHar), 6.96 (dd, Jdd = 1.49 and 8.06 Hz, CHar), 7.55 (dd, Jdd = 1.51 and 7.93 Hz, CHar), 8.40 (s, H, aminic NH)), 11.41 (s, 1H, ArOH)), and 11.42 ppm (s, 1H, hydrazinic-NH)). 13C (400 MHz; DMSO-d6); δ = 176.86 (C=S), 147.86 (CH=N), 145.88 (-COCH3ar), 139.00 (PhOH), 120.82 (CCH=Nar), 118.91 (CHar), 118.10 (CHar), 112.66 (CHar), 58.85 (OCH3), 38.28 (CH2), 14.60 (CH3).
Synthesis of acetylethTSC
2-Acetylthiazole (0.636 g, 0.518 ml 5.0 mmol) was placed in a 100 ml round bottom flask, followed by anhydrous methanol (25 ml). 4-Ethyl-3-thiocarbazide (0.596 g, 5.0 mmol) in anhydrous methanol (25 ml) was slowly added to the solution of 2-acetylthiazole, followed by a few drops of concentrated hydrochloric acid were added. The reaction mixture was refluxed with stirring for two hours and then evaporated to a minimum volume to form a yellow solid. The mixture was filtered, and the residue was washed with ether and air-dried. Yield = 0.750 g (66%). A single crystal for X-ray crystallography was grown by slow evaporation from methanol. FT IR (ν/cm-1): 3164 (m) (-N1H), 3054 (m) (-N2H), 1059 (m) (N-N), 1543 (m) (TSC (C=N)), (1296) (m) and (813) (m) (C=S). 1H NMR (400 MHz; DMSO-d6); δH = 1.16 (t, Jt = 7.10 Hz, 3H, -CH3), 2.42 (s, 3H, CH3CH=N), 3.61 (d, 2H, CH2), 7.79 (d, Jd = 3.21 Hz, CHar), 7.88 (d, Jd = 3.19 Hz, CHar), 8.08 (d, Jd = 3.24 Hz, CHar), 8.30 (s, H, aminic NH), 10.58 (s, 1H, hydrazinic-NH), and 13.2 (s, H-bonded hydrazinic-NH) ppm.
Synthesis of the reduced Schiff base (2-(2-hydroxybenzylamino)-3-(1H-indol-3-yl)propanoic acid)
The reduced Schiff base was prepared using a known procedure involving salicylaldehyde, amino acids, and NaBH4, but with L-tryptophan using the following synthetic procedure:[10] L-tryptophan (2.04 g, 10.0 mmol) and potassium hydroxide (0.56 g, 10.0 mmol) were added to a 125 ml Erlenmeyer flask, followed by deionised water (10 ml). Salicylaldehyde (1.029 ml, 10.0 mmol) in absolute ethanol (10 mL) was slowly added to the mixture. The yellow solution was stirred for thirty minutes prior to cooling in an ice bath.
The intermediate Schiff base which was produced in situ was reduced with an excess of sodium borohydride (0.46 g, 12 mmol) in water (5 ml) containing 10 drops of 2 M sodium hydroxide. The solution was stirred for ten minutes, and the yellow colour was slowly discharged. The solution was then acidified with concentrated HCl to pH 4.68. The resulting solid was filtered off, washed with ethanol (2 × 30 ml) and diethyl ether (2 × 30 ml) and dried. The white solid obtained was recrystalized twice from water/ethanol(1:1). Yield = 1.08 g (35%). m/z (ESI) (+ve mode) 311.08 (100.00%, [M + H]+), 242.42 (13.04%, [M − CHO3 + H]+), 620.36 (12.77%, [2M + H]+), 930.39 (5.02%, [3M + H]+), 1240.92 (3.92%, [4M + H]+). FT IR (ν/cm-1): 3392 (m) (indolic NH), 3055 (br) (OH), 1208 (m) (C-O)), 1494 (m) (COOs) and 1592 (m) (COOas). 1H NMR (400 MHz; DMSO-d6); δH = 2.67 (d, 2H), 3.21 (d, 2H), 3.05 (d, 2H), 5.91 (t, 1H), 6.15 (d, 1H), 6.31 (d, 1H), 6.62 (m, 1H), 6.62 (m, 1H), 6.62 (m, 1H), 6.62 (m, 1H,), 6.90 (d, 1H) and 7.18 ppm (d, 1H). 13C (400 MHz; DMSO-d6); δ = 28.57, 46.72, 63.61, 110.66, 111.57, 113.46, 118.50, 118.69, 121.37, 123.85, 123.89, 126.95, 127.78, 128.16, 128.34, 135.97, 164.55, and 182.05 ppm.
Synthesis of K[(Sal-L-tryp)]
This complex was prepared using an analogous synthesis as reported by Vanco et al.[37] To a mixture of L-tryptophan (3.07 g, 15.0 mmol) and potassium hydroxide (0.85 g, 15.0 mmol) dissolved in water (5 ml), the solution of salicylaldehyde (1.57 ml, 15.0 mmol) in ethanol (10 ml) was added with stirring. The resulting yellow solution was stirred at room temperature for 24 hours; then the solution was diluted by ethanol (25 ml). Upon slow evaporation of solvent, a yellow oil was formed which was then triturated with diethyl ether. The mixture was then filtered to give a yellow powder which was dried under vacuum. Yield = 4.85 g (93%). m/z (ESI) (+ve mode) 344.05 (39.65%, [M + H]+), 300.03 (100.00%, [M − OH − K + H]+), 281.14 (46.08%, [M − OH − O − K + H]+), 262.12 (1.51%, [M − COO − K + H]+), 242.62 (64.65%, [M − COO − OH − K + H]+) and 205.20 (50.43%, [M − C7H6O − K + H]+). FT IR (ν/cm-1): 3407 (m) (indolic NH), 3169 (br) (OH), 1194 (m) (C-O)), 1521 (m) (COOs), 1604 (m) (COOas) and 1570 (m) (TSC (C=N)). 1H NMR (400 MHz; DMSO-d6); δH = 3.13 (dd, Jdd = 8.9 and 16 Hz, 2H), 3.49 (s, 1H), 4.07 (s, 1H), 6.59 (s, 1H), 6.72 - 7.06 (m, 1H), 6.72 - 7.06 (m, 1H), 7.20 (s, 1H), 7.20 (m, 1H), 7.20 (m, 1H), 7.20 (m, 1H), 7.35 - 7.58 (m, H), 7.35 - 7.58 (m, 1H), and 11.17 (s, 1H) ppm. 13C (400 MHz; DMSO-d6); δ = 30.16, 64.82, 72.43, 111.39, 111.56, 115.74, 117.62, 117.99, 118.31, 118.33, 120.59, 123.38, 127.36, 131.77, 132.01, 163.27, and 172.88 ppm.
Synthesis of [VO(Sal-L-tryp)(H2O)] 1
This complex was prepared as by Costa Pessoa et al.[11] L-tryptophan (1.14 g, 5.60 mmol) and sodium acetate trihydrate (1.47 g, 10.8 mmol) were placed in a 250 ml round bottomed flask with deionized water (100 ml). The mixture was stirred and heated at 50 °C to completely dissolve the L-tryptophan. Salicylaldehyde (0.59 ml, 5.6 mmol) in absolute ethanol (14 ml) was slowly added to the mixture. The resulting yellow solution was stirred vigorously on a magnetic stirrer and an aqueous solution of VOSO4.×H2O (0.78 g, 4.8 mmol) in water (2 ml) was added dropwise to the solution. The solution was stirred for 30 minutes. A deep dark brown precipitate formed and the resulting solution was then filtered under vacuum. The grey solid formed was washed with water (30 ml) followed by a ethanol-water mixture (50:50) (30 ml) and air-dried. Yield = 1.19 g (63%). Calc. for C18H16N2O5V: C, 55.25; H, 4.12; N, 7.16. Found: C, 55.16; H, 4.31; N, 6.87 (lit.[11] Calc. for C18H16N2O5V: C, 55.25; H, 4.12; N, 7.16. Found: C, 55.2; H, 4.2; N, 7.1). FT IR (ν/cm-1): 3478 (m) (indolic NH), 3014 (br) (OH), 1540 (m) (C-O), 1490 (m) (COOs), 1597 (m) (COOas), 1600 (m) ([(Sal-L-tryp)] C=N), and 997 (s) (V=O) (lit.[11] 3480 (m) (indolic NH), 3065 (br) (OH), 1630 (m) ([(Sal-L-tryp)] (C=N)), 1600 (m) (C-O)) and 970 (s) (V=O)). UV-visible spectrum (MeOH), λmax./nm (ε/M-1 cm-1): 558 (44) and 758 (27); (lit.:[11] UV-visible spectrum (DMSO), λmax./nm (ε/M-1 cm-1): 275 (15400) and 375 (4850), 520 (45), and 730 (20)).
Synthesis of [VO(Sal-L-tryp)(MeATSC)].1.5 C2H5OH 2
(E)-2-(Anthracen-9-ylmethylene)-N-methylhydrazinecarbothioamide (0.15 g, 0.51 mmol) and [VO(Sal-L-tryp)(H2O)] (0.20 g, 0.51 mmol) were placed in a dried 250 ml round bottom flask. Absolute ethanol (100 ml) was added to the flask and the solution was refluxed with stirring under argon for two hours in an oil bath at 110 °C. The orange-blue solution which was produced was then rotaryevaporated to dryness. Diethyl ether was added to the product and the mixture was filtered under vacuum, and the green residue was collected and air-dried. Yield = 0.23 g (68%). Calc. for C38H38N5O5.5SV: C, 62.03; H, 5.21; N, 9.52. Found: C, 62.62; H, 5.25; N, 9.33. m/z (ESI) 666.17 (100.00%, [M + H]+), 577.08 (33.10%, [[M − C2H5N2S] + H]+, 405.14 (31.86%, [[M − C17H17N3] + H]+), 292.14 (54.60%, [MeATSC + H]+). FT IR (ν/cm-1): 3308 (m) (indolic NH), 3340 (m) (-N1H), 2976 (m) (-N2H), 1148 (m) (N-N), 1544 (m) (C-O)), 1491 (m) (COOs), 1600 (s) (COOas), 1624 (m) (TSC (C=N)), 1590 (m) ([(Sal-L-tryp)] (C=N)), (1225) (m), (829) (m) (C=S), (455) (m) (V-S), and 980 (s) (V=O). UV-visible spectrum (DMSO), λmax./nm (ε/M-1cm-1): 226 (74835), 292 (sh) (17954), 316 (sh) (10046), 388 (15013), 550 (54), 592 (sh) (45), and 756 (28).
Synthesis of [VO(Sal-L-tryp)(N-Ethhymethohcarbthio)].H2O 3
This was prepared in a similar manner to 2 using (E)-2-(4-hydroxy-3-methoxybenzylidene)-N methylhydrazinecarbothioamide (0.065 g, 0.26 mmol) and [VO(Sal-L-tryp)(H2O)] (0.10 g, 0.26 mmol). Yield = 0.12 g (78%). Calc. for C29H30N4O7 SV: C, 53.33; H, 4.64; N, 11.11. Found: C, 53.18; H, 5.21; N, 11.09. m/z (ESI) 624.87 (100.00%, [M − H]-), 431.90 (64.75%, [M − C11H10N2O − H]-), 407.85 (20.99%, [M − C11H15N3O2S - H]-), 287.88 (12.70%, [[C11H15N3O2S − C8H7N] - H]-), 252.00 (25.74%, [N-Ethhymethohcarbthio - H]-). FT IR (ν/cm-1): 3210 (m) (indolic NH), 3306 (m) (-N1H), 2970 (m) (-N2H), 3010 (br) (OH), 1154 (m) (N-N), 1540 (m) (C-O)), 1480 (m) (COOs), 1600 (s) (COOas), 1620 (m) (TSC (C=N)), 1580 (m) ([(Sal-L-tryp)] (C=N)), (1270) (m), (802) (m) (C=S), (454) (m) (V-S) and 976 (s) (V=O). UV-visible spectrum (DMSO), λmax./nm (ε/M-1cm-1): 270 (sh) (10984), 288 (sh) (13874), 330 (24837), 390 (sh) (18250), and 620 (852).
Synthesis of [VO(Sal-L-tryp)(acetylethTSC)].C2H5OH 4
This was prepared in a similar manner to compound 2 using 2-acetylthiozole-4,4’-dimethylthiosemicarbazone (0.12 g, 0.51 mmol) and [VO(Sal-L-tryp)(H2O)] (0.20 g, 0.51 mmol). Yield = 0.18 g (57%). Calc. for C28H32N6O5S2V: C, 51.93; H, 4.98; N, 12.98. Found: C, 52.49; H, 4.72; N, 10.91. m/z (ESI) 600.25 (2.71%, [M + H]+), 513.57 (100.00%, [M − C3H7NS + H]+), 482.38 (37.11%, [M − C7H5NO] + H]+), and 336.06 (4.13%, [[M − C17H15N2O] + H]+). FT IR (ν/cm-1): 3229 (m) (indolic NH), 3310 (m) (-N1H), 2976 (m) (-N2H), 1149 (m) (N-N), 1545 (m) (C-O)), 1480 (m) (COOs), 1600 (s) (COOas), 1624 (m) (TSC (C=N)), 1580 (m) ([(Sal-L-tryp)] (C=N)), (1287) (m), (819) (m) (C=S), (455) (m) (V-S) and 982 (V=O). UV-visible spectrum (DMSO), λmax./nm (ε/M-1cm-1): 276 (21969), 352 (14227), 558 (80), 590 (sh) (70), and 756 (38).
Oxidation of compounds 1 and 3 in DMSO
[VO(Sal-L-tryp)(H2O)] 1 (0.1 g, 0.26 mmol) was added to an evaporating dish. Dimethylsulfoxide (10 ml) was added and the resulting solution was left to evaporate for 32 days to leave a solid. Yield = 0.09 g. m/z (ESI) (+ve mode) 468.60 (1.67%, [M + H]+), 242.41 (100.00%, [M − OH − DMSO − C8H7N + H]+), 451.05 (48.25%, [M − OH + H]+), 763.68 (30.06%, [[V2O3(Sal-L-tryp)2] + H]+), 776.77 (49.36%, [[V2O3(Sal-L-tryp)2(H2O)] + H]+) and 918.28 (3.13%, [[V2O3(Sal-L-tryp)2(DMSO)2] + H]+). FT IR (ν/cm-1): 3185 (m) (indolic NH), 3194 (br)(OH), 1551 (m) (C-O)), 1497 (m) (COOs), 1618 (s) (COOas), 1667 (m) (TSC (C=N)), 1581 (m) ([(Sal-L-tryp)] (C=N)), 1164 (m), 1213 (m) (S=O), 949 (s) (V=O). 1H NMR (400 MHz; DMSO-d6); δH = 2.52 (d, 3H), 2.52 (d, 3H), 10.89 (s, 1H, 7.06 (s, 1H), 7.22 (m, 1H), 7.22 (m, 1H), 7.52 (d, 1H, 8.06 (d, 2H), 3.36 (s, 1H), 3.00 (s, 2H), 7.22 (m, 1H), 7.22 (m, 1H), 7.22 (m, 1H), 7.22 (m, 1H), 9.93 (s, 1H), and 7.06 ppm (s, 1H).
[VO(Sal-L-tryp)(N-Ethhymethohcarbthio)].H2O 3 (0.1 g, 0.16 mmol) was oxidised in an analogous manner. Yield = 0.09 g. m/z (ESI) (+ve mode) 470.01 (0.50%, [M + H]+), 252.26 (100.00%, [N-Ethhymethohcarbthio + H]+), 451.96 (2.97%, [M − OH + H]+), 702.75 (2.15%, [M − OH + N-Ethhymethohcarbthio + H]+), 763.22 (0.74%, [[V2O3(Sal-L-tryp)2 + H]+), 775.40 (1.08%, [[V2O3(Sal-L-tryp)2(H2O)] + H]+) and 916.44 (0.75%, [[V2O3(Sal-L-tryp)2(DMSO)2] + H]+). FT IR (ν/cm-1): 3193 (br)(OH), 1666 (m) (C=N), 1600 (C=O), 1550 (m) (C-O)), 1133 (m), 1157 (m) (S=O), 946 (s) (V=O). 1H NMR (400 MHz; DMSO-d6); δH = 2.52 (d, 3H), 2.52 (d, 3H), 10.43 (s, 1H), 6.96 (s, 1H), 7.22 (m, 1H), 7.22 (m, 1H), 7.42 (d, 1H), 8.00 (d, 2H), 3.34 (s, 1H), 3.00 (s, 2H), 7.22 (m, 1H), 7.22 (m, 1H), 7.22 (m, 1H), 7.22 (m, 1H), 9.95 (s, 1H), and 6.96 ppm (s, 1H).
Pharmacology
Cell culture
Three human colon cancer cells, HT-29 (human colon adenocarcinoma), HCT-116 (human colon carcinoma) and Caco-2 (human epithelial colorectal adenocarcinoma)_as well as non-cancerous human colon cells, CCD-18Co (human colon fibroblasts) were used in this study. All cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD, U.S.A.) and maintained at the University of Rhode Island, U.S.A. Caco-2 cells were grown in EMEM medium supplemented with 10% v/v fetal bovine serum, 1% v/v non-essential amino acids, 1% v/v L-glutamine and 1% v/v antibiotic solution (Sigma-Aldrich). HT-29 and HCT-116 cells were grown in McCoy’s 5a medium supplemented with 10% v/v fetal bovine serum, 1% v/v non-essential amino acids, 2% v/v HEPES and 1% v/v antibiotic solution. CCD-18Co cells were grown in EMEM medium supplemented with 10% v/v fetal bovine serum, 1% v/v non-essential amino acids, 1% v/v L-glutamine and 1% v/v antibiotic solution and were used from PDL = 26 to PDL = 35 for all experiments. Cells were maintained at 37 °C in an incubator under a 5% CO2/95% air atmosphere at constant humidity and maintained in the linear phase of growth. The pH of the culture medium was determined using pH indicator paper (pHydrionTM Brilliant, pH 5.5-9.0, Micro Essential Laboratory, NY, U.S.A.) inside the incubator. All test samples were dissolved in DMSO (<0.5 % in the culture medium) by sonication; then filter sterilised (0.2 μm) prior to addition to the culture media. Control cells were also carried out in parallel sequences, and subjected to the same changes in medium with a 0.5 % DMSO.
Cytotoxicity assay
This assay was carried out as described previously[38] in order to measure the IC50 values for samples. Briefly, the in vitro cytotoxicity of samples were assessed in tumour cells by a tetrazolium-based colorimetric assay, which takes advantage of the metabolic conversion of MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfenyl)-2H-tetrazolium, inner salt] to a reduced form that absorbs light at 490 nm. Cells were counted using a hemacytometer and were plated at 2000-5,000 cells per well, depending on the cell line, in a 96-well format for 24 hours prior to drug addition. Test samples and a positive control, etoposide 4 mg ml-1 (Sigma-Aldrich), were dissolved in DMSO by sonication. All samples were diluted with media to the desired treatment concentration and the final DMSO concentration per well did not exceed 0.5%. Control wells were also included on all plates. Following a 24, 48, or 72 hour drug-incubation period at 37 °C with serially diluted test compounds, MTS, in combination with the electron coupling agent, phenazine methosulphate, was added to the wells and cells were incubated at 37 °C in a humidified incubator for three hours. Absorbances at 490 nm (OD490) were acquired on a spectrophotometer (SpectraMax M2, Molecular Devices Corp., operated by SoftmaxPro v.4.6 software, Sunnyvale, CA, U.S.A.) to obtain the number of surviving cells relative to control populations. The results are expressed as the median cytotoxic concentrations (IC50 values) and were calculated from six-point dose response curves using 4-fold serial dilutions. Each point on the curve was tested in.
Statistics
Data are expressed as mean ± SE for three replications on each cell line.
Supplementary Material
Supporting Information (see footnote on the first page of this article): …. Crystallographic data for acetylethTSC has been deposited at Cambridge Crystallographic Data Centre as supplementary publication number CCDC No. = 812348. Copies of available material can be obtained on application to CCDC, 12 Union Road, Cambridge CB2 1Ez, UK (fax: 044 1223336 033 or deposit@ccdc.cam.ac.uk).
Schemes S1-S5; mass spectra, Figures S1-S9; FT IR spectra, Figures S10-S18; 1H and 13C NMR spectra, Figures S19-S30; UV-visible spectra, Figures S31-S34; and UV-visible and fluorescence spectra, Figures S35-S37. X-ray crystallographic data, Tables S1-S5.
Acknowledgments
This work was supported in part by P20RR016476 (the Mississippi INBRE funded by the National Center for Research Resources/NIH). The project described was also supported in part by Award Number P20RR16460 from the National Institutes of Health to FAB. The authors also acknowledge the NSF for funding our ESI and MALDI-ToF mass spectrometers (Grant CHE 0639208).
We are also grateful for the use of our EMXmicro ESR spectrometer, which was funded by the NSF CRIF:MU Award # 0741991; also our new 400 MHZ NMR spectrometer, which was funded by the NSF CRIF:MU Award # 0840390. AAH is also grateful for the USM Lucas Endowment Grant and funding from ExxonMobil Research and Engineering Company through Dr. John Robbins. We would like to thank Dr. Vijayaraghavan Rangachari and his research group for the use of their Cary Eclipse fluorescence spectrophotometer. We would also like to thank Professor Glen Shearer for his constant positive motivation which helped mobilised this project.
We thank Dr. Andrew Ozarowski of the National High Magnetic Field Laboratory (NHMFL, Tallahassee, FL, U.S.A.) for providing us with the ESR simulation programme used in this study.
Footnotes
Supporting information for this article is available on the WWW under http://www.eurjic.org/ or from the author.
References
- 1.Wong E, Giandomenico CM. Chem Rev. 1999;99:2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 2.a) Galanski M, Arion VB, Jakupec MA, Keppler BK. Curr Pharm Des. 2003;9:2078–2089. doi: 10.2174/1381612033454180. [DOI] [PubMed] [Google Scholar]; b) Jakupec MA, Galanski M, Keppler BK. Rev Physiol Biochem Pharm. 2003;146:1–54. doi: 10.1007/s10254-002-0001-x. [DOI] [PubMed] [Google Scholar]
- 3.Ott I, Gust R. Archiv der Pharmazie - Chemistry in Life Sciences. 2007;340:117–126. doi: 10.1002/ardp.200600151. [DOI] [PubMed] [Google Scholar]
- 4.a) Clarke MJ. Coord Chem Rev. 2003;203:209–233. [Google Scholar]; b) Clarke MJ, Zhu F, Frasca DR. Chem Rev. 1999;99:2511–2533. doi: 10.1021/cr9804238. [DOI] [PubMed] [Google Scholar]; c) Armitage B. Chem Rev. 1998;98:1171–1200. doi: 10.1021/cr960428+. [DOI] [PubMed] [Google Scholar]
- 5.Silva DdO. Anti-Cancer Agents Med Chem. 2010;10:312–323. doi: 10.2174/187152010791162333. [DOI] [PubMed] [Google Scholar]
- 6.Beckford FA, Shaloski M, Jr, Leblanc G, Thessing J, Lewis-Alleyne LC, Holder AA, Li L, Seeram NP. Dalton Trans. 2009:10757–10764. doi: 10.1039/b915081a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Tarasconi P, Capacchi S, Pelosi G, Cornia M, Albertini R, Bonati A, Dall’Aglio PP, Lunghi P, Pinelli S. Bioorg Med Chem. 2000;8:157–162. doi: 10.1016/s0968-0896(99)00260-6. [DOI] [PubMed] [Google Scholar]; b) West DX, Swearingen JK, Valdes-Martinez J, Hernandez-Ortega S, El-Sawaf AK, Van Meurs F, Castineiras A, Garcia I, Bermejo E. Polyhedron. 1999;18:2919–2929. [Google Scholar]
- 8.Noblia P, Vieites M, Parajon-Costa BS, Baran EJ, Cerecetto H, Draper P, Gonzalez M, Piro OE, Castellano EE, Azqueta A, Lopez de Cerain A, Monge-Vega A, Gambino D. J Inorg Biochem. 2005;99:443–451. doi: 10.1016/j.jinorgbio.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 9.a) D’Cruz OJ, Dong Y, Uckun FM. Anti-Cancer Drugs. 2000;11:849–858. doi: 10.1097/00001813-200011000-00009. [DOI] [PubMed] [Google Scholar]; b) Narla RK, Dong Y, Ghosh P, Thoen K, Uckun FM. Drugs of the Future. 2000;25:1053–1068. [Google Scholar]; c) D’Cruz OJ, Uckun FM. Expert Opin Invest Drugs. 2002;11:1829–1836. doi: 10.1517/13543784.11.12.1829. [DOI] [PubMed] [Google Scholar]; d) Fichtner I, Claffey J, Deally A, Gleeson B, Hogan M, Markelova MR, Mueller-Bunz H, Weber H, Tacke M. J Organomet Chem. 2010;695:1175–1181. [Google Scholar]; e) Rehder D. Bioinorganic Vanadium Chemistry. John Wiley & Sons, Ltd.; Chichester, West Sussex: 2008. p. 176. [Google Scholar]
- 10.Koh LL, Ranford J, Robinson W, Svensson JO, Tan ALC, Wu D. Inorg Chem. 1996;35:6466–6472. doi: 10.1021/ic9606441. [DOI] [PubMed] [Google Scholar]
- 11.Pessoa JC, Duarte MT, Gillard RD, Madeira C, Matias PM, Tomaza I. J Chem Soc Dalton Trans. 1998:4015–4020. [Google Scholar]
- 12.Cavaco I, Pessoa JC, Duarte MT, Henriques RT, Matias PM, Gillard RD. J Chem Soc Dalton Trans. 1996:1989–1996. [Google Scholar]
- 13.Viñuelas-Zahínos E, Luna-Giles F, Torres-García P, Fernández-Calderón MC. Eur J Med Chem. 2011;46:150–159. doi: 10.1016/j.ejmech.2010.10.030. [DOI] [PubMed] [Google Scholar]
- 14.Venkatraman R, Ameera H, Sitole L, Ellis E, Fronczek FR, Valente EJ. J Chem Crystallogr. 2009;39:711–718. [Google Scholar]
- 15.Lobana TS, Sharma R, Bawa G, Khanna S. Coord Chem Rev. 2009;253:977–1055. [Google Scholar]
- 16.a) Mostafa MM, El-Hammid A, Shallaby M, El-Asmay AA. Transition Met Chem. 1981;6:303. [Google Scholar]; b) West DX, Liberta AE, Padhye SB, Chikate RC, Sonawane PB, Kumbher AS, Yerande RG. Coord Chem Rev. 1993;123:49. [Google Scholar]
- 17.Beraldo H, Nacif WF, Teixeira LR, Reboucas JS. Transition Met Chem. 2002;27:85–88. [Google Scholar]
- 18.a) Demirci TB, Koseoglu Y, Guner S, Ulkuseven B. Cent Eur J Chem. 2006;4:149–159. [Google Scholar]; b) Zhang H, Thomas R, Oupicky D, Peng F. J Biol Inorg Chem. 2008;13:47–55. doi: 10.1007/s00775-007-0299-6. [DOI] [PubMed] [Google Scholar]
- 19.a) Mendes IC, Botion LM, Ferreira AVM, Castellano EE, Beraldo H. Inorg Chim Acta. 2009;362:414. [Google Scholar]; b) Pessoa JC, Cavaco I, Correia I, Tomaz I, Duarte T, Matias PM. J Inorg Biochem. 2000;80:35–39. doi: 10.1016/s0162-0134(00)00037-4. [DOI] [PubMed] [Google Scholar]
- 20.Raposo MMM, Garcia-Acosta B, Abalos T, Calero P, Martinez-Manez R, Ros-Lis JV, Soto J. J Org Chem. 2010;75:2922–2933. doi: 10.1021/jo100082k. [DOI] [PubMed] [Google Scholar]
- 21.Afrasiabi Z, Sinn E, Kulkarni PP, Ambike V, Padhye S, Deobagakar D, Heron M, Gabbutt C, Anson CE, Powell AK. Inorg Chim Acta. 2005;358:2023–2030. [Google Scholar]
- 22.Mahalingam V, Chitrapriya N, Fronczek FR, Natarajan K. Polyhedron. 2008;27:2743–2750. [Google Scholar]
- 23.Baruah B, Das S, Chakravorty A. Inorg Chem. 2002;41:4502–4508. doi: 10.1021/ic020259d. [DOI] [PubMed] [Google Scholar]
- 24.Maurya MR, Khurana S, Rehder D. Transition Met Chem. 2003;28:511–517. [Google Scholar]
- 25.Nakajima K, Kojima M, Toriumi K, Saito K, Fujita J. Bull Chem Soc Jpn. 1989;62:760–767. [Google Scholar]
- 26.Kanoongo N, Singh R, Tandon JP. Transition Met Chem. 1987;12:271–273. [Google Scholar]
- 27.a) Sasmal PK, Patra AK, Nethaji M, Chakravarty AR. Inorg Chem. 2007;46:11112–11121. doi: 10.1021/ic7011793. [DOI] [PubMed] [Google Scholar]; b) Toshima K, Takano R, Ozawa T, Matsumura S. Chem Commun. 2002:212–213. doi: 10.1039/b107829c. [DOI] [PubMed] [Google Scholar]
- 28.a) Rath SP, Ghosh T, Mondal S. Polyhedron. 1997;16:4179–4186. [Google Scholar]; b) Bonadies JA, Carrano CJ. J Am Chem Soc. 1986;108:4088–4095. [Google Scholar]
- 29.Ozarowski A, Lee HM, Balch AL. J Am Chem Soc. 2003;125:12606–12614. doi: 10.1021/ja030221f. [DOI] [PubMed] [Google Scholar]
- 30.a) Branca M, Micera G, Dessi A, Sanna D, Raymond KN. Inorg Chem. 1990;29:1586–1589. [Google Scholar]; b) Chasteen ND, Lord EM, Thompson HJ, Grady JK. Biochim Biophys Acta Gen Subj. 1986;884:84–92. doi: 10.1016/0304-4165(86)90230-8. [DOI] [PubMed] [Google Scholar]; c) Crans DC, Khan AR, Mahroof-Tahir M, Mondal S, Miller SM, la Cour A, Anderson OP, Jakusch T, Kiss T. J Chem Soc Dalton Trans. 2001:3337–3345. [Google Scholar]; d) Efthimiadou Eleni K, Psomas G, Sanakis Y, Katsaros N, Karaliota A. J Inorg Biochem. 2007;101:525–535. doi: 10.1016/j.jinorgbio.2006.11.020. [DOI] [PubMed] [Google Scholar]; e) Ghosh SK, Patra R, Rath SP. Inorg Chem. 2008;47:9848–9856. doi: 10.1021/ic800714w. [DOI] [PubMed] [Google Scholar]; f) Mahroof-Tahir M, Keramidas AD, Goldfarb RB, Anderson OP, Miller MM, Crans DC. Inorg Chem. 1997;36:1657–1668. doi: 10.1021/ic9610243. [DOI] [PubMed] [Google Scholar]; g) Paine TK, Weyhermueller T, Slep LD, Neese F, Bill E, Bothe E, Wieghardt K, Chaudhuri P. Inorg Chem. 2004;43:7324–7338. doi: 10.1021/ic040052f. [DOI] [PubMed] [Google Scholar]; h) Xu YZ, Shi S. Appl Magn Reson. 1996;11:1–6. [Google Scholar]
- 31.a) Micera G, Garribba E. J Comput Chem. 2011;32:2822–2835. doi: 10.1002/jcc.21862. [DOI] [PubMed] [Google Scholar]; b) Micera G, Pecoraro VL, Garribba E. Inorg Chem. 2009;48:5790–5796. doi: 10.1021/ic9001779. [DOI] [PubMed] [Google Scholar]; c) Micera G, Garribba E. Dalton Trans. 2009:1914–1918. doi: 10.1039/b819620f. [DOI] [PubMed] [Google Scholar]; d) Garribba E, Lodyga-Chruscinska E, Micera G, Panzanelli A, Sanna D. Eur J Inorg Chem. 2005:1369–1382. [Google Scholar]
- 32.Kan WL, Cho CH, Rudd JA, Lin G. J Ethnopharmacol. 2008;120:36–43. doi: 10.1016/j.jep.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 33.Puckett CA, Ernst RJ, Barton JK. Dalton Trans. 2010;39:1159–1170. doi: 10.1039/b922209j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crans DC, Holder AA, Saha TK, Prakash GK, Yousufuddin M, Kultyshev R, Ismail R, Goodman MF, Borden J, Florián F. Inorg Chem. 2007;46:6723–6732. doi: 10.1021/ic062484r. [DOI] [PubMed] [Google Scholar]
- 35.Sheldrick GM. Acta Crystallogr Sect A: Foundations of Crystallography. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 36.a) Sheldrick GM. NATO ASI Series Series E: Applied Sciences. 1997;347:219–230. [Google Scholar]; b) Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 37.Vanco J, Svajlenova O, Marek J. Acta Crystallogr Sect C: Cryst Struct Commun. 2003;C59:m190–m192. doi: 10.1107/s0108270103006760. [DOI] [PubMed] [Google Scholar]
- 38.Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information (see footnote on the first page of this article): …. Crystallographic data for acetylethTSC has been deposited at Cambridge Crystallographic Data Centre as supplementary publication number CCDC No. = 812348. Copies of available material can be obtained on application to CCDC, 12 Union Road, Cambridge CB2 1Ez, UK (fax: 044 1223336 033 or deposit@ccdc.cam.ac.uk).
Schemes S1-S5; mass spectra, Figures S1-S9; FT IR spectra, Figures S10-S18; 1H and 13C NMR spectra, Figures S19-S30; UV-visible spectra, Figures S31-S34; and UV-visible and fluorescence spectra, Figures S35-S37. X-ray crystallographic data, Tables S1-S5.