

Abstract
Viruses that selectively replicate in cancer cells, leading to the death of the cell, are being studied for their potential as cancer therapies. Some of these viruses are naturally occurring but cause little if any illness in humans; others have been engineered to make them specifically able to kill cancer cells while sparing normal cells. These oncolytic viruses may be selective for cancer cells because viral receptors are over-expressed on the surface of cancer cells or because antiviral pathways are distorted in cancer cells. Additionally, when oncolytic viruses kill cancer cells, it can stimulate an antitumour immune response from the host that can enhance efficacy. Numerous early phase trials of at least six oncolytic viruses have been reported with no evidence of concerning toxicity either as single agents or in combination with chemotherapies and radiotherapy. Three oncolytic viruses have reached randomized testing in cancer patients; reolysin in head and neck cancer and JX594 in hepatocellular cancers, while results from the first-phase III trial of T-vec in metastatic melanoma are expected shortly.
Keywords: oncolytic virotherapy, immunotherapy, delivery, cancer
Introduction
Remarkably, cancer-killing or ‘oncolytic’ viruses (OV) have appeared in the medical literature repeatedly over the last 100 years. Despite their undoubted antitumour effects, OV have been regarded as medical curiosities rather than credible cancer therapies. However, our improved understanding of both the molecular pathology of common malignancies, as well as basic viral biology, has renewed interest over the last decade.1 The true measure of relevance of any medical innovation is progression to a randomized phase III trial platform and, thereby, potential registration. There are currently two OV that have reached this stage: a modified herpes virus for the treatment of malignant melanoma (talimogene laherperepvec [T-Vec] NCT00769704) and a naturally occurring reovirus for the treatment of head and neck cancer (pelareorep NCT01166542, www.Clinicaltrials.gov). This review will give an overview of the advantages and disadvantages of oncolytic viruses as treatments for cancer including a review of clinical trials to date.
Methods
This review is intended to serve as an overview of the field for fellow clinicians, with particular attention to the clinical data. Medline, PubMed, ClinicalTrials.gov and the authors’ collective familiarity with the literature were used to retrieve the relevant references.
What are oncolytic viruses and how are they suited for cancer therapy?
It is a perverse irony that pathogens responsible for untold human suffering and mortality may hold the key to a new generation of cancer therapeutics. Viruses capable of killing cancer cells, or OV, may be naturally occurring, but can also be genetically engineered for attenuation or to encode additional genes to deliver a ‘therapeutic payload’. Both types of agent specifically replicate in cancer cells leading to their death, while sparing normal cells. In cancer cells, OV usurp cellular machinery for their own reproduction, such that the cell dies as the virus replicates, and daughter virions are released to spread and infect neighbouring cells (Figure 1).2 The basis for OV selectivity against malignant cells centres around three main key concepts: first, cancer cells are unable to generate an antiviral interferon response on infection by OV whereas normal cells can; second, genetic dysregulation in cancer cells (e.g. in the Ras oncogene pathway) supports viral replication; and finally targeting of cancer cells generates an antitumour immune response – in other words, OV-mediated tumour cell death generates a therapeutic vaccine in situ.3,4
Figure 1.
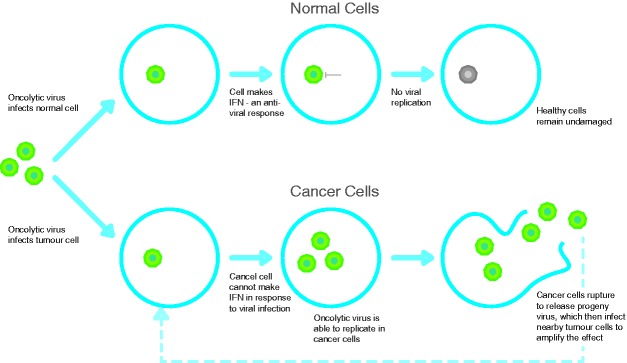
One mechanism by which oncolytic viruses may replicate selectively in cancer cells. The normal response to viral infection is the triggering of specific molecules inside the cell leading to the production of type 1 interferons. These cytokines have a direct antiviral effect and switch off viral replication. In cancer cells, genetic mutations result in the loss of this interferon inhibition.
Advantages of oncolytic viruses over conventional therapeutics
Theoretically, OV have a number of positive attributes as cancer therapies: the replication of viruses selectively in cancer cells potentially provides a high-therapeutic index; upon systemic delivery, viruses can efficiently traffic to tumour tissue using specific or ubiquitous cell receptors to gain entry; viruses have tropism for cancer-associated blood vessels and distinct mechanisms of tumour cell-killing reduce the likelihood of cross-resistance to other anticancer modalities.2 All these mechanisms have been exploited in a burgeoning number of preclinical models as oncolytic virotherapy has developed. However, these studies have also highlighted a number of limitations, particularly related to the body’s highly evolved capacity to deal with viral infections. Hence, OV may be rapidly cleared by antibodies and complement or become trapped in the liver or spleen, before they can reach their intended tumour targets. Even after OV reach a tumour deposit, particular features of the tumour stroma and microenvironment may limit access to, and spread between, tumour cells.5 To address these problems and improve responses, a range of preclinical strategies has been employed in mouse models, including combining OV with other agents, such as immunomodulators and drugs to enhance viral penetration and spread within solid tumours.
Testing oncolytic viruses in humans – clinical trials
Twenty viruses have been or are currently under preclinical evaluation. A dozen or so viruses have been used in clinical trials over the last century. In the current era, there are seven viruses in clinical trial, including three in randomized studies. The prototype OV were adenoviruses, but despite elegant genetic engineering to increase tumour selectivity, they have been problematic due to inadequate infection of cancer cells, lack of specificity and the vigorous antiadenoviral antibody immune response. ONYX 015 was the first live-engineered OV evaluated in humans and, although responses were disappointing, a similar agent has now been licensed in China for the treatment of head and neck cancer in combination with chemotherapy.
In the UK, the largest clinical programmes of systemic and local delivery have involved reovirus (Respiratory Enteric Orphan Virus) and herpes simplex virus (HSV). Reoviruses are naturally occurring and ubiquitous non-pathogenic viruses. Malignant cells harbouring mutations or activation of the Ras oncogene (a defect in around half of all cancer) are susceptible to reovirus replication and killing. This is because Ras-activated cancer cells, unlike normal cells, are unable to activate protein kinase R, an intracellular protein present in all cells, which senses the presence of viral RNA and shuts down viral replication. Clinical evaluation of intravenous reovirus is the largest OV programme currently ongoing worldwide, and the manufacturers estimate that over 2000 viral doses have been delivered to patients as part of over 30 trials (http://www.oncolyticsbiotech.com/). Evidence of antitumour activity has been documented radiologically and histologically in tumour biopsies taken following therapy. Importantly, replicating virus has recently been isolated from tumour sites days after intravenous infusion.6 There were concerns that the potential of reovirus and other systemically delivered OV would be seriously limited by the generation of existing or induced neutralizing antibody (NAB) responses, as had been shown in animal models. However, we recently showed that after systemic delivery, even in the presence of pre-existing NAB, reovirus can ‘hitch hike’ on white blood cells and platelets, evade humoural immunity and efficiently access tumour sites with cytotoxic effect (Figure 2).7 Following a phase I/II study that demonstrated an encouraging response rate, reovirus is currently in a phase III randomized trial in combination with paclitaxel and carboplatin chemotherapy for patients with refractory head and neck cancers.8
Figure 2.
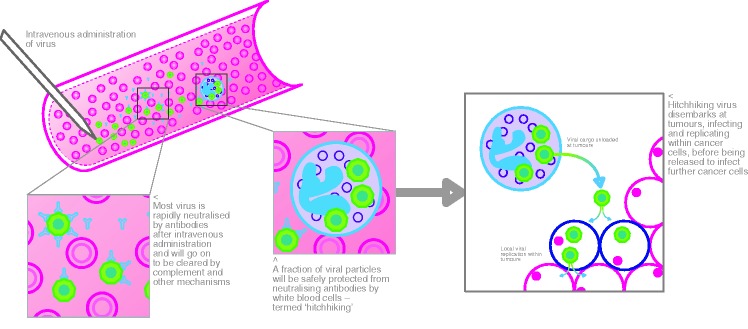
Reovirus ‘hitch hikes’ onto white blood cells where it is protected from neutralizing antibodies and delivered to the tumour.
Although four strains of engineered HSV type 1 (HSV-1) have progressed to clinical trial, the most advanced programme is using T-Vec (talimogene laherparepvec). To enhance the safety of this virus, the gene responsible for neurovirulence has been replaced with a gene encoding the cytokine GM-CSF (granulocyte macrophage colony stimulating factor), intended to boost the immune response against the tumour in conjunction with virus-induced tumour destruction. Repeated intratumoural injection of single-agent T-Vec in melanoma metastases has been shown to result in local tumour kill, but also generation of systemic antitumour immune responses, with radiological evidence of tumour shrinkage in distant, uninjected metastases; complete response evaluation criteria in solid tumours (RECIST) responses were seen in eight of the 50 patients treated in a phase II trial and partial responses in a further five patients.9 These results led to a randomized phase III study in patients with metastatic melanoma, which has recently completed and is expected to report this year.
The third OV to have progressed to randomized testing in patients with hepatocellular cancer is a Wyeth strain of vaccinia, JX-594. The virus has been genetically modified by the deletion of viral thymidine kinase gene (to switch off phosphorylation of nucleotides and prevent viral replication in normal cells) and incorporation again of immunostimulatory GM-CSF similar to T-Vec. In addition to direct and immune-mediated killing of tumour cells, a human study of JX-594 demonstrated an additional potential mechanism of OV anticancer effect, namely the selective targeting of tumour vasculature shutting down tumour perfusion.10
Challenges facing oncolytic virotherapy
Based on findings from phase I and phase II studies, the clinical data to date for OV have been promising enough for progression to randomized trials and significant investment from the pharmaceutical industry.11 Importantly, virotherapy has been remarkably well tolerated with no serious safety or toxicity issues reported to date in treated patients, their families or attending medical staff. The most common adverse effects of OV therapy have been transient flu-like symptoms, as might be expected from a viral infection. These do not overlap with the toxicities of chemotherapy and/or radiotherapy, so that the further development of combination strategies is unlikely to be limited by toxicity. On the negative side, there are a number of factors that still challenge the development of OV therapy. The UK’s robust regulatory framework presents several practical hurdles for widespread routine use in all cancer centres. Handling genetically modified agents raises concerns within the NHS clinical infrastructure but is certainly feasible. There is no evidence to date of virus shedding by patients after treatment that poses a risk to other patients, medical staff or their families. There may be a necessity for the adaptation of current pharmacy facilities, but many centres already have experience handling biological agents. In our experience, defining where, in the hospital, the agents will be administered and how they must be contained, has been an initial, albeit surmountable challenge. Other issues centre around appropriate radiological evaluation of response, since conventional imaging modalities and interpretation of response have been defined in the context of radiotherapy and chemotherapy, while OV result in local tumour destruction with immune-mediated effects. Both these features may be slow to develop and may be highly significant but without shrinkage in tumour size by imaging. OV-specific refinement of radiological RECIST may therefore be required in a similar way to recent adaptations to response definition in cancer immunotherapy.
Producing virus to the high concentrations and purity required for virotherapy is also challenging, and it remains to be seen what the cost of these agents will be if they do prove to be effective therapies.12
The future for OV therapy – next steps
As with many anticancer agents, OV are likely to work better in combination with other treatment modalities rather than as single agents. Several synergistic mechanisms have been identified such as the enhancement of apoptotic pathways when chemotherapy and OV are combined or increased viral replication in irradiated cells.13,14 Preclinical and clinical data already support combinatorial strategies, and it is reassuring that there is no evidence that co-administration of OV with chemotherapy or radiotherapy damages the virus or significantly increases toxicity.15,16 With the expanding portfolio of both new OV agents and novel targeted drugs entering the clinical arena, the number of potential combinations is huge, making rational clinical development a challenge. If, as seems likely, an OV is soon shown to be effective in a phase III setting, there will be a need to carefully prioritize the next tranche of studies to address how best to exploit specific biological features of the tumour (e.g. induction of new cancer-associated vasculature and angiogenesis), the OV (e.g. JX-594 noted for its vascular tropism) and the combination agent (e.g. tyrosine kinase inhibitors with antiangiogeneic properties). It should, ultimately, be possible to define optimal combinations, disease targets, dose and response assessment. Both preclinical models and clinical trials accompanied by meticulous translational scientific analysis will inform us how best to move forward via iterative ‘bench to bedside and back again’ research. In this way, OV may truly become an established and familiar part of the anticancer armamentarium.
Declarations
Competing interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). Hardev Pandha, Hevin Harrington and Alan Melcher have projects funded by Oncolytics Biotech, but declare no conflict of interest. Oliver Donnelly declares no conflicts.
Funding
None declared
Ethical approval
Not applicable
Guarantor
HP
Contributorship
HP conceived the article and along with OD developed the first draft. AM, KH, HP and OD then made a series of revisions to produce the finalized article. KH obtained the patient’s story, along with consent for it to be used for publication. OD and AM prepared Figure 2 and HP prepared Figure 1.
Acknowledgements
OD gratefully acknowledges that he was a recipient of the Ellison-Cliffe Travelling Fellowship from the Royal Society of Medicine (London).
Provenance
Submitted; peer-reviewed by Ketan Shah and John Bell.
References
- 1.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther 2007; 15: 651–5 [DOI] [PubMed] [Google Scholar]
- 2.Vähä-Koskela MJ, Heikkilä JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett 2007; 254: 178–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prestwich RJ, Errington F, Diaz RM, et al. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther 2009; 20: 1119–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White CL, Twigger KR, Vidal L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther 2008; 15: 911–20 [DOI] [PubMed] [Google Scholar]
- 5.Wojton J, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev 2010; 21: 127–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidal L, Pandha HS, Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res 2008; 14: 7127–37 [DOI] [PubMed] [Google Scholar]
- 7.Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med 2012; 4: 138ra77–138ra77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karapanagiotou EM, Roulstone V, Twigger K, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res 2012; 18: 2080–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 2009; 27: 5763–71 [DOI] [PubMed] [Google Scholar]
- 10.Liu TC, Hwang T, Park BH, Bell J, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther 2008; 16: 1637–42 [DOI] [PubMed] [Google Scholar]
- 11. See http://www.amgen.com/media/media_pr_detail.jsp?releaseID=1519312.
- 12.Langfield KK, Wegman TR, Walker HJ, et al. Production and purification of measles virus for oncolytic virotherapy clinical trials. Mol Ther 2004; 9: S31–S31 [Google Scholar]
- 13.Roulstone V, Twigger K, Zaidi S, et al. Synergistic cytotoxicity of oncolytic reovirus in combination with cisplatin–paclitaxel doublet chemotherapy. Gene Ther 2013; 20: 521–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touchefeu Y, Vassaux G, Harrington KJ. Oncolytic viruses in radiation oncology. Radiother Oncol 2011; 99: 262–70 [DOI] [PubMed] [Google Scholar]
- 15.Richard-Fiardo P, Franken PR, Harrington KJ, Vassaux G, Cambien B. The use of molecular imaging of gene expression by radiotracers in gene therapy. Expert Opin Biol Ther 2011; 11: 1273–85 [DOI] [PubMed] [Google Scholar]
- 16.Pandha HS, Heinemann L, Simpson GR, et al. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res 2009; 15: 6158–66 [DOI] [PubMed] [Google Scholar]