

Abstract
Standard cartilage tissue engineering approaches, for example, matrix-induced autologous chondrocyte implantation (MACI), consist of the implantation of cell-based constructs whose survival and further development first depend on the degree of graft maturity at the time of surgery (e.g., matrix production) and, subsequently, on initial host reaction. Indeed, blood vessel ingrowth and macrophage migration within the implant may endanger graft stability of immature constructs; so, control of angiogenesis was proposed as an adjuvant of cellular therapy for the treatment of cartilage defects. In this study, we hypothesized that engineered constructs with no in vitro precultivation, but functionalized to block angiogenesis right on implantation, might result in better survival, as well as superior long-term cartilaginous quality. Here, we propose a clinically compatible fibrin/hyaluronan scaffold seeded with nasal chondrocytes (NC) and functionalized with an FDA-approved anti-angiogenic drug (bevacizumab; Avastin®), which sequestrates vascular endothelial growth factor from the surrounding environment. Our results show that the sustained bevacizumab release from NC-loaded scaffolds after subcutaneous implantation in nude mice efficiently blocked host vessels ingrowth (five times lower CD31+ cells infiltration vs. control group, at 3 weeks after implant), and enhanced constructs survival rate (75% vs. 18% for the control, at 6 weeks after implant). In vitro assays, developed to elucidate the role of specific construct components in the in vivo remodeling, allowed to determine that fibrin degradation products enhanced the in vitro endothelial cell proliferation, as well as the macrophage migration; whereas the presence of bevacizumab was capable of counteracting these effects. The proposed pharmacological control of angiogenesis by a therapeutic drug released from a scaffold might enhance cartilage regeneration by MACI approaches, possibly allowing it to bypass the complex and costly phase of graft preculture to gain increased functionality.
Introduction
Damaged articular cartilage has a limited capacity of self-repair due to its avascular nature and low cellular mitotic activity.1–4 Cell-based repair techniques, such as matrix-induced autologous chondrocyte implantation, showed positive clinical outcomes, despite the formation of a fibrocartilaginous/fibrous repair tissue that is characterized by inferior mechanical properties and limited durability.5 Most likely, the lack of essential extracellular matrix (ECM) components, including high-molecular-weight hyaluronic acid, and other anti-angiogenic factors—such as chondromodulin, endostatin, and angiostatin6—exposes the not fully mature engineered tissues to an early blood vessel invasion. Such a host reaction might lead to final poor cartilaginous quality7 and, eventually, to premature implant degradation. Thus, control of angiogenesis might be crucial for both the development and the maintenance of physiological articular cartilage.8 In particular, it has been demonstrated that vascular endothelial growth factor (VEGF), one of the most potent angiogenic factors, plays an essential role in the ossification process at the level of the growth plate, modulating cartilage vascularization and hypertrophy.9 Recent data also reveal that chondrocyte-derived VEGF promotes catabolic pathways in the osteoarthritic cartilage.10
Neo-angiogenesis is also accompanied by the massive infiltration of mononuclear cells, such as monocytes. VEGF acts as a powerful chemoattractant for monocytes,11 which could potentially lead to a fast macrophage-driven in vivo resorption of the implanted engineered cartilage. Taken together, these aspects strongly underline the importance to control angiogenesis, and, in particular, the signaling of VEGF in cartilage tissue engineering (CTE). To this extent, cell-based anti-angiogenic gene therapies for cartilage regeneration have been already successfully investigated by inducing overexpression of either endostatin12,13 or chondromodulin.14 In addition, overexpression of soluble VEGF receptor-1 combined with the release of growth factors belonging to the transforming growth factor beta (TGF-β) superfamily enhanced cartilage regeneration in both rat osteoarthritic10 and osteochondral defect models.15
We hypothesized that VEGF blockade by using a biomaterial-based anti-angiogenic drug release system could provide—right upon implantation—an appropriate environment for the formation of stable cartilage by freshly seeded engineered constructs. In particular, we developed a hyaluronan/fibrin-based porous scaffold which was functionalized by the incorporation of a humanized monoclonal anti-VEGF antibody (bevacizumab)16 that binds to human VEGF17 and is currently used as an anti-angiogenic therapeutic drug in the treatment of metastatic colorectal cancer, metastatic kidney cancer, and glioblastoma. The use of a drug-eluting scaffold would overcome the limitations of gene therapy in terms of a direct clinical translation.18
High-molecular-weight hyaluronan and fibrin were chosen in virtue of their biocompatibility, chondro-supportive nature,1,4 and extensive clinical use.2,19–21
Among the promising cell sources for CTE, we opted for nasal chondrocytes (NC), as they represent one of the most interesting candidates for clinical application in virtue of (1) the relative ease and low morbidity of the harvest procedure22; (2) a better retained capacity on cell expansion to re-differentiate and generate hyaline-like tissue23 as compared with chondrocytes of other origin; and (3) their capacity to properly respond to mechanical forces which are typically associated with joint loading.24
Despite the orthotopic model being a more clinically relevant approach, in this study, we decided to use a subcutaneous implantation in nude mice, as it represents the most efficacious model that is used for testing the intrinsic capacity of constructs to form stable cartilage tissue,7 being characterized by a more vascularized and hostile microenvironment and, therefore, representing a more arduous testing ground for our purposes.
Materials and Methods
All reagents were purchased from Sigma Aldrich, unless otherwise stated, and were used without further purification. Culture media and supplements were from Gibco (Invitrogen).
Bevacizumab activity and dosage
A set of preliminary experiments was performed on human umbilical vein endothelial cells (HUVEC) in order to determine the suitable bevacizumab concentration to be loaded into the scaffolds. HUVEC were cultured at a density of 5000 cells/well overnight in growth medium (GM, M199 supplemented with 20% fetal bovine serum [FBS], 100 μg/mL endothelial cells growth supplement, 50 IU/mL heparin, 100 IU/mL penicillin, and 100 μg/mL streptomycin). The next day, GM was replaced with low-FBS medium (assay medium [AM], consisting of M199 with 5% FBS, 10 IU/mL heparin, and 1% penicillin/streptomycin), supplemented with recombinant human VEGF (R&D Systems) at different titers between 0 and 10 ng/mL. Bevacizumab (Avastin®; Roche) at different concentrations in the 1–5 μg/mL range was added to AM.
After 2 days, HUVEC metabolic activity was measured by MTS assay (Cell Titer 96®; Promega).
Scaffold preparation
High-molecular-weight sodium hyaluronate (10 mg/mL) and fibrinogen (20 mg/mL) were separately dissolved in saline (0.9% w/v of NaCl) and mixed using the same volume.25 Aprotinin (3000 KIU per mL of solution) and fibrin-stabilizing factor XIIIa (Abnova; 50 ng per mg fibrinogen) were added to the solution under mild stirring. Addition of bevacizumab was performed at two different concentrations with regard to the total volume, selected on the basis of the dosage experiments described earlier: 3.75 μg/mL (hereinafter named HA-FIB-B3.75) and 5 μg/mL (HA-FIB-B5). Samples without bevacizumab were also synthesized (HA-FIB). Polymerization of fibrinogen was achieved by the addition of a 100 IU/mL thrombin solution (0.5 IU per mg fibrinogen). Solution was transferred into a 96-well plate and incubated at 37°C for 30 min until gelation. Cylindrical porous scaffolds (6 mm diameter and 8 mm height) were obtained after freeze drying the gels.
An additional set of materials without hyaluronan, either supplemented (hereinafter FIB-B3.75) or not (FIB) with 3.75 μg/mL bevacizumab, was synthesized in the same experimental conditions to be used as a control for further in vitro assays.
Degradation and water uptake assays
Weight loss of the scaffolds was monitored as a function of incubation time in cell culture medium. Scaffolds (n=3 for each experimental condition, weighing ∼10 mg each) were incubated at 37°C into sealed tubes containing 1 mL of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. Specimens were retrieved at selected time points for approximately 3 weeks, blot dried, and weighed (Wt). Medium was replaced every 2 days. Scaffold weight loss ratio was defined as (W0−Wt)/W0%, where W0 is the initial wet weight and Wt is the wet weight at a given time point. Scaffold water uptake ratio was calculated as UR=(Wwt−Wd0)/Wd0, where Wd0 is the scaffold initial dry weight and Wwt is its wet weight after 1 h incubation in saline solution at 37°C.26
Scanning electron microscopy and mercury intrusion porosimetry
Gold-sputtered scaffold specimens underwent microstructural investigation by Field Emission Scanning Electron Microscopy (FE-SEM, Leo Supra 1535; LEO Electron Microscopy). Scaffold pore size distribution was determined by mercury intrusion porosimetry (Porosimeter 2000; Carlo Erba Instruments) according to the Washburn equation:
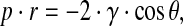 |
where p is the applied pressure, r is the pore radius, γ is the surface tension of mercury (480 mN/m), and θ (141.3°) is the contact angle of mercury.
Compressive mechanical test
Previously, hydrated scaffolds were tested in triplicate under unconfined compression using a mechanical testing system equipped with a 10 N load cell (model 3365; Instron) at a strain rate of 0.05 min−1. The compressive modulus (E*) was calculated as the slope in the linear range of the stress–strain curve.26,27
ELISA quantification of bevacizumab release
HA-FIB-B3.75 and HA-FIB-B5 constructs were incubated in DMEM at 37°C and 5% CO2, and supernatants were withdrawn at fixed time points, for approximately 3 weeks. 96-well plates were incubated overnight at 4°C with 50 μL of supernatants, and solutions with different concentrations of bevacizumab in order to obtain the calibration curve. Unspecific protein-binding sites were saturated by 2 h incubation at room temperature (RT) with phosphate-buffered saline supplemented with 0.05% Tween 20 and 1% bovine serum albumin. After rinsing, horseradish peroxidase-conjugated anti-mouse IgG (H+L) antibody (Southern Biotech; dilution 1:3000) was added for 90 min at RT followed by o-phenylenediamine dihydrochloride substrate (Sigma Aldrich). The reaction was stopped by adding 10% sulfuric acid and, after incubation for 30 min, absorbance was read at 492 nm on a microplate reader (Infinite M200; Tecan).
Cell isolation and expansion
Biopsies were harvested from the nasal cartilage septum of four cadavers (mean age: 51 years; range 31–84 years), following informed consent by relatives and in accordance with the Local Ethical Committee. NC were isolated by digesting minced cartilage in 0.15% type II collagenase for 22 h at 37°C. NC were expanded in DMEM containing 10% FBS, 4.5 mg/mL d-glucose, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 mM HEPES buffer, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 0.29 mg/mL l-glutamate (complete medium) supplemented with 1 ng/mL of TGF-β1 (R&D Systems) and 5 ng/mL of fibroblast growth factor-2 (FGF-2; R&D Systems)24 up to passage 3.
Cytotoxicity test
Cytotoxicity tests were performed on scaffolds seeded with NC (5×103 cells/well) using Vybrant Cytotoxicity Assay Kit® (Life Technologies) at 4, 8, and 24 h according to the manufacturer's protocol.
HUVEC proliferation assay
Scaffolds were incubated in complete medium. Supernatants were timely collected for approximately 7 days of incubation at 37°C and 5% CO2 and stored at−20°C until analysis.
HUVEC proliferation assays (n=3) were performed as previously described,28 adding to VEGF-supplemented AM (0–10 ng/mL VEGF) the previously collected supernatants (mixed using the same volume). Control experiments were performed by supplementing AM with bevacizumab (1.5 or 3.75 μg/mL) or with high-molecular-weight HA (500 μg/mL). After 2 days, HUVEC metabolic activity was measured by MTS assay (Cell Titer 96®; Promega). Data were normalized versus proliferation of HUVEC cultured in GM.
Monocyte migration assay
Scaffolds were incubated in serum-free medium (SFM) for approximately 7 days at 37°C and 5% CO2. Supernatants were timely collected and stored at −20°C until analysis. Peripheral blood mononuclear cells were isolated from peripheral blood of healthy donors (n=2) by gradient centrifugation. Monocytes were purified using the MACS CD14 isolation kit (Miltenyi Biotec), according to the manufacturer's protocol. CD14+ monocytes were cultured in the upper chamber of an HTS-Transwell-24-well microplate (Corning) in SFM and incubated with the previously collected supernatants loaded in the lower chamber for 20 h at 37°C and 5% CO2.29 SFM and SFM supplemented with 10 ng/mL of VEGF11 were used as negative and positive controls, respectively. SFM with 10 ng/mL VEGF and bevacizumab (at the optimal stoichiometric ratio of 2.6:1 with regard to VEGF16) was used as an additional control. Cell migration was measured in terms of DNA quantification using CYQuant® cell proliferation assay kit (Life Technologies) and expressed as the percentage of the migrated monocytes of a given experimental condition with regard to the SFM group.
In vitro pellet culture model
Chondrogenic re-differentiation capacity of two NC primaries (at least three replicates per condition per assay) was tested in a pellet culture model.30 Briefly, pellets were generated by 5×105 cells per pellet and cultured for 2 weeks in SFM supplemented with 1% ITS+1 liquid media supplement, 1.25 mg/mL human serum albumin, 0.1 mM l-ascorbic acid 2-phosphate sesquimagnesium salt, 10−7 M dexamethasone, and 10 ng/mL TGF-β1, with or without the addition of 3.75 μg/mL bevacizumab.
In vitro culture on scaffolds
HA-FIB-B3.75, HA-FIB-B5, HA-FIB, and FIB-lyophilized scaffolds were placed in agarose-coated six-well plates and seeded with NC (two donors with at least three replicates per condition per assay) by adding 10 μL of cell suspension (at a density of 1.5×107 cells/cm3) from the top. After an incubation of 2 h at 37°C, 3 mL of complete medium supplemented with 10 mg/mL insulin (Actrapid HM; Novo Nordisk Pharma AG), 0.1 mM ascorbic acid 2-phosphate, 10 ng/mL TGF-β3 (Novartis), and 3000 KIU/mL aprotinin31,32 was added, and constructs were cultured for 2 weeks by changing the medium twice a week.
Glycosaminoglycan and DNA quantification
Pellets and/or in vitro constructs were digested with protease K 1 mg/mL protease K in 50 mM Tris with 1 mM EDTA, 1 mM iodoacetamide, and 10 mg/mL pepstatin-A for 15 h at 56°C. Glycosaminoglycan (GAG) amount was measured spectrophotometrically using 1,9-dimethylmethylene blue chloride dye.33 Results were normalized by DNA levels, which were assessed by CyQuant® cell proliferation assay kit (Molecular Probes, Invitrogen).32
Real-time quantitative reverse-transcriptase polymerase chain reaction
Total RNA was extracted from pellets and/or in vitro constructs using TRIzol® (Life Technologies), and cDNA was generated as previously described.34 TaqMan® Gene Expression or on-Demand assays (Life Technologies) were used on a ABI 7900 Fast Realtime PCR System (Life Technologies) for gene expression measurements of type I (Hs00164004_m1) and II (Hs00264051_m1) collagen, Sox-9 (Hs00165814_m1), VEGF (Hs00900055_m1), and chondromodulin-I (Lect-1, Hs0017087_m1), using GAPDH (Hs99999905_m1) as the housekeeping gene.
In vivo ectopic mouse model
HA-FIB-B3.75, HA-FIB-B5, and HA-FIB-lyophilized scaffolds were seeded as described earlier. After overnight incubation in DMEM containing 10% FBS, 4.5 mg/mL D-glucose, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 mM HEPES buffer, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 0.29 mg/mL L glutamate, fresh NC constructs (four donors with at least three replicates per condition per assay) were implanted in the back of nude mice (CD1 nu/nu, athymic, 5-week-old females; Charles River Laboratories) in pockets between excised muscle fascia and subcutaneous tissue for 1, 3, and 6 weeks. All the animals in this study were treated according to institutional guidelines.
Histological analysis
Explanted constructs underwent fixation and routine histological processing. Stainings were performed for Safranin O and Alizarin red. Immunohistochemistry on 5-μm-thick paraffin sections was performed for mouse anti-human collagen (types I, II, and X), matrix metalloproteinase 13 (MMP-13), and bone sialoprotein (BSP) (MP Biomedicals),35 using hematoxylin as a nuclear counterstain. Endogenous mouse immunoglobulins in the tissue were blocked using a M.O.M.™ kit (Vector Laboratories) according to the manufacturer's instructions. Images were acquired using an Olympus BX-61 microscope.
Immunofluorescence (IF) for rat anti-mouse CD31 and F4/80 (BD) was performed on 10-μm-thick cryosections of in vivo generated constructs, using DAPI (BD) as a nuclear counterstain. Images were acquired with a confocal microscope (LSM710; Zeiss).
The percentage of area infiltrated by either vessels (CD31+) or macrophages (F4/80+) was calculated on cross-sections of the implant, excluding the outer fibrotic capsule (n=5 per experimental group) using Image J software (NIH; Bethesda).
The quality (by Safranin O staining), calcification (by Alizarin red staining), and host vessels ingrowth (by IF for CD31) of the engineered cartilaginous tissues (n≥3 per each experimental group) were assessed blindly using an ad hoc scoring system, which combines the already used Bern36 and ICRS II37 scores (Table 1). The proposed score was validated by calculating the maximum and overall variability (σmax, σov) of the scores among five blind observers as follows:
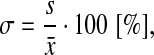 |
Table 1.
Histological Scoring System
| Scoring categories | Score |
|---|---|
| 1. Intensity of Safranin O-fast green stain36 | |
| No stain | 0 |
| Weak staining of poorly formed matrix | 1 |
| Moderately even staining | 2 |
| Even dark stain | 3 |
| 2. Distance between cells/amount of matrix accumulated36 | |
| High cell density with no matrix in between (no spacing between cells) | 0 |
| High cell density with little matrix in between (cells <1 cell-size apart) | 1 |
| Moderate cell density with matrix (cells ∼1 cell-size apart) | 2 |
| Low cell density (cells >1 cell-size apart) with an extensive matrix | 3 |
| 3. Cell morphologies36 | |
| Condensed/necrotic/pyknotic bodies | 0 |
| Spindle/fibrous | 1 |
| Mixed spindle/fibrous with rounded chondrogenic morphology | 2 |
| Majority of rounded/chondrogenic | 3 |
| 4. Uniformity of Safranin O staining37 | |
| Complete disorganization of stained areas | 0 |
| Less than 50% of Safranin O stained areas with regard to the total volume | 1 |
| 50%–80% of Safranin O stained areas with regard to the total volume of the section | 2 |
| Overall homogeneity of the staining | 3 |
| 5. Abnormal calcifications within the cartilage newly formed37 | |
| Strong presence of abnormal calcifications inside the neo-formed cartilage | 0 |
| Presence of some calcification spots inside the newly formed cartilage | 1 |
| Presence of very few calcification spots inside the repaired cartilage | 2 |
| Complete absence of calcifications throughout the newly formed cartilage | 3 |
| 6. Vessels ingrowth (within the newly formed tissue)37 | |
| Vessels ingrowth inside the core of the repaired cartilage | 0 |
| Vessels ingrowth until the inner rim of the neo-formed cartilage (approximately two thirds of newly formed cartilage's total volume) | 1 |
| Vessels ingrowth until the outer rim of the neo-formed cartilage (approximately one third of newly formed cartilage's total volume) | 2 |
| No presence of vessels or vessels confined in the external fibrotic capsule | 3 |
| 7. Final score | |
| Not-formed cartilage. Basically, just fibrotic tissue | 0–6 |
| Inhomogeneous and fibrocartilagineous tissue, having just a few and nonconnected areas of a good newly formed cartilage | 6.1–11 |
| Moderately good tissue, having less than 60%—with regard to the total volume—of a good newly formed cartilage | 11.1–15 |
| Very good quality of homogeneity of newly formed cartilage | 15.1–18 |
where s is the variance and 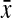 is the average. These values were calculated to be 21.43% and 8.16% for σmax and σov, respectively.
is the average. These values were calculated to be 21.43% and 8.16% for σmax and σov, respectively.
Statistical analysis
Data are presented as means±standard deviation, SD (n≥3). Statistical analysis was performed using the unpaired or nonparametric t-test, taking into account the normal distribution of the collected data by means of Prism® software (GraphPad Software); p-values lower than 0.05 indicate statistically significant differences.
Results
Scaffold characterization
The synthetic route for the preparation of hyaluronan/fibrin-based scaffolds resulted in highly reproducible incorporation of bevacizumab that did not affect their final microstructure, pore size, and distribution (Fig. 1A, B). Scaffolds displayed a similar porosity (70%±5%) regardless of the concentration of bevacizumab, with a similar pore size in the range of 20–220 μm.
FIG. 1.

Scaffold characterization. Scanning electron microscopy micrographs of scaffolds functionalized (A) or not (B) with bevacizumab. Scale bar=100 μm; insets show higher magnification pictures. (C) Bevacizumab cumulative release curve over 3 weeks of in vitro culture, expressed as% with regard to the total amount; (D) bevacizumab activity and dosage measured via MTS assay. Human umbilical vein endothelial cells (HUVEC) showed a vascular endothelial growth factor (VEGF) dose-dependent proliferation rate reaching a plateau at a concentration of 5 ng/mL (assay medium [AM] curve). Bevacizumab, at both tested concentrations (1.5 and 3.75 μg/mL), efficiently and significantly reduced HUVEC metabolic activity for approximately 10 ng/mL of VEGF (*p<0.05, °p<0.01, +p<0.001).
The compressive modulus (E*) was similar in all the experimental groups, ranging from 8.35±0.04 to 8.39±0.08 kPa.
NC viability was higher than 99.2% for all scaffold compositions, demonstrating their noncytotoxicity. HA-FIB, HA-FIB-B3.75, and HA-FIB-B5 showed similar behavior in terms of weight loss and water uptake, suggesting that the addition of bevacizumab did not affect the chemo-physical properties of the final scaffolds. Scaffolds lost ∼50% of their total weight in the first 48 h, whereas the remaining 45% was slowly degraded in the next 3 weeks of culture. Water uptake ratio was calculated to be ∼7.6 w/w for all scaffolds.
Cumulative bevacizumab release profile was about one fourth of the initial amount during the first 48 h; while from day 4 to 10, its rate slowed down until complete elution during the third week of in vitro soaking (Fig. 1C).
Bevacizumab activity and dosage
HUVEC showed a VEGF dose-dependent proliferation rate, reaching a plateau at a concentration of 5 ng/mL (Fig. 1D, positive control curve). Bevacizumab, at a concentration as low as 1.5 μg/mL, efficiently and significantly reduced HUVEC metabolic activity for approximately 10 ng/mL of VEGF (Fig. 1D). At a higher concentration of 3.75 μg/mL, bevacizumab was more effective in blocking the VEGF-induced HUVEC proliferation. Therefore, this was selected as a suitable concentration for scaffold composition (HA-FIB-B3.75). In addition, a higher-level bevacizumab concentration of 5 μg/mL—the highest concentration not affecting the final scaffold microstructure—was chosen (HA-FIB-B5) for the in vivo experiments.
In vitro 3D pellet culture
Supplementation of bevacizumab in the culture medium did not affect the in vitro chondrogenic differentiation potential of NC. Safranin O staining showed no major differences, in terms of staining intensity and uniformity, chondrocyte morphology, and no statistically significant difference was found in the GAG content for pellets generated by NC treated or not with bevacizumab (Fig. 2A). The expression at mRNA level of Sox9, type I collagen, and VEGF was similar in the two experimental groups (Fig. 2B); whereas a slight down-regulation in type II collagen was observed. However, this effect did not alter the expression of type II collagen at protein level, as confirmed by immunohistochemistry for this marker (data not shown).
FIG. 2.

In vitro 3D pellet and scaffold-based culture systems. (A, B) In vitro pellet culture. (A) Quantification of the glycosaminoglycan (GAG)/DNA ratio for pellets generated by nasal chondrocytes (NC) cultured with chondrogenic medium supplemented or not with 3.75 μg/mL of bevacizumab. (B) Analysis at mRNA level of Sox9, collagen (types I and II), and VEGF genes. (C, D) In vitro scaffold-based culture. (C) Quantification of the GAG/DNA ratio of cartilaginous constructs generated by NC loaded on FIB control scaffold, HA-FIB either alone, or functionalized by the supplementation of 3.75 (HA-FIB-B3.75) or 5 (HA-FIB-B5) μg/mL of bevacizumab. (D) Analysis at mRNA level was also performed for Sox9, collagen (types I, II, and X), VEGF, and chondromodulin genes. *p<0.01.
In vitro 3D culture on scaffolds
Release of bevacizumab from the scaffolds did not affect NC chondrogenic differentiation capacity, confirming the findings of the 3D pellet culture model. We, indeed, observed a quite similar cartilaginous ECM, intensively stained for GAG with chondrocyte-like cell morphology in all hyaluronan-containing scaffolds regardless of bevacizumab supplementation. Interestingly, FIB scaffolds did not result in being chondrosupportive as those containing HA, as the deposited ECM was negatively stained for GAGs (data not shown). A comparison with nonseeded HA-FIB scaffolds enabled the exclusion of potential biases or artifacts in histological and biochemical assays.
As compared with the engineered tissues generated with FIB scaffolds, the constructs engineered with scaffolds and also containing HA deposited a significantly higher amount of GAG (Fig. 2C). The presence of HA gave rise to a significantly increased expression of genes of interest, such as type II collagen and chondromodulin (Fig. 2D). The addition of bevacizumab did not affect the expressions of cartilaginous markers.
In vivo ectopic mouse model
After subcutaneous implantation, NC-based constructs generated without bevacizumab were mostly resorbed (Fig. 3). Indeed, the HA-FIB constructs that persisted at 6 weeks in vivo were as low as 18.2% (2 out of 11). On the contrary, the engineered tissues generated with HA-FIB-B3.75 and HA-FIB-B5 scaffolds showed higher survival rates at 6 weeks in vivo, corresponding to 60% (6 out of 10) and 75% (3 out of 4), respectively. Interestingly, the percentage of not-resorbed constructs remained unaltered after 3 weeks. These data remarkably underline the role of the early blocking of angiogenesis for the successful implantation of an immature graft in vivo.
FIG. 3.

In vivo construct degradation—percentage of the non-resorbed engineered tissues with regard to the total number of implants.
Despite the different survival rate, no major differences were observed in terms of cartilage quality between HA-FIB, HA-FIB-B3.75, and HA-FIB-B5 constructs once an initial critical phase of tissue formation has been overcome, demonstrating the self-sustaining capacity of the neo-formed engineered cartilage. Since no remarkable differences were found between the groups at different bevacizumab concentrations (3.75 or 5 μg/mL), histological results are presented only for the HA-FIB-B3.75 group (Fig. 4). After just 1 week, we observed the formation of a cartilaginous ECM, positively stained for Safranin O and type II collagen with chondrocyte-like morphology in all the experimental conditions for the not-resorbed constructs (Fig. 4A, B, D, E), although type I collagen staining was intense and uniform throughout the whole construct (Fig. 4C, F). After 6 weeks in vivo, the cartilaginous ECM showed an even more intense and uniform staining for GAG (Fig. 4G, L) and type II collagen (Fig. 4H, M); whereas type I collagen was present only in the outer edges of the implants (Fig. 4I, N). Cells were mainly showing a typical chondrocyte morphology, and no necrotic or pyknotic cells were found in all the experimental conditions.
FIG. 4.

In vivo chondrogenesis. Safranin O staining: (A) HA-FIB, 1 week, (D) HA-FIB-B3.75, 1 week, (G) HA-FIB, 6 weeks, (L) HA-FIB-B3.75 6 weeks. Immunohistochemistry for type II collagen: (B) HA-FIB, 1 week, (E) HA-FIB-B3.75, 1 week, (H) HA-FIB, 6 weeks, and (M) HA-FIB-B3.75 6 weeks. Immunohistochemistry for type I collagen: (C) HA-FIB, 1 week, (F) HA-FIB-B3.75, 1 week, (I) HA-FIB, 6 weeks, and (N) HA-FIB-B3.75, 6 weeks. Scale bar: 50 μm. Color images available online at www.liebertpub.com/tea
The stability of the chondrogenic phenotype reached by the NC in vivo was assessed by a histological analysis for hypertrophic markers (such as type X collagen) and other key molecules involved in cartilage remodeling (such as MMP-13 and bonesialoprotein), which were negative in all experimental groups over time (data not shown).
At 1 week, in all the groups, no CD31+ cells were found inside the neo-formed cartilaginous ECM, but only in the host fibrotic capsule surrounding the implant. At 3 weeks, host vessels infiltration toward the center of the construct was reported only for the HA-FIB group, giving rise to a process eventually leading to the breakdown of the tissue-engineered cartilage at later time points (Fig. 3). On the contrary, CD31-positive vessel structures remained confined at the outer edges of the engineered tissue for both HA-FIB-B3.75 (Fig. 5B) and HA-FIB-B5 (Fig. 5C), which is consistent with the capacity of the eluted bevacizumab to effectively inhibit neo-angiogenesis. Quantification of the percentage of CD31+ areas showed a statistically significant difference (p<0.05) between HA-FIB constructs and bevacizumab-containing constructs HA-FIB-B3.75 and HA-FIB-B5 (Fig. 5D).
FIG. 5.
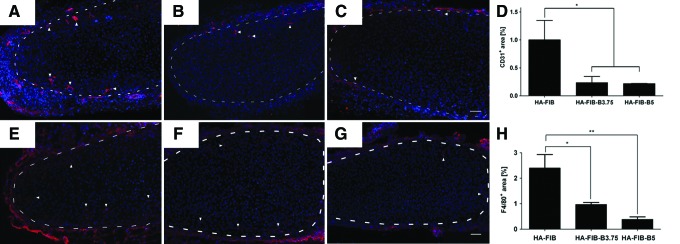
In vivo host vessel ingrowth. Immunofluorescence (IF) for CD31 at 3 weeks' time point of (A) HA-FIB, (B) HA-FIB-B3.75, (C) HA-FIB-B5; (D) vessels quantification, calculated as the percentage of CD31+ area with regard to the total picture area (n=5 per experimental group). Inflammatory response. IF for F4/80 at 3 weeks' time point of (E) HA-FIB, (F) HA-FIB-B3.75, and (G) HA-FIB-B5 samples; (H) migrated monocytes quantification, calculated as the percentage of F4/80+ area with regard to the total picture area (n=5 per experimental group). Scale bar: 50 μm. *p<0.01, **p<0.001. Color images available online at www.liebertpub.com/tea
These observations matched with the results of the histological scoring. Statistically significant differences were, indeed, observed, particularly with regard to the constructs vascularization at 3 weeks between HA-FIB and HA-FIB-B3.75 (p<0.05), and at 6 weeks between HA-FIB and both HA-FIB-B3.75 (p<0.05) and HA-FIB-B5 (p<0.001) (Table 2).
Table 2.
Histological Scoring Evaluation
|
Sample |
HA-FIB |
HA-FIB-B3.75 |
HA-FIB-B5 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Time point | 1 week | 3 week | 6 week | 1 week | 3 week | 6 week | 1 week | 3 week | 6 week |
| Scoring categories | |||||||||
| 1. Intensity | 0.82±0.98 | 1.82±0.87 | 2.70±0.48 | 0.67±0.78 | 1.82±0.60 | 3.00±0.00 | 1.00±1.00 | 1.91±0.54 | 2.64±0.50 |
| 2. Density | 0.55±0.69 | 1.36±0.50 | 2.10±0.57 | 1.00±0.95 | 1.64±0.47 | 2.27±0.65 | 1.00±0.89 | 1.91±0.83 | 2.45±0.69 |
| 3. Morphology | 1.27±1.35 | 1.82±0.40 | 2.50±0.53 | 1.08±1.00 | 1.73±0.67 | 2.91±0.30 | 1.36±1.29 | 2.00±0.63 | 2.55±0.69 |
| 4. Uniformity | 0.55±0.69 | 1.64±0.50 | 2.20±0.63 | 0.92±1.00 | 1.64±0.50 | 2.55±0.69 | 0.91±0.94 | 1.36±0.50 | 2.00±1.00 |
| 5. Calcifications | 3.00±0.00 | 3.00±0.00 | 2.40±0.52 | 3.00±0.00 | 3.00±0.00 | 2.91±0.30 | 3.00±0.00 | 3.00±0.00 | 3.00±0.00 |
| 6. Vascularization | 2.67±0.71 | 1.22±0.67 | 1.38±0.74 | 3.00±0.00 | 1.78±0.67a | 2.78±0.44b | 2.78±0.44 | 1.89±0.60a | 2.67±0.50b |
| Final score | 8.85±2.06 | 10.86±1.39 | 13.28±1.43 | 9.76±1.87 | 11.6±1.32 | 16.41±1.13c | 10.05±2.13 | 12.07±1.41a | 15.3±1.57b |
Statistical differences evaluated between the scaffolds with and without bevacizumab at each time point.
p<0.05.
p<0.01.
p<0.001.
At 3 weeks, the infiltration toward the center of the cartilaginous ECM of F4/80+ murine macrophages appeared to be stronger in constructs without bevacizumab as compared with both HA-FIB-B3.75 and HA-FIB-B5 (Fig. 5E–G). The quantification of F4/80+ cells, indeed, showed a significantly higher number of monocytes in the HA-FIB group as compared with HA-FIB-B3.75 and HA-FIB-B5 groups (Fig. 5H).
In order to elucidate the reported differences in terms of host vessels and monocytes invasion in vivo, all the scaffold groups were tested in vitro against HUVEC proliferation (Fig. 6A) and monocytes migration (Fig. 6B).
FIG. 6.

Scaffold degradation products characterization. The role of each scaffold component was tested by using a HUVEC proliferation assay (A) and an ad hoc developed monocytes migration assay (B). (A) For HUVEC proliferation assay, high-molecular-weight HA (HMW-HA), AM, and growth medium (GM) were used as controls. HUVEC metabolic activity is presented as the ratio between the given experimental condition and the GM condition (fold increase). (B) Monocytes migration assay performed on FIB degradation products, calculated as the percentage of the migrated monocytes of a given experimental condition with regard to the serum-free medium group. Supplementation of 10 ng/mL of VEGF alone or in combination with bevacizumab (using the optimal stoichiometric ration 1:2.615), was used as a control.
Effect of bevacizumab and scaffold degradation products on HUVEC proliferation
Figure 6A shows the results of HUVEC proliferation assay for scaffolds at different compositions, as the ratio between the given experimental condition and the GM condition (fold increase). FIB degradation products were found to strongly enhance HUVEC proliferation, with a 1.71±0.09-fold increase with regard to HUVEC cultured in GM. HA release from HA-FIB scaffolds appeared to mitigate this proliferative effect (1.02±0.09-fold vs. GM group), which was reliably due to the reported anti-angiogenic effect of high-molecular-weight hyaluronan.38
Elution of bevacizumab from FIB-B3.75 scaffolds (not containing hyaluronan) induced a progressive reduction of the HUVEC metabolic activity, which was similar to the levels obtained with HA-FIB scaffolds, and, therefore, similar to the GM control. It should be noted, in HA-FIB-B3.75 scaffolds, that the synergistic effect of HA and bevacizumab contributed toward reducing the HUVEC proliferation rate since day 1. At day 7, HA and bevacizumab were able to reduce HUVEC proliferation to the threshold level represented by the negative control (AM with no VEGF).
Effect of bevacizumab and scaffold degradation products on monocyte migration
Figure 6B shows the results of monocyte migration assay for FIB scaffolds at different time points, as the percentage of the migrated monocytes of a given experimental condition with regard to the SFM group. VEGF supplement to SFM strongly increased the migration of monocytes (+78.07%±1.14% vs. SFM group) (Fig. 6B), as previously shown by Zentilin et al.,11 while the addition of bevacizumab to the VEGF-containing medium completely suppressed their migration (−10% vs. SFM). It should be noted that degradation products obtained from FIB scaffolds increased the migration of monocytes to +12.28%±1.18% vs. the SFM group at day 7. On the contrary, no significant differences were observed between HA-FIB and SFM groups.
Discussion
Our study showed that an anti-angiogenic approach, applied to mimic the avascular nature of hyaline cartilage and to block host reaction, may support the formation of long-term, stable, and high-quality cartilaginous engineered tissue without any in vitro preconditioning.
Despite the promising results recently obtained in CTE field, new strategies are still warranted to overcome the intrinsic limitations to the extensive, costly, and time-consuming in vitro conditioning of the grafts.5 One possible approach relies on the direct implantation of cell-loaded 3D scaffolds, with no previous chondrogenic stimulus.39 To this extent, here we proposed to streamline and overcome the in vitro preconditioning by using a 3D scaffold that, while supporting in vivo chondrogenesis, inhibits host vessel ingrowth while preventing implant early resorption. We developed a scaffold based on well-established, FDA-approved materials for CTE, that is, fibrin and hyaluronan. In this system, fibrin provided the 3D porous structure, while hyaluronan represented the chondrogenic stimulus, which resulted in being a key requirement for inducing in vitro NC chondrogenic re-differentiation. The proposed scaffold displayed a strong chondrosupportive capacity particularly in vivo, leading to the formation of newly formed hyaline-like cartilage already after 1 week. In the last few years, several groups adopted gene therapy-based strategies to block angiogenesis for supporting cartilage regeneration. Progenitor cells were transduced to over-express autologous anti-angiogenic molecules, such as soluble Flt-1,10 endostatin,12,13 and chondromodulin.14 Nagai et al. proposed an alternative approach consisting of the systemic administration of bevacizumab (Avastin), which resulted in the augmented capacity of cartilage self-healing in rabbit osteochondral lesions, thus strengthening the key role of anti-angiogenic signaling in cartilage homeostasis.40 The latter approach holds less concerns in terms of safety and processing time, compared with gene therapy, but still requires systemic drug administration; while, for a clinically compliant articular regeneration strategy, a local treatment is desirable. Indeed, the systemic use of bevacizumab induces severe drawbacks, such as gastrointestinal perforation, serious bleeding, in addition to inherent/acquired resistance and induction of tumor invasiveness.41
The proposed work showed that anti-angiogenesis during cartilage regeneration can be pursued using a direct and clinically oriented approach by means of time-controlled, topical administration of an anti-angiogenic drug (i.e., bevacizumab, Avastin) and by paving the way for the use of freshly isolated patients' cells with regard to the extensively expanded and genetically modified cells.
The release of bevacizumab from implanted grafts during the first 2/3 weeks appeared to effectively block human VEGF released by the implanted NC and, therefore, inhibited host vessel ingrowth,42 as shown by histological images, likely preserving the constructs from resorption and leading to a long-term stability. Indeed, ∼82% of scaffolds without bevacizumab (HA-FIB experimental group) completely degraded after 6 weeks in vivo, which was reasonably due to newly formed matrix remodeling and resorption on host vessel and monocyte invasion, which was already evident at 3 weeks on implantation. Francioli et al.43 and Luo et al.44 demonstrated how the level of maturation of cartilaginous tissues generated in vitro is correlated with a different profile of inflammatory chemokine production (e.g., MCP-1, IL-8), and, in particular, how the in vitro preconditioning can mitigate the postimplantation inflammatory reaction. In particular, Luo et al.44 showed how a 2 week preculture of chondrocyte-based constructs results in a mild inflammatory response in the host tissue, with no vessels ingrowth found in the middle of the constructs and a fewer number of leukocytes in comparison with the “direct implantation group.” Therefore, we concluded that our strategy based on blocking VEGF signal by temporal release of bevacizumab on the graft implantation can streamline CTE applications (1) by overcoming the in vitro preconditioning phase, thus starting with freshly seeded scaffolds; (2) by limiting the inflammatory response; (3) by protecting immature cartilaginous matrix from remodeling; (4) by guaranteeing further tissue development (e.g., endogenous production of antiangiogenic molecules that usually self-protect cartilage from macrophage invasion and modulate the postimplantation inflammatory response); and, finally, (5) by allowing long-term stability of the cell-based grafts.
In order to better study how chondrogenesis is affected by the antiangiogenic drug bevacizumab, further investigations by using an inert and not degradable scaffold are necessary. Indeed, we have reported that fibrin degradation products remarkably enhance endothelial cell proliferation, as well as monocytes migration, and that bevacizumab and high-molecular-weight HA incorporated within the scaffolds counteract that angiogenic effect. Moreover, no major differences in terms of the quality of the neo-formed engineered cartilage were found among the not resorbed constructs, regardless of the presence of bevacizumab. This aspect is probably due both to the intrinsic chondrogenic potential of the NC primary culture and to the good chondrosupportive nature of the proposed scaffold.
Future studies will also aim at investigating the chondrogenic potential of the constructs engineered by using the bevacizumab-functionalized scaffold in an orthotopic and immunocompetent model, as well as at better elucidating the complex system of host reaction and mechanical stimulations. Besides showing the indirect effect of blocking VEGF, and therefore vessel ingrowth, on monocyte infiltration, our preliminary results might also suggest a possible direct effect of bevacizumab on monocyte proliferation, differentiation, and migration. Moreover, the use of bone marrow-derived mesenchymal stromal cells (BMSCs) might be contemplated to broaden the clinical scenario and cell source choice. It has been recently shown that BMSCs undergo hypertrophy, followed by formation of micro-ossicles after ectopic transplantation in immunodeficient mice.45,46 Since VEGF plays an essential role in these mechanisms, future experiments will investigate whether bevacizumab-induced VEGF blockade could affect BMSC endochondral fate and thus be instrumental for CTE with BMSC.
Conclusions
Our findings suggest that blocking angiogenesis in a chondrosupportive immature graft supports the formation of a long-term stable engineered cartilage, as it effectively preserves its avascular nature and prevents its resorption. The scaffold-based approach here that is proposed to limit spatially and temporally the delivery of an anti-angiogenic drug might represent a step forward in the current CTE scenario, offering a valid alternative to conventional biomaterial-induced autologous implantation techniques. The use of all FDA-approved materials for its synthesis, including the anti-angiogenic drug, and its validation with a chondrogenic and clinically relevant cell source, namely NC, are expected to allow a straightforward translation to a clinical setting.
Acknowledgments
The authors are grateful to Beatrice Tonnarelli for her expert contribution in the ICRS scoring process; and to Emanuele Trella and Marco Lepore for their valuable help in the experimental design of monocytes migration assay. This work was partially funded by the MIUR-FIRB (Grant RBAP06SPK5/2006) to CIR–“Università Campus Bio-Medico di Roma”, and by Swiss National Science Foundation (Grant 310030_126965/1) to A.B. M.C. was supported by a mobility grant funded by the Italian Ministry of University and Research.
Disclosure Statement
There are no conflicts of interest to declare. The writing of this article was the sole responsibility of the authors.
References
- 1.Wu S.C. Chang J.K. Wang C.K. Wang G.J. Ho M.L. Enhancement of chondrogenesis of human adipose derived stem cells in a hyaluronan-enriched microenvironment. Biomaterials. 2010;31:631. doi: 10.1016/j.biomaterials.2009.09.089. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen L.H. Kudva A.K. Guckert N.L. Linse K.D. Roy K. Unique biomaterial compositions direct bone marrow stem cells into specific chondrocytic phenotypes corresponding to the various zones of articular cartilage. Biomaterials. 2010;32:1327. doi: 10.1016/j.biomaterials.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 3.Ho S.T.B. Cool S.M. Hui S.M. Hutmacher D.W. The influence of fibrin based hydrogels on the chondrogenic differentiation of human bone marrow stromal cells. Biomaterials. 2010;31:38. doi: 10.1016/j.biomaterials.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 4.Ahmed T. Hincke M.T. Strategies for articular cartilage lesion repair and functional restoration. Tissue Eng Part B Rev. 2010;16:305. doi: 10.1089/ten.TEB.2009.0590. [DOI] [PubMed] [Google Scholar]
- 5.Pelttari K. Wixmerten A. Martin I. Do we really need cartilage tissue engineering? Swiss Med Weekly. 2009;139:602. doi: 10.4414/smw.2009.12742. [DOI] [PubMed] [Google Scholar]
- 6.Nyberg P. Xie L. Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 7.Moretti M. Wendt D.J. Dickinson S.C. Sims T.J. Hollander A.P. Kelly D.J. Prendergast P.J. Heberer M. Martin I. Effects of in vitro preculture on in vivo development of human engineered cartilage in an ectopic model. Tissue Eng. 2005;11:1421. doi: 10.1089/ten.2005.11.1421. [DOI] [PubMed] [Google Scholar]
- 8.Takita H. Kikuchi M. Sato Y. Kuboki Y. Inhibition of BMP-induced ectopic bone formation by an antiangiogenic agent (Epigallocatechin 3-Gallate) Conn Tissue Res. 2002;43:520. doi: 10.1080/03008200290000772. [DOI] [PubMed] [Google Scholar]
- 9.Gerber H.P. Vu T.H. Ryan A.M. Kowalski J. Werb Z. Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto T. Cooper G.M. Gharaibeh B. Meszaros L.B. Li G. Usas A. Fu F.H. Huard J. Cartilage repair in a rat model of osteoarthritis through intraarticular transplantation of muscle-derived stem cells expressing bone morphogenetic protein 4 and soluble Flt-1. Arthritis Rheum. 2009;60:1390. doi: 10.1002/art.24443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zentilin L. Tafuro S. Zacchigna S. Arsic N. Pattarini L. Sinigaglia M. Giacca M. Bone marrow mononuclear cells are recruited to the sites of VEGF-induced neovascularization but are not incorporated into the newly formed vessels. Blood. 2006;107:3546. doi: 10.1182/blood-2005-08-3215. [DOI] [PubMed] [Google Scholar]
- 12.Jeng L. Olsen B.R. Spector M. Engineering endostatin-producing cartilaginous constructs for cartilage repair using nonviral transfection of chondrocyte-seeded and mesenchymal-stem-cell-seeded collagen scaffolds. Tissue Eng Part A. 2010;16:3011. doi: 10.1089/ten.tea.2009.0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeng L. Olsen B.R. Spector M. Engineering endostatin-expressing cartilaginous constructs using injectable biopolymer hydrogels. Acta Biomater. 2012;8:2203. doi: 10.1016/j.actbio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 14.Klinger P. Surmann-Schmitt C. Brem M. Swoboda B. Distler J. Carl H.D. von der Mark K. Hennig F.F. Gelse K. Chondromodulin-I stabilizes the chondrocytes phenotype and inhibits endochondral ossification of cartilage repair tissue. Arthritis Rheum. 2011;63:2721. doi: 10.1002/art.30335. [DOI] [PubMed] [Google Scholar]
- 15.Kubo S. Cooper G.M. Matsumoto T. Phillippi J.A. Corsi K.A. Usas A. Li G. Fu F.H. Huard J. Blocking vascular endothelial growth factor with soluble Flt-1 improves the chondrogenic potential of mouse skeletal muscle-derived stem cells. Arthritis Rheum. 2009;60:155. doi: 10.1002/art.24153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y. Fei D. Vanderlaan M. Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335. doi: 10.1007/s10456-004-8272-2. [DOI] [PubMed] [Google Scholar]
- 17.Yu L. Wu X. Cheng Z. Lee C.V. LeCouter J. Campa C. Fuh G. Lowman H. Ferrara N. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest Ophthalmol Vis Sci. 2008;49:522. doi: 10.1167/iovs.07-1175. [DOI] [PubMed] [Google Scholar]
- 18.Mulligan R.C. The basic science of gene therapy. Science. 1993;260:926. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- 19.Spiller K.L. Maher S.A. Lowman A.M. Hydrogels for the repair of articular cartilage defects. Tissue Eng Part B Rev. 2011;17:281. doi: 10.1089/ten.teb.2011.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visna P. Pasa L. Cizmar I. Hart R. Hoch J. Treatment of deep cartilage defects of the knee using autologous chondrograft transplantation and by abrasive techniques—a randomized controlled study. Acta Chir Belg. 2004;104:709. doi: 10.1080/00015458.2004.11679648. [DOI] [PubMed] [Google Scholar]
- 21.Marcacci M. Berruto M. Brocchetta D. Delcogliano A. Ghinelli D. Gobbi A. Kon E. Pederzini L. Rosa D. Sacchetti, G.L., Stefani G., and Zanasi, S. Articular cartilage engineering with Hyalograft C: 3-year clinical results. Clin Orthop Relat Res. 2005;435:96. doi: 10.1097/01.blo.0000165737.87628.5b. [DOI] [PubMed] [Google Scholar]
- 22.Siegel N.S. Gliklich R.E. Taghizadeh F. Chang Y. Outcomes of septoplasty. Otolaryngol Head Neck Surg. 2000;122:228. doi: 10.1016/S0194-5998(00)70244-0. [DOI] [PubMed] [Google Scholar]
- 23.Tay A.G. Farhadi J. Suetterlin R. Pierer G. Heberer M. Martin I. Cell yield, proliferation, and postexpansion differentiation capacity of human ear, nasal, and rib chondrocytes. Tissue Eng. 2004;10:762. doi: 10.1089/1076327041348572. [DOI] [PubMed] [Google Scholar]
- 24.Candrian C. Vonwil D. Barbero A. Bonacina E. Miot S. Farhadi J. Wirz D. Dickinson S. Hollander A. Jakob M. Li Z. Alini M. Heberer M. Martin I. Engineered cartilage generated by nasal chondrocytes is responsive to physical forces resembling joint loading. Arthritis Rheum. 2008;58:197. doi: 10.1002/art.23155. [DOI] [PubMed] [Google Scholar]
- 25.Park S.H. Cui J.H. Park S.R. Min B.H. Potential of fortified fibrin/hyaluronic acid composite gel as a cell delivery vehicle for chondrocytes. Artif Org. 2009;33:439. doi: 10.1111/j.1525-1594.2009.00744.x. [DOI] [PubMed] [Google Scholar]
- 26.Davidenko N. Campbell J.J. Thian E.S. Watson C.J. Cameron R.E. Collagen–hyaluronic acid scaffolds for adipose tissue engineering. Acta Biomater. 2010;6:3957. doi: 10.1016/j.actbio.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Tan H. Wu J. Lao L. Gao C. Gelatin/chitosan/hyaluronan scaffold integrated with PLGA microspheres for cartilage tissue engineering. Acta Biomater. 2009;5:328. doi: 10.1016/j.actbio.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 28.Witzenbichler B. Asahara T. Murohara T. Silver M. Spyridopoulos I. Magner M. Principe N. Kearney M. Hu JS. Isner J.M. Vascular endothelial growth factor-C (VEGF-C/VEGF-2) promotes angiogenesis in the setting of tissue ischemia. Am J Pathol. 1998;153:381. doi: 10.1016/S0002-9440(10)65582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albini A. Benelli R. The chemoinvasion assay: a method to assess tumor and endothelial cell invasion and its modulation. Nat Prot. 2007;2:504. doi: 10.1038/nprot.2006.466. [DOI] [PubMed] [Google Scholar]
- 30.Francioli S.E. Martin I. Sie C.P. Hagg R. Tommasini R. Candrian C. Heberer M. Barbero A. Growth factors for clinical-scale expansion of human articular chondrocytes: relevance for automated bioreactor systems. Tissue Eng. 2007;13:1227. doi: 10.1089/ten.2006.0342. [DOI] [PubMed] [Google Scholar]
- 31.Wendt D. Marsano A. Jakob M. Heberer M. Martin I. Oscillating perfusion of cell suspensions through three-dimensional scaffolds enhances cell seeding efficiency and uniformity. Biotechnol Bioeng. 2003;84:205. doi: 10.1002/bit.10759. [DOI] [PubMed] [Google Scholar]
- 32.Miot S. Gianni-Barrera R. Pelttari K. Acharya C. Mainil-Varlet P. Juelke H. Jaquiery C. Candrian C. Barbero A. Martin I. In vitro and in vivo validation of human and goat chondrocyte labeling by green fluorescent protein lentivirus transduction. Tissue Eng Part C Methods. 2010;16:11. doi: 10.1089/ten.TEC.2008.0698. [DOI] [PubMed] [Google Scholar]
- 33.Farndale R.W. Buttle D.J. Barrett A.J. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- 34.Strobel S. Loparic M. Wendt D. Schenk A.D. Candrian C. Lindberg R.L.P. Moldovan F. Barbero A. Martin I. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res Ther. 2010;12:R34. doi: 10.1186/ar2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grogan S.P. Rieser F. Winkelmann V. Berardi S. Mainil-Varlet P. A static, closed and scaffold-free bioreactor system that permits chondrogenesis in vitro. Osteoarthritis Cartilage. 2003;11:403. doi: 10.1016/s1063-4584(03)00053-0. [DOI] [PubMed] [Google Scholar]
- 36.Grogan S.P. Barbero A. Winkelmann V. Rieser F. Fitzsimmons J.S. O'Driscoll S. Martin I. Mainil-Varlet P. Visual histological grading system for the evaluation of in vitro-generated neocartilage. Tissue Eng. 2006;12:2141. doi: 10.1089/ten.2006.12.2141. [DOI] [PubMed] [Google Scholar]
- 37.Mainil-Varlet P. Van Damme B. Nesic D. Knutsen G. Kandel R. Roberts S. A new histological scoring system for the assessment of the quality of human cartilage repair: ICRS II. Am J Sports Med. 2010;38:880. doi: 10.1177/0363546509359068. [DOI] [PubMed] [Google Scholar]
- 38.Pardue E.L. Ibrahim S. Ramamurthi A. Role of hyaluronan in angiogenesis and its utility to angiogenic tissue engineering. Organogenesis. 2008;4:203. doi: 10.4161/org.4.4.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niemeyer P. Lenz P. Kreuz P.C. Salzmann G.M. Sudkamp N.P. Schmal H. Steinwachs M. Chondrocyte-seeded type I/III collagen membrane for autologous chondrocyte transplantation: prospective 2-year results in patients with cartilage defects of the knee joint. Arthroscopy. 2010;26:1074. doi: 10.1016/j.arthro.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 40.Nagai T. Sato M. Kutsuna T. Kokubo M. Ebihara G. Ohta N. Mochida J. Intravenous administration of anti-vascular endothelial growth factor humanized monoclonal antibody bevacizumab improves articular cartilage repair. Arthritis Res Ther. 2010;12:R178. doi: 10.1186/ar3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320:130. doi: 10.1016/j.canlet.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 42.Mujagic E. Gianni-Barrera R. Trani M. Patel A. Gürke L. Heberer M. Wolff T. Banfi A. Induction of aberrant vascular growth, but not of normal angiogenesis, by cell-based expression of different doses of human and mouse VEGF is species-dependent. Hum Gene Ther Methods. 2013;24:28. doi: 10.1089/hgtb.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Francioli S. Cavallo C. Grigolo B. Martin I. Barbero A. Engineered cartilage maturation regulates cytokine production and interleukin-1β response. Clin Orthop Relat Res. 2011;469:2773. doi: 10.1007/s11999-011-1826-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo X. Zhou G. Liu W. Zhang W.J. Cen L. Cui L. Cao Y. In vitro precultivation alleviates post-implantation inflammation and enhances development of tissue-engineered tubular cartilage. Biomed Mat. 2009;4:025006. doi: 10.1088/1748-6041/4/2/025006. [DOI] [PubMed] [Google Scholar]
- 45.Pelttari K. Winter A. Steck E. Goetzke K. Hennig T. Ochs B.G. Aigner T. Richter W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006;54:3254. doi: 10.1002/art.22136. [DOI] [PubMed] [Google Scholar]
- 46.Scotti C. Tonnarelli B. Papadimitropolous A. Scherberich A. Schaeren S. Schauerte A. Lopez-Rios J. Zeller R. Barbero A. Martin I. Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. PNAS. 2010;107:7251. doi: 10.1073/pnas.1000302107. [DOI] [PMC free article] [PubMed] [Google Scholar]