

Abstract
Aluminum (Al3+) is the most prevalent metal in the earth's crust, and is a known human neurotoxicant. Al3+ has been shown to accumulate in the substantia nigra of Parkinson's disease (PD) patients, and epidemiological studies suggest correlations between Al3+ exposure and the propensity to develop both PD and the amyloid plaque-associated disorder Alzheimer's disease (AD). Although Al3+ exposures have been associated with the development of the most common neurodegenerative disorders, the molecular mechanism involved in Al3+ transport in neurons and subsequent cellular death has remained elusive. In this study we show that a brief exposure to Al3+ decreases mitochondrial membrane potential and cellular ATP levels, and confers dopamine (DA) neuron degeneration in the genetically tractable nematode Caenorhabditis elegans (C. elegans). Al3+ exposure also exacerbates DA neuronal death conferred by the human PD-associated protein α-synuclein. DA neurodegeneration is dependent on SMF-3, a homologue to the human divalent metal transporter (DMT-1), as a functional null mutation partially inhibits the cell death. We also show that SMF-3 is expressed in DA neurons, Al3+ exposure results in a significant decrease in protein levels, and the neurodegeneration is partially dependent on the PD-associated transcription factor Nrf2/SKN-1 and caspase Apaf1/CED-4. Furthermore we provide evidence that the deletion of SMF-3 confers Al3+-resistance due to sequestration of Al3+ into an intracellular compartment. This study describes a novel model for Al3+-induced DA neurodegeneration and provides the first molecular evidence of an animal Al3+ transporter.
Keywords: C. elegans, Parkinson's disease, Nrf2/Skn-1
Introduction
Al3+ is a highly abundant and ubiquitously distributed environmental and industrial toxicant (Vasudevaraju et al. 2008). Human exposure to Al3+ occurs through a number of mechanisms including soil and fertilizers, cookware, water from purification systems, as well as pharmaceutical and cosmetic preparations (Verstraeten et al. 2008). Al3+ is readily absorbed in the gastrointestinal tract, skin, and olfactory system, and can interact with a number of biological systems including the liver, kidney, bone, and brain. Bone can contain approximately 70% of the total Al3+ in the body (Crisponi et al. 2011). A physiological requirement for Al3+ has not been identified (Verstraeten et al. 2008).
High exposure to Al3+ is toxic to the central nervous system, and epidemiological and molecular evidence suggests a link between exposures, cell Al3+ concentrations, and the propensity to develop several neurological disorders (Vasudevaraju et al. 2008, Yokel 2006). Al3+ exposure has been reported to be a risk factor for the development of the most common neurodegenerative disorder Alzheimer's disease (AD). Al3+ has been found in senile plaques and neurofibrillary tangles in degenerating neurons in AD brains, and exposures can induce neurofibrillary tangles in vertebrate AD animal models (Vasudevaraju et al. 2008, Kawahara & Kato-Negishi 2011). Al3+ exposures have been associated with the development of Parkinson's disease (PD), which is characterized by motor deficits and the loss of dopamine (DA) neurons in the substantia nigra (SN) (Zayed et al. 1990a). Combinatorial long-term exposure of Al3+, Fe2+, and Mn2+ has also been associated with increasing the probability of developing PD (Zayed et al. 1990a). Al3+ has been shown to accumulate in the SN in PD and parkinsonism patients, affect tyrosine hydroxylase (TH) activity and inhibit DA synthesis (Erazi et al. 2011). Furthermore, the formation of the toxic moiety of the PD-associated protein α-synuclein has shown to be accelerated in the presence of Al3+, and PD Lewy bodies have been shown to contain Al3+ (Uversky et al. 2001).
Although the molecular basis of Al3+-induced toxicity is not well defined, oxidative stress appears to play a significant role in the cellular pathology (Verstraeten et al. 2008, Sanchez-Iglesias et al. 2009). Exposure of cells to Al3+ can increase reactive oxygen species (ROS) formation by interfering directly with mitochondrial function, or by increasing the redox activity of Fe2+ through the Fenton reaction (Vasudevaraju et al. 2008). Al3+ exposure can also increase oxidative stress by causing protein misfolding, reduced microtubule transport of neuronal vesicles, DNA damage, and lipid peroxidation resulting in apoptosis (Kumar et al. 2009, Yokel 2006).
The molecular basis of Al3+ transport into neurons and between intracellular compartments is largely unknown (Yokel 2002, Yokel 2006). Al3+ may bind to proteins and the plasma membrane and potentially enter the cell through diffusion, although in vitro transport kinetics do not support this mechanism. The physiochemical similarities (ionic radius and hydration shells) between Fe2+ and Al3+ suggest that Al3+ may bind to Fe2+-containing proteins and Fe+2 transporters (Martin 1986, Vasudevaraju et al. 2008). Al3+ has also been proposed to enter cells through transferrin receptor mediated endocytosis (TfR-ME) yet this does not appear to be the primary route of Al3+ influx (Yokel 2002, Yokel 2006). In vertebrates, the natural resistance-associated macrophage protein (NRAMP) family consists of Nramp1 and Nramp2 (divalent metal transporter 1, DMT-1) that are involved in the transport of divalent metals across plasma and intracellular membranes (Cellier et al. 1995). DMT-1 has been implicated in metal-associated DA neuronal death in PD (Salazar et al. 2008). In yeast, the DMT-1 homologues Smf2p and Smf3p transport Mn2+ across intracellular membranes (Portnoy et al. 2000). Recently a DMT-1 homologue in rice, Nrat1, has been shown to transport Al3+, and is involved in Al3+-associated cellular toxicity (Xia et al. 2010). Caenorhabditis elegans (C. elegans) contains 3 homologues to the vertebrate DMT-1, SMF-1–3 (Settivari et al. 2009). SMF-1 and SMF-2 are expressed in DA neurons and play a role in PD-associated 6-hydroxydopamine (6-OHDA) and Mn2+-induced DA neuron degeneration (Settivari et al. 2009). SMF-3 GFP fusions indicate expression in a variety of tissues in the nematode, including neurons, although localization to specific neuronal types has not been previously described (Bandyopadhyay et al. 2009). SMF-3 does not play a role in Mn2+-induced DA neuron degeneration (Settivari et al. 2009).
The nematode Caenorhabditis elegans (C. elegans) is a useful model system to explore the molecular basis of PD and DA neuron vulnerability to metals (Nass et al. 2008, Nass & Settivari 2008). All of the genes responsible for DA neurotransmission as well as apoptosis-associated genes are present in the nematode, and DA neuron specific expression of the green fluorescent protein (GFP) allows the eight DA neurons to be clearly visible in vivo under a fluorescent dissecting microscope (Nass & Blakely 2003). The DA neurons are also sensitive to PD-associated toxicants, and the expression of human α-synuclein confers DA neuronal death (Nass et al. 2001, Nass et al. 2002, Lakso et al. 2003, Cooper et al. 2006, Vartiainen et al. 2006). The DA neurons also express the PD-associated transcription factor Nrf2/SKN-1 that inhibits MeHg-induced neuronal death (Vanduyn et al. 2010).
Considering the strong conservation of genes and proteins between C. elegans and humans, and the high similarities on the molecular level in how both organisms respond to neurotoxicants, we asked whether the nematode C. elegans may be a useful genetic model to explore the molecular basis of Al3+-induced DA neurodegeneration in vivo. In this study we show that C. elegans recapitulates key molecular attributes of Al3+-induced vertebrate DA neuron toxicity, identify Al3+-associated DA neuron death effectors, and identify and characterize a novel Al3+ transporter that modulates intracellular Al3+ and inhibits DA neurodegeneration.
Methods
C. elegans strains and maintenance
The following strains have been described previously: BY250 (Pdat-1∷GFP); RJ907 (Pdat-1∷GFP; smf-1(eh5)); RJ938 (Pdat-1∷GFP; smf-2(gk133)); RJ934 (Pdat-1∷GFP; smf-3(ok1035)); BY273 (Pdat-1∷GFP; Pdat-1∷WTα-synuclein); and RJ928 (Pdat-1∷GFP; rrf-3(pk1426)) (Settivari et al. 2009, Nass et al. 2002, Lakso et al. 2003). C. elegans strains were cultured on OP50 or NA22 bacterial lawns on NGM or 8P plates, respectively, at 20°C according to standard methods (Brenner 1974, Hope 1999).
Toxicant exposures
To obtain synchronized L1 stage worms, gravid adults were treated with hypochlorite and the embryos were incubated in M9 buffer for 18 h and washed 3× in dH20 using standard protocols (Nass et al. 2002, Nass & Hamza 2007). For acute exposures, L1 stage worms (10 worms/μl) were incubated with dH20 +/− aluminum chloride (AlCl3; Fisher Scientific, Fair Lawn, NJ), as previously described (Nass & Hamza 2007) for 30 min at room temperature (~ 22°C) with gentle mixing every 10 min. After exposure, the worms were placed onto NGM/OP50 plates and allowed to recover for 72 h at 20°C. After recovery, 50–60 worms were immobilized on 2% agarose pads with 2% sodium azide and were scored for DA neurodegeneration under a fluorescent microscope (Leica MZ 16FA, Switzerland). Worms were scored positive for DA neuron degeneration when GFP in any part of the four cephalic dendrites (CEP; which run from nerve ring to tip of the nose) was absent (Nass et al. 2002, Settivari et al. 2009). Each of the experiments was performed at least in triplicate.
Mitochondrial membrane potential analysis
Mitochondrial membrane potential was measured using the lipophilic cationic dye tetramethyl rhodamine ethyl ester (TMRE, Sigma, St. Louis, MO) as previously described (Yoneda et al. 2004, Ehrenberg et al. 1988, Settivari et al. 2009). L1 stage worms were exposed to 100 μM AlCl3 for 30 min and then were allowed to recover on NGM plates containing 0.1 μM TMRE for 48 h. A Leica MZ 16FA fluorescent microscope was used to obtain fluorescent images of the head region of each animal (from tip of nose to posterior end of terminal bulb) and the amount of dye accumulated was quantified using Image Pro Plus v6.2 software (Media Cybernetics, MD). The membrane potential of at least 20 live animals was evaluated in triplicate for each experimental condition.
ATP analysis
ATP levels were determined using the ATP determination kit (Molecular Probes, Eugene, OR) following the manufacturer's instructions with minor modifications. Briefly, synchronized L1 stage nematodes were exposed to water or 100 μM AlCl3 for 30 min and allowed to recover on NGM plates for 24 h at 20°C. The nematodes were collected from the plates and washed three times. The worm pellet was immediately frozen in liquid nitrogen, thawed, and sonicated in TE buffer (100 mM Tris-Cl, pH 7.6, 4 mM EDTA). ATPase activity was inhibited by incubating the lysate at 85°C for 7 min. The lysate was spun at 14000 g for 30 min, the supernatant was collected and protein concentration was determined using the Bradford assay with bovine gamma globulin as the standard. 10 μl of the supernatant was added to 100 μl of the standard reaction solution (provided with the kit) and light intensities were measured using a luminometer (Spectra Fluor Plus, Tecan). ATP levels within each sample were background subtracted and normalized to the protein content. All assays were performed in triplicate.
Antibodies and Western analysis
An antibody to amino acids 100–154 from the putative C. elegans SMF-3 protein (WP:CE36370) was generated using Genomic Antibody Technology at Strategic Diagnostics Inc. (SDI, Newark, DE). Rabbit polyclonal antibodies were further purified at SDI. GAPDH (ab36840 Abcam, Cambridge, MA) was used as a loading control for Western analysis. To prepare protein for Western blot analysis, synchronized L1 stage worms were exposed to 100 μM AlCl3 for 30 min and allowed to recover for 24 h on NGM plates. Following recovery, worms were pelleted from media plates as described above. 150 μl of mito buffer (20 mM HEPES, pH 7.5, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 0.1 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 2 μg/ml aprotinin) was added to 300–400 μl of pelleted worms and the tubes were frozen at −20°C until protein purification. Worm samples stored at −20°C were thawed and homogenized on ice with 50–60 strokes with in a 2 ml glass homogenizer. The lysate was spun at 400 g at 4°C for 4 min, the supernatant was collected in a sterile tube, and protein concentration was determined using the Bradford assay with bovine gamma globulin as the standard. The samples were diluted in NuPAGE LDS buffer (Invitrogen, Carlsbad, CA), heated at 85°C for 15 min, and total cell lysates (50 μg protein) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% non-fat dry milk dissolved in TBST (tris-buffered saline, 0.1% Tween-20) for 2 h at room temperature, followed by incubation with the appropriate primary antibody dilution (anti-SMF-3 at 1:80,000; anti-GAPDH at 1:20,000) at 4°C overnight. The membranes were washed 3 times at room temperature for 15 min, and incubated with HRP-conjugated secondary anti-rabbit IgG (611–1302 Rockland, Gilbertsville, PA). The membrane was developed using enhanced chemiluminescence (ECL) (Amersham Biosciences, Pittsburgh, PA), captured using Bio-Rad ChemiDoc XRS, and total protein intensities were measured using QuantityOne software (Bio-Rad, Hercules, CA).
Immunohistochemistry and intracellular localization analysis
Primary C. elegans cultures were prepared as previously described with slight modifications (Bianchi & Driscoll 2006, Settivari et al. 2009). Briefly, BY250 gravid adults were lysed with the synchronization solution, and the egg pellet was washed using egg buffer (118 mM NaCl, 48 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM HEPES). The eggs were then separated from the debris using a 60% sucrose solution, digested using 4 mg/ml chitinase (Sigma, St. Louis, MO), and the embryonic cells were dissociated using a syringe. The embryonic cells were then resuspended in L-15 medium (containing 10% FBS and 1% pen/strep) and grown on polylysine-coated cover slips at 20°C. Following growth for 72 h, cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and incubated in blocking buffer containing 2% BSA and 20% normal goat serum for 1 h at room temperature. The cells were then incubated with SMF-3 primary antibody (1:100) at 4°C overnight (14 h), followed by incubation with an Alexa Fluor 594 conjugated goat anti-rabbit secondary antibody (Invitrogen; 1:200) at room temperature for 1 h. Images were captured using confocal microscopy (Olympus FV1000-MPE Confocal/Multiphoton Microscope). To confirm intracellular localization of SMF-3 staining we performed spatial overlap analysis using the Measure Colocalization Application in MetaMorph imaging software (Molecular Devices, Downingtown, PA). A projection image was created from Z-stack slices and the region of interest was chosen to include the GFP expressing dopamine neuron. The area of overlap between GFP (green) and SMF-3 (red) channels was calculated as a percentage of red pixels above threshold colocalizing with green pixels above threshold and the percentage of green pixels above threshold that colocalize with red pixels above threshold. SMF-3 immunofluorescence was analyzed in three primary GFP expressing DA neurons.
RNA interference
RNA-mediated interference using the RNAi sensitive strain RJ928 was carried out on NGM plates containing 1 mM isopropyl β-d-thiogalactoside (IPTG) and 100 μg/ml ampicillin and seeded with HT115 (DE3), an RNase III-deficient E. coli strain carrying L4440 vector with the gene fragment (skn-1 or ced-4) (GeneService, Source BioScience, PLC, Nottingham, UK) or empty vector (Addgene, Cambridge, MA) (Timmons & Fire 1998). Synchronized L1 stage RJ928 worms were transferred onto RNAi plates and grown to adulthood following the feeding protocol with slight modifications (Kamath & Ahringer 2003). Gravid adults grown on RNAi bacteria were treated with hypochlorite to obtain synchronized L1 stage worms. The L1 staged worms were exposed to 100 μM AlCl3 as described above and allowed to recover on fresh RNAi plates for 72 h and dopamine neuronal death was evaluated as previously described (Nass et al., 2002). As skn-1 knockdown results in embryonic lethality, first-generation synchronized L1 larvae were exposed to AlCl3 and DA neuron vulnerability was evaluated following 72 h recovery.
ICP-MS analysis
Wild type (BY250) and smf-3 mutant (RJ934) synchronized L1 stage worms were obtained by hypochlorite treatment of gravid adults followed by incubation of the embryos in M9 buffer for 18 h. L1s were washed 3 times with H2O, placed on NGM plates and grown at 20°C for 48 h. After 48 h, L4 stage worms were washed off of the plates with W5 H2O (Fisher Scientific, submicron filtered HPLC grade H2O to minimize contamination of aluminum from diH2O) and washed three times to remove bacteria. Worms of each strain were divided among 6 tubes and exposed to W5 H2O or 100 μM AlCl3 for 30 min at room temperature. Following the exposure, ice-cold W5 H2O was added to each tube to stop the reaction, worms were moved to pre-weighed tubes, and additionally washed 4 more times with ice-cold W5 H2O. After the last wash, all water was removed from the top of the worm pellet and the worms were frozen at −80°C. To prepare the samples for analysis, worms were digested using the MARS Xpress system (CEM, Matthews, NC). Worm samples were dried overnight (12–14 h) at 60°C and the dry weight of each sample was determined and then added to the digestion vessel. Samples were digested in 45% HNO3, 5% HCl at 200°C for 15min. Digested samples were diluted with W5 H2O to achieve a solution containing 2% acid. Total aluminum content of each sample was determined by analysis with the X Series ICP-MS (Thermo Fisher Scientific), using beryllium and gallium as internal standards, and data was normalized to the dry weight.
Statistics
All data are expressed as the mean ± SEM. The difference between control and treated groups was evaluated with a student's t-test, or one-way anova for multiple treatment groups. Two-way anova was used for experiments involving treatment of wild type (WT) and mutant animals. Statistical analysis was carried out with GraphPad Prism software. Differences were considered statistically significant when p < 0.05, unless otherwise indicated.
Results
Al3+ exposure decreases mitochondrial membrane potential and ATP levels in C. elegans
In order to determine whether a brief 30 min exposure to Al3+ may be toxic to C.elegans mitochondria, we exposed WT first larval stage animals (L1) to 100 μM AlCl3, and evaluated mitochondrial membrane potential changes in the head region following growth on bacteria and tetramethylrhodamine ethyl ester (TMRE) for 48 h (Settivari et al. 2009). TMRE is a mitochondrial-specific fluorescent dye whose rate of uptake is dependent on the mitochondria membrane potential (Yoneda et al. 2004). Under these exposure conditions the animals appeared to develop normally and there was no decrease in animal viability. A single exposure to 100 μM AlCl3 resulted in a significant decrease in mitochondria membrane potential within the head relative to non-exposed nematodes (Fig. 1a). These results suggest that as in vertebrate systems, Al3+ may impair mitochondria function. To further explore whether Al3+ may be toxic to mitochondria in C. elegans, we determined total ATP levels in control and Al3+ exposed animals. Exposure to 100 μM AlCl3 resulted in a 40% decrease in cellular ATP levels relative to controls (Fig. 1b). These results indicate that exposure of C. elegans to Al3+ is deleterious to mitochondria, and suggests that Al3+ may also be toxic to cells that are sensitive to mitochondria-associated neurotoxicants.
Figure 1. Al3+ exposure decreases mitochondrial membrane potential and ATP production, and confers DA neuron degeneration.

Synchronized L1 nematodes were exposed to 100 μM AlCl3 for 30 min and allowed to recover on plates containing TMRE for 48 h. The mitochondrial membrane potential was evaluated by measuring the fluorescence intensity per area for the head region of at least 20 animals per replicate and the experiment was performed three times. Shown are mean values ± SEM of three individual replicates. p values were calculated using t test analysis. Asterisk indicates p ≤ 0.05 (a). ATP levels were evaluated following a 30 min exposure of L1 stage worms to 100 μM AlCl3 and a 24h recovery on NGM plates. Asterisk indicates p ≤ 0.05 (b). ATP was quantified by a luminescent ATP determination kit and normalized to protein content for three independent replicates. To determine the effect of Al3+ exposure on DA neurons, L1 stage worms were exposed to 100 μM AlCl3 for 30 min and allowed to recover for 72 h before examining the integrity of GFP-expressing dopamine neurons (CEPs). Asterisk indicates p ≤ 0.01 (c). Transgenic worms expressing human α-synuclein are more sensitive to Al3+ exposure as indicated by a significant increase in DA neuron degeneration with 100 μM AlCl3. Asterisk indicates a difference between WT and α-synuclein, # indicates a difference between control and AlCl3 exposure, p ≤ 0.001 (d).
Exposure to sub-lethal concentrations of Al3+ confers DA neurodegeneration in WT and human α-synuclein expressing animals
The DA neurons in vertebrates are particularly sensitive to oxidative stress and heavy metals, and exposures have been associated with the development of Parkinson's disease and parkinsonism. We previously generated transgenic worms that express the green fluorescent protein (GFP) in the eight DA neurons in the hermaphrodite that allows clear visualization of the neurons under a fluorescent dissecting microscope (Nass et al. 2002). Our studies and others have shown that brief exposures to PD-associated neurotoxicants including 6-OHDA, MPP+, rotenone, and Mn2+ can result in DA neuron cell death (Settivari et al. 2009, Nass et al. 2002, Nass & Settivari 2008). To determine whether a brief exposure to Al3+ can induce DA neuron degeneration in C. elegans, we exposed the animals to various concentrations of toxicant ranging from 0 to 500 μM for 30 min and transferred the animals to agar plates. We scored these animals similar to our earlier studies in which the CEP processes that cannot be visually followed from the cell body to the tip of the nose are considered to have degeneration (Nass et al. 2002, Settivari et al. 2009, Vanduyn et al. 2010). We find that a brief 30 min exposure to Al3+ caused a loss of DA neurons within 72 h at all concentrations tested, and approximately 15% of the animals displayed DA neuron degeneration following exposure to 100 μM AlCl3 (Fig. 1c). This exposure does not result in any apparent long-term changes in whole animal morphology or behavior, which suggests there is not large-scale cell death, and the DA neurodegeneration appears similar to our earlier studies in which we characterized 6-OHDA-induced DA cell death by loss of dendritic GFP and loss of neuronal integrity by electron microscopy (Nass et al. 2002). Al3+ exposure has been associated with the development of PD, and has been shown to increase PD-associated α-synuclein aggregation and fibrillation in vitro which suggests that the toxicant may interact with α-synuclein to increase DA neuron dysfunction and pathology (Uversky et al. 2001, Zayed et al. 1990b). In order to determine whether Al3+ may amplify the toxicity of α-synuclein-induced DA neuronal death in vivo, we exposed nematodes expressing human WT α-synuclein to 100 μM AlCl3 for 30 min, and found a significant increase in DA neuronal death (Fig. 1d). These results indicate that short-term Al3+ exposure can confer DA neuron degeneration in C. elegans, and α-synuclein expression amplifies the cell death.
SMF-3 is highly conserved with the rice Al3+ transporter Nrat1 and the yeast intracellular metal transporter Smf2p
A number of divalent metals enter eukaryotic cells and are transported between cellular compartments through the DMTs (Papp-Wallace & Maguire 2006, Portnoy et al. 2000). Recently a DMT-1 homologue, Nramp aluminum transporter 1 (Nrat1) in rice was shown to function as a trivalent Al3+ transporter (Xia et al. 2010). A BLAST search with the rice Nrat1, and a sequence alignment of the results using ClustalW2 indicated that the C. elegans SMF-3 is highly conserved with Nrat1 (53% similar, 34% identical) (Fig. 2) (Altschul 1991, Larkin et al. 2007). There is also high sequence homology between SMF-3 and the yeast intracellular DMT Smf2p (49% similar, 31% identical) (Portnoy et al. 2000, Portnoy et al. 2002). We have previously shown that SMF-3 is highly homologous with the human DMT-1 (Settivari et al. 2009), and the strong conservation of SMF-3 with Nrat1 and Smfp2 suggests that SMF-3 may also transport intracellular Al3+ to facilitate Al3+-induced DA neurodegeneration.
Figure 2. Sequence alignment of the rice Al3+ transporter Nrat1, yeast and mammalian DMTs, and C. elegans SMF-3.

ClustalW was used to align the protein sequences of the rice aluminum transporter Nrat1 (accession BAJ22943.1), human DMT-1 (accession NP_000608), yeast Smf2p (accession NP_011917) and C. elegans SMF-3 (accession NP_500235). Putative transmembrane domains are highlighted in gray with the arrow indicating the orientation relative to the membrane (right-directed arrowhead, N-terminal to C-terminal, extracellular to intracellular). The consensus transport sequence is highlighted in black. An asterisk indicates that the amino acids in that column are identical, a colon indicates that the amino acids are highly conserved and a period indicates that the amino acids are similar.
SMF-3 is expressed within DA neurons
To determine the location of SMF-3 expression in C. elegans, we generated antibodies to the nematode SMF-3. The antigenic sequence is unique to SMF-3 and spans between transmembrane domains two and four. To determine whether the DA neurons express SMF-3, we generated primary cultures from BY250, since GFP is strongly expressed in DA neurons both in vivo and in vitro (Carvelli et al. 2004, Nass et al. 2002). We incorporated affinity-purified anti-SMF-3 to evaluate cellular SMF-3 expression levels. As can be seen in Figure 3c, SMF-3 immunoreactivity is observed in all DA neurons, as well as other cell types (Fig. 3a–d, data not shown). To determine whether SMF-3 may be expressed intracellularly, we utilized the Measure Colocalization Application in MetaMorph imaging software. The degree of overlap between GFP and SMF-3 signal was calculated. On average 96% of SMF-3 immunofluorescence overlapped with GFP fluorescence, while only 74% of GFP overlapped with SMF-3 fluorescence (data not shown). Based on protein sequence, SMF-3 is likely a membrane protein. Furthermore, GFP is a soluble protein and would likely be found only in the cytoplasm and intracellular compartments (ie, not the plasma membrane). Considering that nearly all of the SMF-3 signal overlaps with the GFP signal suggests that the majority of SMF-3 does not reside on the plasma membrane. Taken together, these results indicate that SMF-3 is expressed in DA neurons and likely in an intracellular compartment.
Figure 3. SMF-3 is expressed in C. elegans DA neurons.
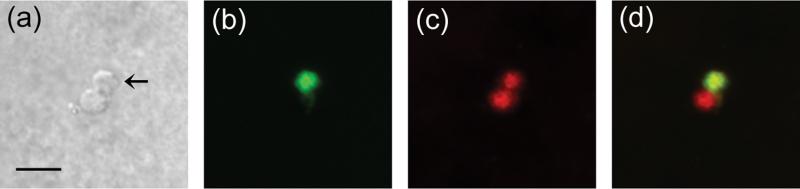
Primary cultures were generated from WT nematodes expressing GFP in the DA neurons. Differential interference contrast (DIC) image of a DA neuron (a) which was identified by the expression of GFP (b). Immunofluorescence was performed with a primary antibody to SMF-3 and an Alexa Fluor 594 secondary antibody to observe the expression of SMF-3 (c). The merge image indicates that SMF-3 is expressed in DA neurons (d). Final images were created in Image J from a Z-stack of images obtained with an Olympus2 confocal microscope. Scale bar represents 5 μm.
SMF-3 contributes to Al3+-induced DA neuron degeneration
Considering that Al3+ confers DA neurodegeneration and SMF-3 is expressed in DAergic neurons, we asked if SMF-3 plays a role in Al3+-associated neuropathology. To determine if SMF-3 contributes to Al3+-associated DA neuron degeneration, we crossed our Pdat-1∷GFP transgenic animals (BY250) into the C. elegans strain that contains a mutation in SMF-3, RB1074 smf-3(ok1035) resulting in strain RJ934 (Pdat-1∷GFP; smf-3(ok1035)). Sequencing of the smf-3(ok1035) mutant revealed a 2,067 bp deletion that spans from amino acid 154 to amino acid 378 and results in a truncation of the protein beginning in transmembrane domain 4 (data not shown). The mutation likely results in a null mutant since at least 80% of the consensus transport sequence as well as the C-terminal of the protein has been deleted. The morphology of the DA neurons in animals containing the deletion mutation appear identical to those of BY250 ((Settivari et al. 2009); data not shown), suggesting that SMF-3 does not play a significant role in maintaining DA neuron integrity. To determine if SMF-3 contributes to Al3+-induced DA neuron degeneration, we exposed the smf-3 mutants to various concentrations of AlCl3 for 30 min and evaluated DA neuronal integrity 72 h later. As can be seen in Figure 4a, the smf-3 mutants were significantly more resistant to 100, 250 and 500 μM AlCl3 relative to WT. We also asked whether SMF-1 or SMF-2 may contribute to Al3+-induced degeneration and we did not find any significant change in Al3+-induced cell death (Fig. 4b). These results indicate that the expression of SMF-3 increases DA neuron vulnerability to Al3+, and suggest that animals containing the deletion likely do not express a functional protein.
Figure 4. SMF-3 contributes to Al3+-induced DA neurodegeneration.

Synchronized L1 stage WT or smf-3 mutant (Δsmf-3) nematodes were exposed to increasing concentrations of AlCl3 for 30 min and allowed to recover for 72 h on NGM plates. DA neuron degeneration was evaluated in WT and mutant animals at each concentration as described in Methods. Asterisk indicates p ≤ 0.01 (a). There is no significant difference in the DA neuron sensitivity to Al3+ in smf-1 or smf-2 mutant worms (b).
SMF-3 protein expression is downregulated following exposure to Al3+
We have previously shown that acute exposure to Mn2+ reduces gene expression of the C. elegans DMTs, likely to protect against oxidative stress (Settivari et al. 2009). In order to determine if Al3+ exposure results in a decrease in SMF-3 protein levels, we examined transporter expression levels in control and Al3+-exposed nematodes. Young nematodes exposed to 100 μM AlCl3 for 30 min showed a dramatic decrease in SMF-3 protein levels within 24 h (Fig. 5a). These results indicate that SMF-3 protein levels are exquisitely sensitive to Al3+. We also do not observe immunoreactive bands in the unexposed or Al3+-exposed mutant at the WT SMF-3 molecular weight size, or lower molecular weights that could represent a truncated protein (Fig. 5a, data not shown). Taken together, these results suggest that the decrease in SMF-3 expression may be an attempt to limit intracellular exposure to Al3+.
Figure 5. SMF-3 protein expression is reduced following Al3+ exposure and contributes to cellular Al levels.

Western blotting with a SMF-3 antibody was used to evaluate protein levels following a 30 min exposure to 100 μM AlCl3 and 24 h recovery. No SMF-3 immunoreactivity is observed in smf-3 mutant worms. GAPDH is used as a loading control (a). Whole animal aluminum levels in L4 stage animals were determined following a 30 min exposure to 100 μM AlCl3 and analyzed on an ICP-MS. smf-3 mutant worms accumulate higher levels of Al than wild type worms. Asterisk indicates p ≤ 0.001 (b).
SMF-3 modulates cellular Al3+ levels
The high homology between the C. elegans SMF-3 protein and the rice Nrat1 and yeast Smf2p/Smf3p transporters suggests that SMF-3 may also be involved in Al3+ homeostasis. In order to determine if SMF-3 expression can modulate whole animal Al3+ levels, we treated L4 WT or smf-3 mutant animals with either water or 100 μM AlCl3 (pH 4.6) for 30 min and determined whole animal Al3+ concentrations by ICP-MS. As can be seen in Figure 5b, an acute Al3+ exposure resulted in a 40% increase in Al in the smf-3 mutant relative to WT. We also found a slightly lower level of Al3+ in the non-Al3+ exposed mutant animals relative to WT. Although this low amount is statistically insignificant from WT animals, this difference may be due changes in general cellular metal homeostasis (eg, Fe2+ or Cu2+ levels) that could compete for the transporter or TfR-Me in a low Al3+ environment (Wu et al. 2012, Yokel 2006, Becaria et al. 2002). Taken together these results indicate the SMF-3 transporter modulates cellular Al3+ levels and is involved in cellular retention of Al3+.
SKN-1 and CED-4 modulate Al3+-induced DA neurodegeneration
SKN-1, the homologue to the PD-associated vertebrate Nrf2, has recently been shown to be expressed in C. elegans DA neurons and to inhibit methylmercury-induced DA neuronal death (Vanduyn et al. 2010). In order to determine if SKN-1 may also inhibit Al3+-induced DA neuron death in C. elegans, we knocked down skn-1 gene expression using RNAi. As genetic knockdown of skn-1 is embryonic lethal, first generation L1 larvae were exposed to 100 μM Al3+ for 30 min, and then allowed to recover on the RNAi bacteria for 72 h before evaluating DA neuron morphology. Animals in which skn-1 gene expression was reduced showed a small yet significant increase in DA neuron death (Fig. 6a), indicating that SKN-1 plays a neuroprotective role in Al3+-induced DA neurodegeneration. In vertebrates, Al3+ has been demonstrated to induce cell death via apoptosis (Banasik et al. 2005, Lukiw et al. 2005). In order to determine if apoptosis may also play a role in Al3+-induced cell death in C. elegans, we knocked down gene expression of the apoptotic gene Apaf1/ced-4 using RNAi as described above except the second generation L1s were exposed to AlCl3. Genetic knockdown resulted in a decrease in DA neuronal death (Fig. 6b), indicating that apoptosis is likely contributing to the Al3+-induced cell death.
Figure 6. skn-1 and ced-4 modulate Al3+-induced DA neuron degeneration.

RNAi was used to knockdown gene expression of skn-1 (a) or ced-4 (b) and worms were exposed to 100 μM AlCl3 for 30 min, and the RNAi feeding was continued during the 72 h recovery period. DA neuron degeneration was evaluated as described in Methods. Asterisk indicates p ≤ 0.05.
DISCUSSION
Overexposure to Al3+ has been associated epidemiologically with the development of a number of human neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Gulf War syndrome, Parkinsonism dementia, AD and PD (Kawahara & Kato-Negishi 2011, Petrik et al. 2007). In PD, post-mortem metal analysis of brains demonstrates increased Al3+ levels in the substantia nigra relative to healthy controls (Yasui et al. 1992, Hirsch et al. 1991). High exposure to Al3+ is reported to cause oxidative stress, mitochondrial dysfunction, ATP depletion, and apoptosis, although the genetic and molecular basis for the toxicity is poorly defined (Kumar et al. 2008, Lemire et al. 2009). A difficulty in identifying the molecular components involved in Al3+-induced neuropathology has been the cellular complexity of vertebrate models and the lack of in vivo genetic models to dissect the molecular pathways involved in the cellular dysfunction. In this study we describe a novel model for Al3+-induced DA neurodegeneration. We show that a brief 30 min exposure of C. elegans to 100 μM Al3+ reduces mitochondrial membrane potential, ATP levels, and confers DA neuron degeneration partially through apoptosis (Fig. 1, 6b). The concentrations of Al3+ that C. elegans were exposed in this study are environmentally relevant and similar to vertebrate studies, as well as the estimated dietary Al levels consumed by Americans (Becaria et al. 2002, Greger 1993, Walton 2012). Taken together, this model recapitulates key characteristics of Al3+-associated toxicity in vertebrates, and suggests the nematode will be a valuable genetic tool to explore the relationship between Al3+ exposures and toxicity and the subsequent neuropathology (Kumar et al. 2009, Sanchez-Iglesias et al. 2009).
Our studies show that Al3+-induced DA neurodegeneration is partially dependent on a homologue of human DMT-1, SMF-3 (Fig 4). Until recently DMTs were believed to exclusively transport divalent cations. The biophysical characteristics of Al3+ suggest though that the metal can interact with divalent metal binding sites. Al3+'s small ionic radius (54 pm) relative to other DMT substrates (74 pm – 175 pm), as well as an ionic radius-to-charge ratio similar to Fe2+ (0.16 vs. 0.17) indicates that Al3+ should be able to compete with Fe2+ for binding on macromolecules (Kawahara & Kato-Negishi 2011, Bharathi & Rao 2008). Consistent with these properties, Al3+ has been shown to compete with divalent metals including Fe2+, Ca2+, Mg2+, and Zn2+ in binding with biomolecules (Martin 1986). The identification of the rice plasma membrane Al3+ transporter Nrat1, which has high amino acid sequence conservation with the C. elegans SMF-3 (Fig. 2), suggests that SMF-3 may also transport Al3+ (Xia et al. 2010). Indeed our studies show a significant increase in whole animal Al-levels in the functional null mutant, consistent with a role of SMF-3 in cellular Al3+ regulation.
A homologue of SMF-3, Smf2p, is expressed in yeast intracellular vesicles and transports Fe2+ and Mn2+ out of vesicles into the cytosol where the metals are subsequently transported into the Golgi or mitochondria (Luk & Culotta 2001, Portnoy et al. 2000, Portnoy et al. 2002). Smf2p is tightly regulated post-translationally, as excess Mn2+ results in Smf2p vacuole-associated degradation (Portnoy et al. 2000, Luk & Culotta 2001). SMF-3 may also be regulated post-translationally as the addition of Al3+ resulted in a dramatic decrease in protein levels within 24 h following Al3+ exposure (Fig. 5a). Furthermore, similar to DMTs and Smf2p, SMF-3 may reside in an intracellular compartment to transport metals into the cytoplasm, as the immunofluorescence data suggests that the vast majority of SMF-3 is located within the cell (Portnoy et al. 2002, Burdo et al. 2001). Consistent with SMF-3 residing in an intracellular compartment, a SMF-3 functional null mutant inhibits Al3+-induced DA neuron degeneration and retains 40% more Al3+ relative to controls. It is unlikely that SMF-1 and SMF-2 also transport Al3+, since a genetic deletion of either transporter does not affect Al3+-induced DA neuron vulnerability (Fig 4), but this cannot be ruled out until a more detailed Al3+ transport study of the cloned gene is performed. Taken together, these results suggest that SMF-3 contributes to Al3+-induced DA neurodegeneration through Al3+ efflux from an intracellular compartment within the DA neuron (Fig 7).
Figure 7. A model for SMF-3-associated Al3+ trafficking and toxicity in C. elegans DA neurons.

SMF-3 resides in an intracellular compartment and is predicted to transport Al3+ into the cytosol. Cytoplasmic Al3+ can increase α-synuclein aggregation, activate stress response pathways through the transcription factor SKN-1, and activate mitochondria associated-cell death pathways. The mechanism by which Al3+ is transported into the DA neurons and the intracellular vesicles is not known, but may involve receptor-mediated endocytosis, simple diffusion, or facilitated diffusion through ion channels (Becaria et al. 2002).
The PD-associated proteins DMT-1, α-synuclein and Nrf2 can interact functionally with each other and can contribute to DA neuron degeneration. DMT-1 expression has been shown to increase in PD patients and with age, and has been shown to increase in PD cell models (Salazar et al. 2008, Zhang et al. 2009). α-synuclein, the presynaptic protein whose aggregation and fibrillation contributes to familial and idiopathic PD, has been shown to facilitate DMT-1-associated DA neuron cell death (Chew et al. 2011). Nrf2 haplotypes have been associated with the development of PD, and Nrf2 deficiency has recently been shown to exacerbate α-synuclein-induced aggregation and cell death (Lastres-Becker et al. 2012). Furthermore, the aggregation and fibrillation of α-synuclein increases dramatically in the presence of Al3+ (Uversky et al. 2001). Our studies are consistent with the role these proteins likely play in Al3+-associated DA neuron vulnerability as overexpression of human α-synuclein or genetic knockdown of Nrf2/SKN-1 results in an increase in Al3+-induced DA neuropathology.
Al3+ induces cell death in vertebrates through the initiation of the apoptotic pathway (Vasudevaraju et al. 2008). Mitochondria are likely an intracellular target of Al3+ as the metal inhibits Na+/Ca2+ exchange resulting in an increase in Ca2+ in the mitochondria and ROS levels, and leads to the release of cytochrome c and subsequent apoptosis (Vasudevaraju et al. 2008, Kawahara & Kato-Negishi 2011). Al3+ has been shown to induce the opening of the mitochondrial transition pore resulting in apoptosis (Toninello et al. 2000). Al3+ also can cause oxidative stress and apoptosis by increasing the free radical damage associated with Fe2+ (Vasudevaraju et al. 2008, Zatta et al. 2003). Our studies are consistent with the role of apoptosis in Al3+-induced cell death in the nematode as a reduction in gene expression of the vertebrate apoptotic caspase homologue Apaf1, ced-4, results in a significant increase in viable DA neurons in the nematode.
In summary, we describe a novel model for Al3+ toxicity and show that the C. elegans transporter SMF-3 plays a significant role in modulating Al3+-induced DA neuron degeneration through the intracellular sequestration of Al3+. We also show that SMF-3 expression is sensitive to Al3+, and the PD-associated proteins α-synuclein, Nrf2/SKN-1, and Apaf1/CED-4 modulate Al3+-associated DA neuron cell death. This novel genetic model should facilitate identification of molecular pathways and potential therapeutic targets involved in Al3+-associated DA neuron pathology.
ACKNOWLEDGMENTS
We gratefully appreciate the technical assistance of Brittany Flores. We also greatly appreciate helpful discussions with Dr. Bob Yokel at University of Kentucky in Lexington, KY and Dr. Malgorzata Kamocka at the Center for Biological Microscopy at IUSM. Some of the strains were provided by the Caenorhabditis Genetics Center, which is supported by the National Institutes of Health Center for Research Resources. This study was partially supported by grants R011ES014459 and ES015559 from the National Institute of Environmental Health Sciences (RN) and an EPA STAR graduate fellowship (NV).
Abbreviations used
- AD
Alzheimer's disease
- DA
dopamine
- DMT
divalent metal transporter
- ECL
enhanced chemiluminescence
- GFP
green fluorescent protein
- ICP-MS
inductively coupled plasma mass spectrometry
- NRAMP
natural resistance-associated macrophage protein
- PAGE
polyacrylamide gel electrophoresis
- PD
Parkinson's disease
- ROS
reactive oxygen species
- SN
substantia nigra
- TfR-ME
transferrin receptor mediated endocytosis
- TH
tyrosine hydroxylase
- TMRE
tetramethyl rhodamine ethyl ester
- WT
wild type
Footnotes
The authors also state that they have no conflict of interest to declare.
References
- Altschul SF. Amino acid substitution matrices from an information theoretic perspective. J Mol Biol. 1991;219:555–565. doi: 10.1016/0022-2836(91)90193-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazi H, Ahboucha S, Gamrani H. Chronic exposure to aluminum reduces tyrosine hydroxylase expression in the substantia nigra and locomotor performance in rats. Neurosci Lett. 2011;487:8–11. doi: 10.1016/j.neulet.2010.09.053. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure. J Biol Chem. 2001;276:44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Brandel JP, Galle P, Javoy-Agid F, Agid Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson's disease: an X-ray microanalysis. J Neurochem. 1991;56:446–451. doi: 10.1111/j.1471-4159.1991.tb08170.x. [DOI] [PubMed] [Google Scholar]
- Hope I. C. elegans: A Practical Approach. Oxford University Press; New York: 1999. [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kato-Negishi M. Link between Aluminum and the Pathogenesis of Alzheimer's Disease: The Integration of the Aluminum and Amyloid Cascade Hypotheses. Int J Alzheimers Dis. 2011;2011:276393. doi: 10.4061/2011/276393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Bal A, Gill KD. Impairment of mitochondrial energy metabolism in different regions of rat brain following chronic exposure to aluminium. Brain Res. 2008;1232:94–103. doi: 10.1016/j.brainres.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Kumar V, Bal A, Gill KD. Aluminium-induced oxidative DNA damage recognition and cell-cycle disruption in different regions of rat brain. Toxicology. 2009;264:137–144. doi: 10.1016/j.tox.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Lakso M, Vartiainen S, Moilanen AM, Sirvio J, Thomas JH, Nass R, Blakely RD, Wong G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. Journal of Neurochemistry. 2003;86:165–172. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rabano A, Kirik D, Cuadrado A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson's disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- Lemire J, Mailloux R, Puiseux-Dao S, Appanna VD. Aluminum-induced defective mitochondrial metabolism perturbs cytoskeletal dynamics in human astrocytoma cells. J Neurosci Res. 2009;87:1474–1483. doi: 10.1002/jnr.21965. [DOI] [PubMed] [Google Scholar]
- Luk EE, Culotta VC. Manganese superoxide dismutase in Saccharomyces cerevisiae acquires its metal co-factor through a pathway involving the Nramp metal transporter, Smf2p. J Biol Chem. 2001;276:47556–47562. doi: 10.1074/jbc.M108923200. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Percy ME, Kruck TP. Nanomolar aluminum induces pro-inflammatory and pro-apoptotic gene expression in human brain cells in primary culture. J Inorg Biochem. 2005;99:1895–1898. doi: 10.1016/j.jinorgbio.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Martin RB. The chemistry of aluminum as related to biology and medicine. Clin Chem. 1986;32:1797–1806. [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass R, Hamza I. The nematode Caenorhabditis elegans as a model to explore toxicology in vivo: Solid and axenic growth culture conditions and compound exposure parameters. Current Protocols in Toxicology. 2007:1.9.1–1.9.18. doi: 10.1002/0471140856.tx0109s31. [DOI] [PubMed] [Google Scholar]
- Nass R, Merchant KM, Ryan T. Caenohabditis elegans in Parkinson's disease drug discovery: addressing an unmet medical need. Mol Interv. 2008;8:284–293. doi: 10.1124/mi.8.6.6. [DOI] [PubMed] [Google Scholar]
- Nass R, Miller DM, Blakely RD. C. elegans: a novel pharmacogenetic model to study Parkinson's disease. Parkinsonism Relat Disord. 2001;7:185–191. doi: 10.1016/s1353-8020(00)00056-0. [DOI] [PubMed] [Google Scholar]
- Nass R, Blakely RD. The Caenorhabditis elegans dopaminergic system: opportunities for insights into dopamine transport and neurodegeneration. Annual Review of Pharmacology & Toxicology. 2003;43:521–544. doi: 10.1146/annurev.pharmtox.43.100901.135934. [DOI] [PubMed] [Google Scholar]
- Nass R, Settivari RS. Caenorhabditis elegans Models of Parkinson's Disease: A Robust Genetic System to Identify and Characterize Endogenous and Environmental Components involved in Dopamine Neuron Degeneration. In: Nass R, Przedborski S, editors. Parkinson's Disease: Molecular and Therapeutic Insights from Model Systems. Elsevier Academic Press; 2008. pp. 347–360. [Google Scholar]
- Papp-Wallace KM, Maguire ME. Manganese transport and the role of manganese in virulence. Annu Rev Microbiol. 2006;60:187–209. doi: 10.1146/annurev.micro.60.080805.142149. [DOI] [PubMed] [Google Scholar]
- Petrik MS, Wong MC, Tabata RC, Garry RF, Shaw CA. Aluminum adjuvant linked to Gulf War illness induces motor neuron death in mice. Neuromolecular Med. 2007;9:83–100. doi: 10.1385/nmm:9:1:83. [DOI] [PubMed] [Google Scholar]
- Portnoy ME, Liu XF, Culotta VC. Saccharomyces cerevisiae expresses three functionally distinct homologues of the nramp family of metal transporters. Mol Cell Biol. 2000;20:7893–7902. doi: 10.1128/mcb.20.21.7893-7902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy ME, Jensen LT, Culotta VC. The distinct methods by which manganese and iron regulate the Nramp transporters in yeast. Biochem J. 2002;362:119–124. doi: 10.1042/0264-6021:3620119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton JR. Cognitive deterioration and associated pathology induced by chronic low-level aluminum ingestion in a translational rat model provides an explanation of Alzheimer's disease, tests for susceptibility and avenues for treatment. Int J Alzheimers Dis. 2012;2012:914947. doi: 10.1155/2012/914947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Du Y, Xue H, Wu Y, Zhou B. Aluminum induces neurodegeneration and its toxicity arises from increased iron accumulation and reactive oxygen species (ROS) production. Neurobiol Aging. 2012;33:199, e191–112. doi: 10.1016/j.neurobiolaging.2010.06.018. [DOI] [PubMed] [Google Scholar]
- Banasik A, Lankoff A, Piskulak A, Adamowska K, Lisowska H, Wojcik A. Aluminum-induced micronuclei and apoptosis in human peripheral-blood lymphocytes treated during different phases of the cell cycle. Environ Toxicol. 2005;20:402–406. doi: 10.1002/tox.20125. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay J, Song HO, Park BJ, Singaravelu G, Sun JL, Ahnn J, Cho JH. Functional assessment of Nramp-like metal transporters and manganese in Caenorhabditis elegans. Biochem Biophys Res Commun. 2009;390:136–141. doi: 10.1016/j.bbrc.2009.09.082. [DOI] [PubMed] [Google Scholar]
- Becaria A, Campbell A, Bondy SC. Aluminum as a toxicant. Toxicol Ind Health. 2002;18:309–320. doi: 10.1191/0748233702th157oa. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Driscoll M. Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses. WormBook. 2006:1–15. doi: 10.1895/wormbook.1.122.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdo JR, Menzies SL, Simpson IA, Garrick LM, Garrick MD, Dolan KG, Haile DJ, Beard JL, Connor JR. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res. 2001;66:1198–1207. doi: 10.1002/jnr.1256. [DOI] [PubMed] [Google Scholar]
- Bharathi, Rao KS. Molecular understanding of copper and iron interaction with alpha-synuclein by fluorescence analysis. J Mol Neurosci. 2008;35:273–281. doi: 10.1007/s12031-008-9076-4. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, Defelice LJ. Dopamine transporters depolarize neurons by a channel mechanism. Proc Natl Acad Sci U S A. 2004;101:16046–16051. doi: 10.1073/pnas.0403299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellier M, Prive G, Belouchi A, Kwan T, Rodrigues V, Chia W, Gros P. Nramp defines a family of membrane proteins. Proc Natl Acad Sci U S A. 1995;92:10089–10093. doi: 10.1073/pnas.92.22.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew KC, Ang ET, Tai YK, et al. Enhanced autophagy from chronic toxicity of iron and mutant A53T alpha-synuclein: implications for neuronal cell death in Parkinson disease. J Biol Chem. 2011;286:33380–33389. doi: 10.1074/jbc.M111.268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisponi G, Nurchi V, Faa F, Remelli M. Human diseases related to aluminium overload. Monatsh Chem. 2011;142:331–340. [Google Scholar]
- Greger JL. Aluminum metabolism. Annu Rev Nutr. 1993;13:43–63. doi: 10.1146/annurev.nu.13.070193.000355. [DOI] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc Natl Acad Sci U S A. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Iglesias S, Mendez-Alvarez E, Iglesias-Gonzalez J, Munoz-Patino A, Sanchez-Sellero I, Labandeira-Garcia JL, Soto-Otero R. Brain oxidative stress and selective behaviour of aluminium in specific areas of rat brain: potential effects in a 6-OHDA-induced model of Parkinson's disease. J Neurochem. 2009;109:879–888. doi: 10.1111/j.1471-4159.2009.06019.x. [DOI] [PubMed] [Google Scholar]
- Settivari R, Levora J, Nass R. The divalent metal transporter homologues SMF-1/2 mediate dopamine neuron sensitivity in caenorhabditis elegans models of manganism and parkinson disease. J Biol Chem. 2009;284:35758–35768. doi: 10.1074/jbc.M109.051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- Toninello A, Clari G, Mancon M, Tognon G, Zatta P. Aluminum as an inducer of the mitochondrial permeability transition. J Biol Inorg Chem. 2000;5:612–623. doi: 10.1007/s007750000144. [DOI] [PubMed] [Google Scholar]
- Vanduyn N, Settivari R, Wong G, Nass R. SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci. 2010;118:613–624. doi: 10.1093/toxsci/kfq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen S, Pehkonen P, Lakso M, Nass R, Wong G. Identification of gene expression changes in transgenic C. elegans overexpressing human alpha-synuclein. Neurobiology of Disease. 2006;22:477–486. doi: 10.1016/j.nbd.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Vasudevaraju P, Govindaraju M, Palanisamy AP, Sambamurti K, Rao KS. Molecular toxicity of aluminium in relation to neurodegeneration. Indian J Med Res. 2008;128:545–556. [PubMed] [Google Scholar]
- Verstraeten SV, Aimo L, Oteiza PI. Aluminium and lead: molecular mechanisms of brain toxicity. Arch Toxicol. 2008;82:789–802. doi: 10.1007/s00204-008-0345-3. [DOI] [PubMed] [Google Scholar]
- Xia J, Yamaji N, Kasai T, Ma JF. Plasma membrane-localized transporter for aluminum in rice. Proc Natl Acad Sci U S A. 2010;107:18381–18385. doi: 10.1073/pnas.1004949107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui M, Kihira T, Ota K. Calcium, magnesium and aluminum concentrations in Parkinson's disease. Neurotoxicology. 1992;13:593–600. [PubMed] [Google Scholar]
- Yokel RA. Brain uptake, retention, and efflux of aluminum and manganese. Environ Health Perspect. 2002;110(Suppl 5):699–704. doi: 10.1289/ehp.02110s5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokel RA. Blood-brain barrier flux of aluminum, manganese, iron and other metals suspected to contribute to metal-induced neurodegeneration. J Alzheimers Dis. 2006;10:223–253. doi: 10.3233/jad-2006-102-309. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- Zatta P, Lucchini R, van Rensburg SJ, Taylor A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bull. 2003;62:15–28. doi: 10.1016/s0361-9230(03)00182-5. [DOI] [PubMed] [Google Scholar]
- Zayed J, Campanella G, Panisset JC, Ducic S, Andre P, Masson H, Roy M. Parkinson disease and environmental factors. Rev Epidemiol Sante Publique. 1990a;38:159–160. [PubMed] [Google Scholar]
- Zayed J, Ducic S, Campanella G, Panisset JC, Andre P, Masson H, Roy M. Environmental factors in the etiology of Parkinson's disease. Can J Neurol Sci. 1990b;17:286–291. [PubMed] [Google Scholar]
- Zhang S, Wang J, Song N, Xie J, Jiang H. Up-regulation of divalent metal transporter 1 is involved in 1-methyl-4-phenylpyridinium (MPP(+))-induced apoptosis in MES23.5 cells. Neurobiol Aging. 2009;30:1466–1476. doi: 10.1016/j.neurobiolaging.2007.11.025. [DOI] [PubMed] [Google Scholar]