

Abstract
Central heterocyclic ring size reduction from piperidinyl to pyrrolidinyl in the vesicular monoamine transporter-2 (VMAT2) inhibitor GZ-793A and its analogs resulted in novel N-propane-1,2(R)-diol analogs 11a–i. These compounds were evaluated for their affinity for the dihydrotetrabenazine (DTBZ) binding site on VMAT2 and for their ability to inhibit vesicular dopamine (DA) uptake. The 4-difluoromethoxyphenethyl analog 11f was the most potent inhibitor of [3H]-DTBZ binding (Ki=560 nM), with 15-fold greater affinity for this site than GZ-793A (Ki=8.29 μM). Analog 11f also showed similar potency of inhibition of [3H]-DA uptake into vesicles (Ki=45 nM) compared to that for GZ-793A (Ki=29 nM). Thus, 11f represents a new water-soluble inhibitor of VMAT function. 2009 Elsevier Ltd. All rights reserved.
Keywords: Pyrrolidine analogs; GZ-793A; Dihydrotetrabenazine binding site; Vesicular monoamine transporter-2; N-propane-1,2(R)-diols
Methamphetamine (METH) abuse is a growing problem in the United States, especially in the rural West and Southwest. To date, there is no approved therapeutic agent for treating METH addiction. (−)-Lobeline (1, Fig. 1), the major alkaloid of Lobelia inflata, has been shown to decrease the rewarding effects of methamphetamine in a rat behavioral model,1–4 and is believed to inhibit dopamine (DA) uptake into synaptic vesicles via an interaction with the tetrabenazine (TBZ) binding site on the vesicular monoamine transporter-2 (VMAT2).5 METH is a substrate at the presynaptic dopamine transporter (DAT), and within the presynaptic terminal, METH interacts with VMAT2 located on synaptic vesicles, inhibiting DA uptake and promoting vesicular DA release, leading to increased cytosolic DA concentrations in the extracellular space.6,7 Hence, VMAT2 has been identified as a valid target for the development of therapeutic agents that inhibit VMAT2 function, counteracting the effects of METH.
Fig 1.
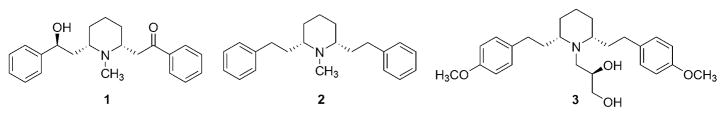
Molecular structures of lobeline (1), lobelane (2), and GZ-793A (3).
The chemically defunctionalized lobeline analogue, lobelane (2S,6R-1-methyl-2,6-diphenethylpiperidine) (2, Fig. 1), and its analogues have recently been reported to have 10–15-fold more potency and selectivity for inhibition of [3H]dopamine ([3H]DA) uptake into synaptic vesicles when compared to lobeline.8–11
Additionally, an extensive study on the variation of methylene linker length between C-6 and C-2 of the piperidine ring and each of the two phenyl rings in the lobelane molecule confirmed that a minimum of two methylene units in the alkane linker moiety is essential for retention of binding affinity at the TBZ binding site on VMAT2.12
Recently, we have identified a novel structurally optimized N-propane-1,2-diol analogue of lobelane (GZ-793A; 3, Fig. 1) which potently and selectively interacts with VMAT2 to inhibit DA uptake.13,14 GZ-793A also blocked METH self-administration (SA) and reward in rats, and had no effect on responding for food.15,16 The physicochemical properties of GZ-793A provide enhanced water-solubility and drug-likeness relative to the majority of previously reported lobelane analogs.
In a previous communication, we have reported on a series of pyrrolidino analogs of lobelane (2), and have shown that VMAT2 inhibition is increased when the piperidine ring of lobelane is replaced by a pyrrolidine ring.10 The main aim of the current study was to determine [3H]DTBZ binding affinity and inhibition of [3H]DA uptake by VMAT2 by, of a series of novel N-1,2(R)-dihydroxylpropyl group (NDHP) substituted pyrrolidine analogs of GZ-793A.
From previous SAR studies, it was found that lobelane analogues containing either fluorophenethyl or methoxyphenethyl moieties were the most potent VMAT2 ligands.14 These observations prompted us to focus on the synthesis and evaluation of N-1,2(R)-dihydroxylpropyl (NDHP) pyrrolidine analogues (11a–i) as inhibitors of VMAT2 function.
The synthesis of analogues 11a–i was achieved via initial reaction of benzotriazole, (S)-phenylglycinol (formed from LAH reduction of (S)-phenylglycine), and succindialdehyde at room temperature to afford the key synthon, (3S,5R, 7aR)-5-(benzotriazol-1-yl)-3-phenyl[2,1-b]oxazolopyrrolidine (6),17 which upon reaction with a variety of substituted phenethyl magnesium halides (7a–i) in tetrahydrofuran afforded a 2:1 mixture of the appropriate cis- and trans-isomers, 8 and 9. These isomers were then separated by silica gel chromatography (8:2 hexane: ethyl acetate) to afford the appropriate 2R,5S- and 2R,5R-diastereomers 8a–i and 9a–i. Compounds 8a–i were hydrogenolyzed by catalytic-transfer hydrogenation with palladium hydroxide-over-carbon, employing ammonium formate as the hydrogen source in refluxing methanol. These conditions afforded a quantitative conversion to the respective N-deprotected products 10a–i within 30 min, as opposed to 12 h utilizing previously reported conditions (i.e., H2/Pd-C).10 The cis-meso-2,5-diarylethyl pyrrolidine free bases were each refluxed with S-glycidol in ethanol to afford a series of substituted cis-meso-2,5-diaryl ethylpyrrolidin-1-yl-propane-1,2(R)-diols (11a–i), which were then converted to their respective HCl salts using ethereal HCl. The products were fully characterized by 1H and 13C-NMR spectroscopy and mass spectrometry; representative examples are provided in the reference section.18
The hydrochloride salts of the pyrrolidino analogues (11a–i) were evaluated for their affinity for the DTBZ binding site on VMAT2 and for their ability to inhibit [3H]DA uptake into synaptic vesicles (functional inhibition of VMAT2) (Table 1).
Table 1.
Inhibition constants (Ki) for GZ-793A (3) analogs at the [3H]DTBZ binding site and [3H]-DA uptake site on VMAT2
| |||
|---|---|---|---|
| Comp | R | [3H]DTBZ Bindinga | VMAT2 [3H]DA uptakec |
| Ki±SEM (μM)b | Ki±SEM (nM) | ||
| 3 | - | 8.29 ± 2.79 | 29 ± 8 |
| 11a | H | 5.91 ± 1.57 | 279 ± 35 |
| 11b | 2-OCH3 | 2.35 ± 0.44 | 270 ± 6 |
| 11c | 3-OCH3 | 2.28 ± 0.44 | 160 ± 10 |
| 11d | 4-OCH3 | 3.94 ± 1.12 | 49 ± 9 |
| 11e | 3-F | 6.76 ± 1.68 | 170 ± 15 |
| 11f | 4-OCHF2 | 0.56 ± 0.23 | 45± 7 |
| 11g | 3,4-methylenedioxy | 8.47 ± 5.19 | 269 ± 28 |
| 11h | 3,4-dimethoxy | > 100 | 3780 ± 230 |
| 11i | 3,5-difluoro | 4.06 ± 0.81 | 200 ± 1 |
All binding experiments have n=3,
Each Ki value represents mean ±SEM.
All uptake experiments represent n=4
Nine GZ-793A analogues were evaluated. The binding affinity of these compounds at the DTBZ binding site on VMAT2 ranged from 560 nM to >100 μM. The most potent compound was the 4-difluoromethoxy substituted 2,5-diphenethylpyrrolidin-1-yl-propane-1,2(R)-diol analogue, 11f (Ki=560 nM) (Table 1,Fig. 2), which had a 15-fold greater affinity for this site than GZ-793A (3, Ki=8.29 μM). The binding affinity of the 2-, 3-, and 4-methoxy analogues 11b–11d also exhibited slightly higher affinity than GZ-793A at the DTBZ binding site on VMAT2.
Fig. 2.

Affinity of Analogue 11f at the TBZ binding site on VMAT2 in the [3H]DTBZ binding assay.
In the vesicular DA uptake assay, the 4-difluoromethoxy analogue, 11f (Ki =45 nM) (Table 1, Fig. 3), and the 4-methoxy analogue, 11d (Ki=49 nM) (Table 1, Fig. 4), both exhibited similar inhibition of [3H]DA uptake when compared to GZ-793A (3, Ki=29 nM). Thus, if one considers the binding affinity and functional data for compounds 11d, 11f, and GZ-793A (3), ring size reduction of the piperidino moiety in GZ-793A appears to lead generally to an improvement in affinity for the DTBZ binding site, but does not alter inhibition of vesicular DA uptake.
Fig. 3.

Inhibition of [3H]DA uptake into rat synaptic vesicles by analogue 11d
Fig. 4.

Inhibition of [3H]DA uptake into rat synaptic vesicles by analogue 11f
Synaptic vesicles were prepared as described previously.19 Briefly, fresh whole brain (excluding cerebellum) was homogenized in 28 mL of ice-cold 0.32 M sucrose using a glass homogenizer (7 up and down strokes of a Teflon pestle, clearance 0.003 in). Homogenates were centrifuged at 1000g for 12 min at 4°C. Resulting supernatants (S1) were centrifuged at 22,000g for 10 min. The resulting pellets (P2), containing the synaptosomes, were resuspended in 18 mL of ice-cold Milli-Q water for 5 min with 7 up and down strokes of the Teflon pestle homogenizer. Osmolarity was restored by immediate addition of 2 mL of 25 mM HEPES and 100 mM K2-tartrate buffer (pH 7.5). Samples were centrifuged at 20,000g for 20 min. MgSO4 (final concentration, 1 mM) was added to the resulting supernatants (S3). Final centrifugations were performed at 100,000g for 45 min. Pellets (P4) were resuspended immediately in ice-cold buffer providing ~15 μg of protein/100 μL.
[3H]DTBZ binding to synaptic vesicle membranes was performed according to previously described procedures.20 Briefly, 100 μL of vesicles suspension was incubated in assay buffer (in 25 mM HEPES, 100 mM K2-tartrate, 5 mM MgSO4, 0.1 mM EDTA, and 0.05 mM EGTA, pH 7.5, 25°C) in the presence of 5 nM [3H]DTBZ and 1 nM-1 mM lobelane analogs (final concentrations) for 30 min at room temperature. Nonspecific binding was determined in the presence of 20 μM TBZ. Assays were performed in duplicate using Unifilter 96-well GF/B filter plates (presoaked in 0.5% polyethylenimine) and terminated by harvesting using a FilterMate harvester. After washing five times with 350 μL of the ice-cold wash buffer (in 25 mM HEPES, 100 mM K2-tartrate, 5 mM MgSO4, and 10 mM NaCl, pH 7.5), filter plates were dried, bottoms were sealed, and each well was filled with 40 μL of Packard’s MicroScint 20 cocktail. Bound [3H]DTBZ was measured using a Packard Top Count NXT scintillation counter and a Packard Windows NT-based operating system.
Inhibition of [3H]DA uptake was conducted as previously described,22 utilizing a striatal synaptic vesicle preparation. Briefly, rat striata were homogenized with 10 up and down strokes of a Teflon pestle homogenizer (clearance ~ 0.003″) in 14 ml of 0.32 M sucrose solution. Homogenate was centrifuged (2,000 g for 10 min at 4°C), and the resulting supernatant was centrifuged again (10,000 g for 30 min at 4°C). The pellet was resuspended in 2 ml of 0.32 M sucrose solution and subjected to osmotic shock by adding 7 ml of ice-cold water to the preparation, followed by the immediate restoration of osmolarity by adding 900 μl of 0.25M HEPES buffer and 900 μL of 1.0 M potassium tartrate solution. Samples were centrifuged (20,000 g for 20 min at 4°C), and the resulting supernatant was centrifuged again (55,000 g for 1 hr at 4°C), followed by the addition of 100 μL of 10 mM MgSO4, 100 μL of 0.25 M HEPES and 100 μL of 1.0 M potassium tartrate solution prior to the final centrifugation (100,000 g for 45 min at 4°C). The final pellet was resuspended in 2.4 mL of assay buffer (25 mM HEPES, 100 mM potassium tartrate, 50 μM EGTA, 100 μM EDTA, 1.7 mM ascorbic acid, 2 mM ATP-Mg2+, pH 7.4). Aliquots of the vesicular suspension (100 μL) were added to tubes containing assay buffer, various concentrations of inhibitor (0.1 nM - 10 mM) and 0.1 μM [3H]DA in a final volume of 500 μL. Nonspecific uptake was determined in the presence of Ro4-1284 (10 μM). Reactions were terminated by filtration, and radioactivity retained by the filters was determined by liquid scintillation spectroscopy (Liquid scintillation analyzer; PerkinElmer Life and Analytical Sciences, Boston, MA).
In summary, reduction in ring size of the central heterocyclic piperidine ring of GZ-793A has led to the discovery of the 4-methoxy and 4-difluoromethoxy cis-meso-2,5-disubstitued pyrrolidine analogues 11d and 11f. These two analogues showed similar inhibitory potency of [3H]DA uptake into vesicles (Ki = 49 and 45 nM, respectively) when compared to that for GZ-793A (Ki =29 nM), and were the most potent analogues in the series in this assay. Analogue 11f was also the most potent inhibitor of [3H]DTBZ binding in the series (Ki =560 nM), and had a 15-fold greater affinity for this site than GZ-793A (Ki =8.29 μM). Thus, 11d and 11f represent new water-soluble analogues of GZ-793A that inhibit VMAT2 function.
Scheme 1.
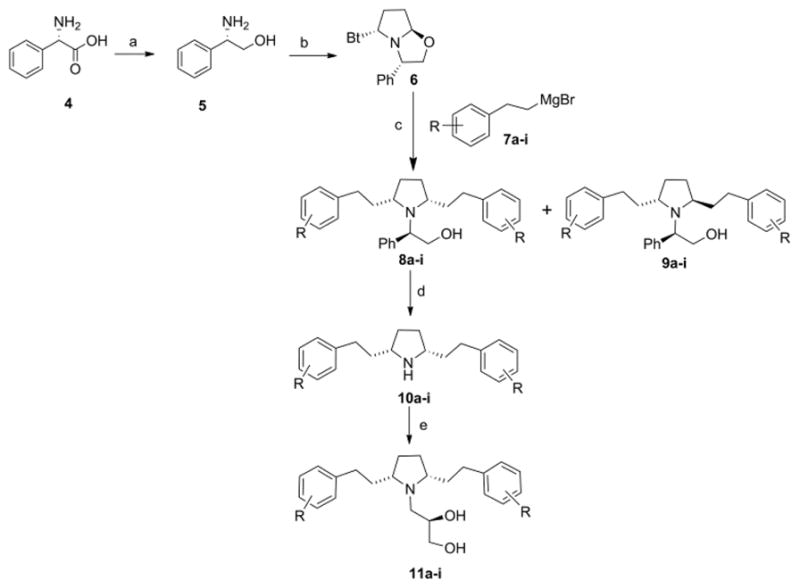
Reagents and conditions: a) LiAlH4, THF, reflux, 24h; b) 2,5 dimethoxy tetrahydrofuran, benzotriazole, 10% aq HCl, rt, 12h: c) Substituted phenethyl magnesium bromides (7a–i), THF, rt, 12h; d) Pd(OH)2, NH4HCO2, MeOH, reflux, 30 min; e) (S)-glycidol, EtOH, reflux, 24h.
Acknowledgments
This research was supported by NIH grant U01 DA13519. The University of Kentucky holds patents on lobeline and the analogues described in the current work. A potential royalty stream to LPD and PAC may occur consistent with University of Kentucky policy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Miller DK, Crooks PA, Teng L, Witkin JM, Munzar P, Goldberg SR, Acri JB, Dwoskin LP. J Pharmacol Exp Ther. 2001;296:1023. [PubMed] [Google Scholar]
- 2.Harrod SB, Dwoskin LP, Crooks PA, Klebaur JE, Bardo MT. J Pharmacol Exp Ther. 2001;298:172. [PubMed] [Google Scholar]
- 3.Miller DK, Harrod SB, Green TA, Wong MY, Bardo MT, Dwoskin LP. Pharmacol Biochem Behav. 2002;74:279. doi: 10.1016/s0091-3057(02)00996-6. [DOI] [PubMed] [Google Scholar]
- 4.Harrod SB, Dwoskin LP, Green TA, Gehrke BJ, Bardo MT. Psychopharmacology. 2003;165:397. doi: 10.1007/s00213-002-1289-6. [DOI] [PubMed] [Google Scholar]
- 5.(a) Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. J Pharmacol Exp Ther. 1997;280:1432. [PubMed] [Google Scholar]; (b) Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. J Neurochem. 1998;71:258. doi: 10.1046/j.1471-4159.1998.71010258.x. [DOI] [PubMed] [Google Scholar]
- 6.Sulzer D, Rayport S. Neuron. 1990;5:797. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- 7.Brown JM, Hanson GR, Fleckenstein AE. J Neurochem. 2000;74:2221. doi: 10.1046/j.1471-4159.2000.0742221.x. [DOI] [PubMed] [Google Scholar]
- 8.Zheng G, Dwoskin LP, Deaciuc AG, Norrholm SD, Crooks PA. J Med Chem. 2005;48:5551. doi: 10.1021/jm0501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nickell JR, Krishnamurthy S, Norrholm S, Deaciuc G, Zheng G, Crooks PA, Dwoskin LP. J Pharmacol Exp Ther. 2009;332:612. doi: 10.1124/jpet.109.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vartak AP, Nickell JR, Chagkutip J, Dwoskin LP, Crooks PA. J Med Chem. 2009;52:7878. doi: 10.1021/jm900770h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vartak AP, Deaciuc AG, Dwoskin LP, Crooks PA. Bioorg Med Chem Lett. 2010;20:3584. doi: 10.1016/j.bmcl.2010.04.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng G, Dwoskin LP, Deaciuc AG, Crooks PA. Bioorg Med Chem Lett. 2008;18:6509. doi: 10.1016/j.bmcl.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horton DB, Kiran BS, Zheng G, Crooks Peter A, Dwoskin LP. J Pharmacol Exp Ther. 2011;339(1):286. doi: 10.1124/jpet.111.184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng G, David BH, Reddy PN, Nickell JR, Culver JP, Deaciuc AG, Dwoskin LP, Crooks PA. Med chem com. 2013;4:564. doi: 10.1039/C3MD20374C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alvers KM, Beckmann JS, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. Psychopharmacology (Berl) 2012;224(2):255. doi: 10.1007/s00213-012-2748-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beckmann JS, Denehy ED, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. Psychopharmacology (Berl) 2012;220:395. doi: 10.1007/s00213-011-2488-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katritzky AR, Cui X, Baozhen Y, Steel PJ. J Org Chem. 1999;64:1979. doi: 10.1021/jo9821426. [DOI] [PubMed] [Google Scholar]
- 18.Spectral data of some potent compounds; Compound 11a: (free base), 1H NMR (300 MHz, CDCl3): δ 7.12–7.40 (m, 10H), 3.90–3.95 (m, 1H), 3.80-3.46 (m, 3H), 3.40-2.95 (m, 4H), 2.72-2.40 (m, 4H), 2.38-1.95 (m, 6H), 1.90-1.82 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3): δ 140.0, 128.8, 128.5, 126.6, 72.1, 69.7, 67.6, 56.1, 32.7, 32.3, 27.8 ppm; MS (EI) m/z 353 (M+). Compound 11c: 1H NMR (300 MHz, CDCl3): δ 10.38 (brs, 1H), 7.16 (t, 2H, J=7.5 Hz), 6.74-6.70 (m, 6H), 4.15 (m, 1H), 3.75 (s, 6H), 3.58-3.35 (m, 2H), 3.13-2.78 (m, 4H), 2.77-2.74 (m, 2H), 2.50-2.38 (m, 6H), 2.16-1.99 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 159.9, 141.2, 129.9, 120.8, 114.3, 112.0, 70.5, 69.32, 67.6, 64.3, 55.5, 32.7, 31.5, 27.6; MS (EI) m/z; 413 (M+). Compound 11d: 1H NMR (300 MHz, CDCl3): δ 9.86 (brs, 1H), 7.05-6.95 (m, 4H), 6.95-6.82 (m, 4H), 4.05 (brs, 1H), 3.80-3.42 (m, 12H), 3.28-2.95 (m, 2H), 2.80-1.79 (m, 10H) ppm; 13C NMR (75 MHz, CDCl3): δ 158.2, 131.7, 129.5, 114.2, 70.3, 69.0, 67.7, 64.4, 55.6, 32.1, 31.9, 27.8 ppm; MS (EI) m/z; 413 (M+). Compound 11f: 1H NMR (300 MHz, CDCl3): δ 10.85 (brs, 1H), 7.35-7.15 (m, 4H), 6.95–7.10 (m, 4H), 6.47 (t, 2H, J = 72.0 Hz), 4.19 (brs, 1H), 3.60-3.32 (m, 6H), 3.15-2.85 (m, 4H), 2.75-1.70 (m, 8H) ppm; 13C NMR (75 MHz, CDCl3): δ 149.7, 137.2, 129.7, 119.7, 115.9, 67.9, 65.9, 64.0, 48.6, 32.9, 31.3, 27.5 ppm; MS (EI) m/z; 485 (M+). Compound 11g: 1H NMR (300 MHz, CDCl3): δ 10.42 (brs, 1H), 6.70-6.59 (m, 6H), 5.89 (s, 4H), 5.32 (brs, 1H), 4.20 (m, 1H), 3.73-3.50 (m, 2H), 2.33–3.98 (m, 3H), 2.82-2.62 (m, 2H), 2.60-2.28 (m, 6H), 2.16-2.00 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 147.9, 146.2, 133.3, 121.3, 108.6, 101.1, 70.6, 69.3, 67.7, 64.3, 57.5, 32.4, 31.8, 27.6; MS (EI) m/z 441 (M+). Compound 11i: 1H NMR (300 MHz, CDCl3): δ 10.92 (brs, 1H), 6.91-6.64 (m, 6H), 4.30-4.21 (m, 1H), 3.65-3.55 (m, 2H), 3.25-2.95 (m, 4H), 2.92-2.83 (m, 4H), 2.62-2.25 (m, 4H), 2.25-1.90 (m, 4H); 13C NMR (75 MHz, DMSO-d6): δ 163.2, 141.3, 112.3, 102.1, 69.3, 67.7, 65.3, 49.5, 32.4, 31.8, 27.6; MS (EI) m/z; 425(M+).
- 19.Nickell JR, Zheng G, Deaciuc G, Crooks PA, Dwoskin LP. J Pharmacol Exp Ther. 2011;336:724. doi: 10.1124/jpet.110.172882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teng L, Crooks PA, Dwoskin LP. J Neurochem. 1998;71:258. doi: 10.1046/j.1471-4159.1998.71010258.x. [DOI] [PubMed] [Google Scholar]