

Abstract
4-Benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8) was previously identified as an antileukemic agent exhibiting no evident toxicity toward normal hematopoietic cells. An SAR study has been carried out to examine the effect of varying the C-2 and C-4-substituents on the thiadiazolidinone ring of TDZD-8 on anti-leukemic activity. These studies resulted in the identification of more druglike analogs that exhibited comparable potency to TDZD-8 in killing acute myelogenous leukemia (AML) cells in culture. Surprisingly, the cell death kinetics induced by several of these novel analogs on MV-411 cells were extremely fast, with commitment to death occurring within 30 minutes. At a concentration of 10 μM, 3f (LD50=3.5 μM) completely eradicated cell viability of MV-411 cells within 2 hours, while analog 3e (LD50=2.0 μM) decimated cell viability within 30 min at a concentration of 10 μM and effectively abolished cell viability at 5 μM within 1-2 hours.
Keywords: 1,2,4-Thiadiazolidine-3,5-diones; antileukemic; rapid cell death kinetics; acute myelogenous leukemia (AML)
Acute myelogenous leukemia (AML) is a hematologic malignancy characterized by the abnormal accumulation of immature white blood cells in the bone marrow (BM), and a poor overall survival rate; 12,330 people were diagnosed with AML in 2010.1 AML is thought to originate and be maintained from a malignant population of cells known as leukemia stem cells (LSCs). It has been shown that LSCs are not effectively eliminated by standard chemotherapy regimens and are likely to result in disease relapse.2 Thus, the development of clinically-viable small-molecule chemotherapeutics that eradicate such stem cells has become a high priority.
TDZD-8 was first identified as a highly selective inhibitor of glycogen synthase kinase 3β (GSK3β);3a-e We have recently demonstrated that 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8) displays unique antileukemic properties.4 It was found that TDZD-8 possesses the ability to eradicate acute and chronic, lymphoid and myeloid forms of leukemia, including the LSC populations, at concentrations of about 20 μM with rapid kinetics (i.e. within two hours of exposure). In this concentration range, TDZD-8 possesses insignificant toxicity toward normal hematopoietic stem cells. The toxicity of TDZD-8 is also specific toward hematologic malignancies, since it was found not to be toxic toward a broad swath of other cancer cell lines in the NCI 60 anticancer screen.4 The precise antileukemic mechanism of action of TDZD-8 is not completely understood and likely involves multiple cellular targets; TDZD-8 also appears to rapidly disrupt membrane integrity and deplete free cellular thiols. The latter effect is not surprising, since we have noted in our own synthetic studies that the 1,2,4-thiadiazolidine-3,5-dione (TDZD) ring is capable of oxidizing both P(III) and Pd(0).5
The current study represents our first efforts identifying TDZD analogs which conserve or improve upon the novel selectivity and cytotoxicity of TDZD-8 toward AML cells, and that also represent more water-soluble molecules compared to the parent compound, TDZD-8. In this report, we have focused on determining the effect of varying the N-2 substituent of TDZD-8 on antileukemic activity in MV-411 cells in culture. In addition, we have investigated structural modifications that introduce a water solubilizing group into the 4-benzyl moiety of the most potent TDZD analogues in order to improve druglikeness.
The oxidative condensation of isothiocyanates with isocyanates via chlorine addition has been the subject of recent reports.3b,c Classically, 1,2,4-thiadiazolidine-3,5-dione analogs are synthesized through oxidative condensation of isothiocyanates with isocyanates in the presence of gaseous chlorine as the oxidizing agent. In our hands, we found that the introduction of chlorine as a solution in CCl4/diethyl ether was more reliable and more controllable, in terms of stoichiometry (Scheme 1, Method 1), than the treatment of a mixture of isothiocyanate and isocyanate with chlorine gas. We have also utilized SO2Cl2 in diethyl ether as an alternative reported method of preparation of 1,2,4-thiadiazolidine-3,5-diones analogs5 (Scheme 1, Method 2). Recently, we have reported that N-chlorosuccinimide is a safe and viable alternative to the use of chlorine gas in the synthesis of 1,2,4-thiadiazolidine-3,5-diones analogs (Scheme 1, Method 3).6 The TDZD analogs 3a-3f described herein were synthesized from readily available starting materials utilizing a combination of the three methodologies outlined in Scheme 1.
Scheme 1.

Methodologies for the preparation of 1,2,4-thiadiazolidine-3,5-dione analogs from isocyanates and isothiocyanates.
Despite promising in vitro antileukemic activity (Table 1), TDZD analogs 3a-3f generally had poor water-solubility. Thus, in another structural iteration, we examined the effect of introducing ionizable hydrophilic groups, such as NH2 and COOH, into the TDZD molcule to improve water-solubility. Five analogs, 10b and 10d-10g, that incorporated a protonatable aminomethyl moiety at either C4 or C3 of the phenyl ring were synthesized via the route illustrated in Scheme 2A. The appropriate Boc-protected aminomethylbenzoic acid 5 was converted to the corresponding benzylamine analog 6, utilizing standard synthetic procedures. Intermediate 6 was then converted into the corresponding benzylisothiocyanate 7 utilizing a modification of the procedure reported by Wong and Dolman7. In this procedure, a triethylammonium thiocarbamate is formed through treatment of the primary amine with 1.0 equivalent of CS2 and triethylamine in anhydrous THF, followed by addition of tosyl chloride with stirring for 3 hours. While following this protocol we experienced difficulty in obtaining a practical yield of the desired isothiocyanate. The reason was traced to the insolubility of the amine in THF, which leads to a very sluggish formation of the intermediate thiocarbamate. The yield improved dramatically when additional equivalents of CS2 were added. Consequently, we conducted the entire reaction in a 10:1 mixture of CH2Cl2/CS2 (10 mL/mmole), which resulted in instantaneous conversion of the primary amine to the thiocarbamic acid (an insoluble product). Subsequent addition of triethylamine caused the dissolution of the precipitated thiocarbamic acid. After cooling the resulting solution to 0 °C, tosyl chloride (1 equivalent) was added in one portion, and the reaction mixture worked up to afford the isothiocyanate in good yield. Reaction of 7 with the requisite isocyanate 2, utilizing Method 3 conditions (Scheme 1), afforded the corresponding Boc-protected aminomethyl TDZD analogs. The TDZD ring in the five resulting Boc-protected aminomethyl analogs was found to be stable to 4N HCl in dioxane and thus amenable to N-deprotection and HCl salt formation to afford 10b and 10d-10g.
Table 1.
LD50 (μM) values of 1,2,4-thiadiazolidine-3,5-diones against cultured MV-411 cells.
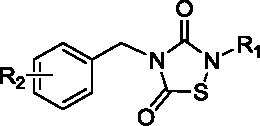
| |||
|---|---|---|---|
|
| |||
| Analog | R2 | R1 | LD50 μ M* |
| 3a (TDZD-8) | 4-H | CH3 | 4.01 |
| 3b | 4-H | CH2CH3 | 9.21 |
| 3c | 4-H | CH2CH2CH3 | 11.6 |
| 3d | 4-H | CH2CH=CH2 | 9.20 |
| 3e | 4-H | CH2CH2Cl | 2.03 |
| 3f | 4-H | CH2CH2Br | 3.50 |
| 10b | 4-CH2NH2•HCl | CH2CH3 | 15.5 |
| 10d | 4-CH2NH2•HCl | CH2CH=CH2 | 6.70 |
| 10e | 4-CH2NH2•HCl | CH2CH2Cl | 3.01 |
| 10f | 4-CH2NH2•HCl | CH2CH2Br | 4.51 |
| 10g | 3-CH2NH2•HCl | CH2CH2Cl | 4.40 |
| 15 | 4-COONa | CH2CH2Cl | >20 |
LD50 (lethal dose) denotes the concentration of analog that causes cell death in 50% of the cell population.
Scheme 2. Synthesis of phenyl substituted 1,2,4-thiadiazolidine-3,5-diones.

For the synthesis of the 4-carboxylate analog 15 (Scheme 2B), the precursor tert-butyl-protected isothiocyanate 14 was synthesized from tert-butyl-4-cyanobenzoate (12)8, which was converted to tert-butyl-4-(aminomethyl)-benzoate (13), as reported by Andrew et al.9, and then subsequently converted to 14 with CS2/CH2Cl2. Reaction of 14 with isocyanate 2e utilizing Method 3 conditions (Scheme 1), followed by deprotection and sodium salt formation afforded 15 (Scheme 2B). NMR and mass spectral characterization data for all TDZD analogs are provided in the reference section10.
Cytotoxicity data (LD50) for compounds 3e-3f against cultured MV-411 cells11 are shown in Table 1 and are compared to TDZD-8 (3a). Generally, increasing the size of the N-2 substituent resulted in compounds with lower potency (3b-3d) compared to TDZD-8. However, the presence of an N-2-(2-halogenoethyl) substituent (3e; LD50 = 2.0 μM, and 3f; LD50 = 3.5 μM) afforded compounds with comparable LD50 values to TDZD-8 (3a; LD50 = 4.0 μM). Evaluation of the more water soluble analogs 10b, 10d-10g, and 15, also afforded several compounds with comparable LD50 values to TDZD-8, indicating that a water solubilizing amino moiety can be introduced into the aromatic ring of the active analogs 3e and 3f without compromising potency. Thus, the HCl salt of the N-2-(2-chloroethyl) and N-2-(2-bromoethyl) analogs 10e and 10f were found to have comparable potency (LD50 = 3.0 μM and 4.5 μM, respectively) to 3e, 3f and TDZD-8 but had improved hydrophilicity and druglikeness. Moving the aminomethyl moiety of 10e from the para-position to the meta-position on the phenyl ring to afford analog 10g had little effect on the LD50 values for these two regiospecific isomers. Unfortunately, introducing a water-solubilizing carboxylate group at the C4 position of the phenyl ring of 3e to afford 15 resulted in loss of antileukemic activity.
As with TDZD-8, none of the active TDZD analogs synthesized in this study was toxic to normal hematopoietic cells. In addition, it was previously observed that the cell death kinetics induced by TDZD-8 on MV-411 cells was extremely fast2, with commitment to death occurring within several hours. We similarly measured the kinetics of cell death caused by TDZD-8 and analogs 3d, 3e, and 3f. Figure 1 shows the time-course to determine the cell-death kinetics of these four compounds. At a concentration of 20 μM, TDZD-8 abolished MV-411 cell viability over a period of 2 hours. At a concentration of 5 μM, the activity of analog 3d was negligible, while at a concentration of 10 μM, the analog caused halving of percentage cell viability over 24 hours. However, at a concentration of 20 μM, 3d caused a 50% decrease in cell viability within 30 minutes, and effectively abolished viability over a period of 2 hours. Analog 3f caused a 50 percent decrease in cell viability within the first 30 min at a concentration of 5 μM. However, the effect was not sustained, and cell viability was retained, even at 2.5 μM after 24 hours. At a concentration of 10 μM, 3f completely eradicated cell viability within the first 2 hours. In contrast, analog 3e decimated cell viability within 30 min at a concentration of 10 μM and effectively abolished cell viability at 5 μM within 1-2 hours, and caused halving of percentage cell viability over 24 hours at a concentration of 2.5 μM.
Figure 1.

Time-course of cell death kinetics for analogs 3d, 3e, and 3f against cultured MV-411 cells when compared to the parent analog, TDZD-8.
From these results, it is clear that determination of the LD50 parameter alone provides an estimation of only the antileukemic effect of the above TDZD analogs in total populations of AML cells using MV4-11 cultures. However, the mechanism of action of each of the TDZD analogs synthesized in this study may be different. It is known that the parent compound, TDZD-8, exhibits several intracellular activities such as oxidation of cellular thiols, disruption of membrane integrity, and inhibition of protein kinases. Differential abilities of the TDZD analogs to act at different sites of action inside the leukemic cell may be responsible for the observed inconsistencies between the LD50 values and the cell death kinetics experiments. One possible mechanism for the antileukemic action of the TDZD analogs 3e, 3f, 10e, 10f, may be nonspecific chemical reactivity, such as the alkylative ability of the N-2 halogenoalkyl substituted analogs, or the aforementioned oxidative potential of the TDZD ring itself. However, our experiments show that these TDZD analogs have negligible cytotoxicity toward normal hematopoietic cells or other tumor cell types, and analog 15, which is also an N-2-(2-chloroethyl) analog, is inactive as an antileukemic agent. This suggests that alkylative or oxidative reactivity is unlikely as a mechanism of antileukemic action for these TDZD analogs, and that their mechanism of inhibition is not related to nonspecific inhibition of rapidly dividing cells.
This study has also provided useful information about the importance of the N-2-substituent in the TDZD-8 molecule for antileukemic activity. Sites for attachment of hydrophilic groups with retention of pharmacological properties have also been identified, leading the way for the the development of more druglike TDZD analogs. Future studies can now focus on a more thorough pharmacological and toxicological evaluation of these analogs. Lastly, the positive, as well as the negative SAR trends, resulting from this study will serve as a guide for further optimization of this unique series of compounds, not only with regard to potency and selectivity, but also for achieving optimal pharmacokinetics and drugability.
Supplementary Material
Acknowledgments
Financial support from the Leukemia-Lymphoma Society, Grant #6077-08, is gratefully acknowledged. M.G is funded by the National Institutes of Health through the NIH Director's New Innovator Award Program, 1-DP2-OD007399-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.The Leukemia-Lymphoma Society. Facts 2010-2011. Leukemia-Lymphoma Society; 2011. Edit. [Google Scholar]
- 2.Jordan CT. Best Pract Res Cl Ha. 2007;20:13. doi: 10.1016/j.beha.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Martinez A, Fernandez E, Castro A, Conde S, Rodriguez Franco I, Banos JE, Badia A. Eur J Med Chem. 2000;35:913. doi: 10.1016/s0223-5234(00)01166-1. [DOI] [PubMed] [Google Scholar]; (b) Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. J Med Chem. 2002;45:1292. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]; (c) Martinez A, Alonso M, Castro A, Dorronsoro I, Gelpi JL, Luque FJ, Perez C, Moreno FJ. J Med Chem. 2005;48:7103. doi: 10.1021/jm040895g. [DOI] [PubMed] [Google Scholar]; (d) Martinez A, Castro A, Dorronsoro I, Alonso M. Med Res Rev. 2002;22:373. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]; (e) Castro A, Encinas A, Gil C, Brase S, Porcal W, Perez C, Moreno JF, Martinez A. Bioorg Med Chem. 2008;16:495. doi: 10.1016/j.bmc.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Guzman ML, Xiaojie L, Corbett CA, Rossi RM, Bushnell T, Liesveld JL, Hebert J, Young F, Jordan CT. Blood. 2007;110:4436. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slomczynska U, Barany G. J Heterocycl Chem. 1984;21:241. [Google Scholar]
- 6.Nasim S, Crooks PA. Tetrahedron Lett. 2009;50:257. [Google Scholar]
- 7.Wong R, Dolman SJ. J Org Chem. 2007;72:39969. doi: 10.1021/jo070246n. [DOI] [PubMed] [Google Scholar]
- 8.Stanton MG, Gagne MR. J Org Chem. 1997;62:8240. doi: 10.1021/jo971138b. [DOI] [PubMed] [Google Scholar]
- 9.Andrew DR, Gaeta FC. U.S. Patent 4,885,293. 1989
- 10.Spectral and analytical data for compounds 3a, 3b and 3e were in agreement with previously reported data.3b,3c,6 2-Propyl-4-benzyl-1,2,4-thiadiazolidine-3,5-dione (3c), clear oil, yield = 72 %; 1H NMR (300 MHz, CDCl3) δ ppm 7.42-7.23 (m, 5H), 4.79 (s, 2H), 3.56 (t, J = 6.5 Hz, 2H), 1.61 (q, J = 6.5 Hz, 2H), 0.92 (t, J = 6.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ ppm 166.0, 153.0, 135.3, 128.9, 128.8, 128.3, 46.8, 46.2, 22.4, 11.4; EI-MS m/z = 250; Anal. (C12H14N2O2S) C, H, N. 2-Allyl-4-benzyl-1,2,4-thiadiazolidine-3,5-dione (3d), clear oil, yield = 60 %; 1H NMR (300 MHz, CDCl3) δ ppm 7.46–7.26 (m, 5H), 5.81 (m, 1H), 5.35 (m, 2H), 4.83 (s, 2H), 4.23 (m, 2H); 13C NMR (75 MHz, CDCl3) δ ppm 165.9, 152.6, 135.2, 131.0, 128.9, 128.7, 128.3, 120.7, 47.4, 46.1; EI-MS m/z = 248. Anal. (C12H12N2O2S) C, H, N. 2-(2′-Bromoethyl)-4-benzyl-1,2,4-thiadiazolidine-3,5-dione (3f), white solid, mp = 76-78 °C, yield = 82 %; 1H NMR (300 MHz, CDCl3) δ ppm 7.45-7.30 (m, 5H), 4.83 (s, 2H), 3.99 (t, J = 6.3 Hz, 2H), 3.52 (t, J = 6.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ ppm 165.7, 153.2, 135.1, 129.0, 128.9, 128.5, 46.9, 46.4, 29.1. Anal. (C11H11BrN2O2S) C, H, N. 2-Ethyl-4-(4′-methylaminobenzyl)-1,2,4-thiadiazolidine-3,5-dione hydrochloride (10b), white solid, mp = 151–153 °C, yield = 73 %; 1H NMR (300 MHz, CD3OD) δ ppm 7.44 (s, 4H), 4.83 (s, 2H), 4.10 (s, 2H), 3.69 (q, J = 6.3 Hz, 2H), 1.23 (t, J = 6.3 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ ppm 166.4, 152.9, 136.5, 134.3, 129.9, 128.3, 45.5, 42.5, 39.8, 14.3. Anal. (C12H16ClN3O2S.0.5H2O) C, H, N. 2-Allyl-4-(4′-methylaminobenzyl)-1,2,4-thiadiazolidine-3,5-dione hydrochloride (10d), white solid, mp = 105–107 °C, yield = 75 %; 1H NMR (300 MHz, CD3OD) δ ppm 7.44 (s, 4H), 5.87 (m, 1H), 5.32 (m, 2H), 4.85 (s, 2H), 4.24 (m, 2H), 4.10 (s, 2H); 13C NMR (75 MHz, CD3OD) δ ppm 167.7, 154.6, 138.4, 134.5, 132.9, 130.6, 130.3, 120.6, 48.4,46.6, 44.3; Anal (C13H16ClN3O2S) C, H, N. 2-(2′-Chloroethyl)-4-(4″-methylaminobenzyl)-1,2,4-thiadiazolidine-3,5-dione hydrochloride (10e), white solid, mp = 103–105 °C, yield = 64 %; 1H NMR (300 MHz, CD3OD) δ ppm 7.42 (s, 4H), 4.82 (s, 2H), 4.15 (s, 2H), 3.90 (t, J = 6.5 Hz, 2H), 3.78 (t, J = 6.5 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ ppm 165.7, 153.2, 137.4, 135.1, 129.0, 128.7, 46.9, 46.4, 44.1, 41.7; Anal. (C12H15Cl2N3O2S) C, H, N. 2-(2′-Bromoethyl)-4-[4″-methylaminobenzyl)-1,2,4-thiadiazolidine-3,5-dione hydrochloride (10f), white solid, mp = 91–93 °C, yield = 75%; 1H NMR (300 MHz, , CD3OD) δ ppm 7.44 (s, 4H), 4.85 (s, 2H), 4.10 (s, 2H), 4.04 (t, J = 6.3 Hz, 2H), 3.63 (t, J = 6.3 Hz, 2H); 13C NMR (75 MHz, , CD3OD) δ ppm 167.3, 154.6, 137.9, 134.2, 130.3, 129.8, 47.5, 46.3, 44.0, 30.5; Anal. (C12H15BrClN3O2S) C, H, N. 2-(2′-Chloroethyl)-4-(3″-methylaminobenzyl)-1,2,4-thiadiazolidine-3,5-dione hydro- chloride (10g), white solid, mp 97–99 °C, yield = 53 %; 1H NMR (300 MHz, CD3OD) δ ppm 7.36 (t, J = 7.6 Hz, 1H), 7.31 (m, 3H), 4.83 (s, 2H), 4.13 (s, 2H), 4.05 (t, J = 5.7 Hz, 2H), 3.81 (t, J = 5.7 Hz, 2H); 13C NMR (75 MHz, CD3OD) δ ppm 165.7, 153.2, 135.1, 129.0, 128.9, 128.7, 128.3, 128.5, 46.5, 46.3, 43.9, 42.1; Anal. (C12H15Cl2N3O2S.H2O) C, H, N. 4-{4′-[2″-(2‴-Chloroethyl)-1,2,4-thiadiazolidine-3,5-dionyl]} sodium benzoate (15), white solid, mp = 170–172 °C, yield = 90 %; 1H NMR (300 MHz, , CD3OD) δ ppm 7.97 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 4.74 (s, 2H), 3.96 (t, J = 5.4 Hz, 2H), 3.73 (t, J = 5.4 Hz, 2H); 13C NMR (75 MHz, , CD3OD) δ ppm 168.9, 167.0, 154.1, 140.9, 131.2, 130.8, 128.8, 47.4, 46.2, 42.6. Anal (C12H10ClN2O4SNa) C, H, N.
- 11.Assessment of LD50for TDZD analogs from MV4-11 cell assays. MV4-11 cells were purchased from the ATCC (American type culture collection). Cells were cultured at 300,000 cells/mL and exposed to at least 4 different concentrations of the various analogs for 24 hours (or for the indicated time period in the time-course experiments). Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% fetal bovine serum (FBS) was used. Cell viability was determined by staining with Annexin V-Fluorescein isothiocyanate (FITC) and 7-aminoactinomycin D (7-AAD). Analyses were performed in the BD-LSRII 15 minutes later. The percent of viable cells was determined relative to the untreated control by gaiting on Annexin V negative/7-AAD negative cells. The calculation of LD50 values was performed using Calcusyn software (Biosoft).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.