

Abstract
Replacing the phenyl groups in the structure of the VMAT2 inhibitor, lobelane with either pyridyl, quinolyl or indolyl groups affords novel analogues with improved water solubility. The synthetic methodologies reported herein also underscore the paucity of hydrogenation methods that offer selectivity in the synthesis of the different classes of heteroaromatic lobelane analogues. The quinolyl group was the only replacement for the phenyl group in lobelane that retained VMAT2 inhibition.
Lobelane (1, N-methyl-cis-2,6-diphenethylpiperidine) is a minor alkaloid present in Lobelia inflata, and a defunctionalized analog of lobeline (2), the major alkaloid of L. inflata, (Figure 1). Mid- 20th century investigations into the pharmacology of lobelane by Foster et al. revealed the peripheral spasmolytic, ‘central stimulating’ action using guinea pigs, and marked respiratory analeptic activity in dogs.1 Lobeline inhibits (+)-methamphetamine (METH) self-administration in rats, and the effect was not surmounted by METH indicating a noncompetitive mechanism of inhibition. Repeated administration of lobeline in rats prior to (+)-METH self-administration also prevents (+)-METH-induced dopaminergic toxicity. 2 The underlying neurochemical mechanism of action of lobeline is believed to be due to reversible inhibition of the vesicular monoamine transporter-2 (VMAT2). Since VMAT2 sequesters cytosolic dopamine (DA) into synaptic vesicles, thereby preventing metabolism by monoamine oxidase (MAO), VMAT2 inhibition by lobeline leads to increased metabolism of DA into dihydroxyphenyl acetic acid (DOPAC) and reduced levels of cytosolic DA.3 METH inhibits VMAT2 and MAO, leading to increased cytosolic DA levels. METH also reverses DA transporter function, leading to increases in extracellular DA levels). The psychological excitation or ‘high’ produced by METH is the result of the increased extracellular DA concentrations as an outcome of actions of METH. One possibility for the lobeline-induced attenuation of METH self-administration is that the reduction in cytosolic DA produced by lobeline renders METH ineffective as a stimulant. As an outcome of these preclinical studies, lobeline is being evaluated in Phase II clinical trials as a treatment for METH abuse.4
Figure 1.
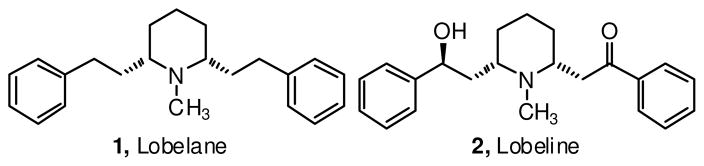
Structures of Lobelane (1) and Lobeline (2).
The development of lobeline as a therapeutic agent to treat METH-addiction is complicated by its additional inhibitory effects on the α4β2* and α6* nicotinic acetylcholine receptors (nAChRs). Glennon et al. reported that deoxygenation and reduction of 2 attenuated its activity at nAChRs, with the fully reduced analog 1 binding with a >10,000-fold lower affinity to nAChRs when compared to lobeline.5 A rigorous study into the pharmacology of 1 and its aryl-substituted analogues revealed that 1 possessed a 5-fold improvement in affinity for the dihydrotetrabenazine (DTBZ) binding site on VMAT2.6,7 Replacement of the phenyl groups in lobelane with anisyl and naphthyl moieties significantly improved affinity for VMAT2 with retention of selectivity for VMAT2.8
Lobelane and its analogues were recently reported to potently inhibit vesicular DA uptake by VMAT2, as well as inhibit METH-evoked DA release, with lobelane being 10–15 fold more potent than lobeline in these studies.9,10 Based on this work, lobelane was considered a lead candidate for the development of VMAT2 inhibitors as therapeutics for psychostimulant abuse. The evaluation of lobelane in vivo for its behavioral effects in rats was limited somewhat by the low water solubility of its salt forms. A subsequent systematic investigation of lobelane analogues was initiated with the aim of discovering analogs possessing improved water solubility, as well inhibition and selectivity at VMAT2.
This report focuses on the substitution of the side-chain phenyl groups of lobelane with various heteroaromatic moieties and a study of the resultant effects on water solubility and VMAT2 inhibition. Initial attempts at synthesizing heteroaromatic analogues of lobelane were focused on the development of an advanced 2,6-disubstituted piperidine intermediate that would be amenable to condensation with a suitably functionalized heteroaromatic ring. Toward this end, the synthesis of either a dialdehyde such as 3, or a phosphonium salt such as 4 was undertaken (Scheme 1).
Scheme 1.
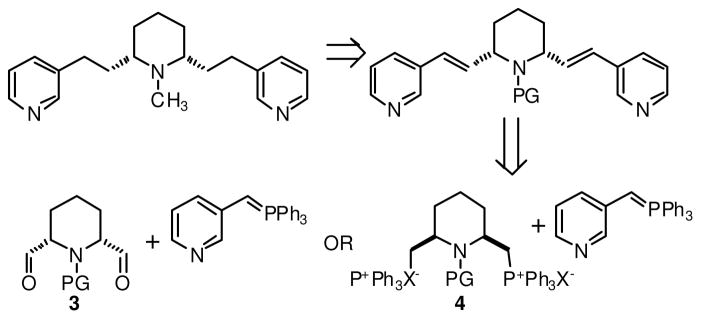
Initial synthetic plan for the preparation of heteroaromatic analogs of lobelane; the 3-pyridyl analog is illustrated as an example.
In the synthesis of 3, the preparation of the key precursor 5 (Scheme 2), was accomplished through an adaptation of the method reported by Chenevert et al.11 Reduction of the ester functions of 5 yielded the diol 6 (Scheme 3), which underwent conversion over 2–3 days at ambient temperature to 7. Exposure of 6 to silica gel during a chromatographic purification attempt led to complete conversion to 7 within 15 min, suggesting a Brønsted acid-mediated 5-exo-tet cyclization involving the ejection of a molecule of H2O and isobutylene. Exposure of 6 to an equivalent of NaOCH3 in MeOH for 60 min led to negligible amounts of 7, which precludes the possibility of a nucleophilic mechanism that involves attack of the hydroxyl function on the carbamate carbonyl in the spontaneous conversion of 6 to 7. Lastly, no t-butyl alcohol was detected in the 1H NMR spectrum of a sample of 6 that had spontaneously converted into 7. Treatment of diol 6 (immediately following its preparation) with tosyl chloride and lutidine led to the crystalline ditosylate 8, which also underwent cyclization in both protic and aprotic solvents at ambient temperature, forming 9 and an equivalent of Ts-OH (Scheme 3). Ditosylate 8 was stable in its solid form, and could be stored in the freezer (−20 °C). Unfortunately, a nucleophilic coupling of 8 with 2- or 3-lithio and 2- or 3-sodio picoline could not be achieved, even in the presence of Cu+ (as CuOTf). Treatment of 8 with Ph3P at ambient temperature did not result in the formation of the required phosphonium salt, and increasing the reaction temperature led to rapid formation of 9. Displacement of the tosylate moieties with halides was also accompanied by cyclization, and Swern or Moffat oxidation of 6 led unfortunately to a mixture of the dl and meso diastereomers of 10. Analogous sets of results were obtained with the Cbz, acetyl and 2,2,2-trichloroethoxycarbonyl (Troc)-protected analogues of compounds 5–10. At this point, it was concluded that the close proximity of the functional groups at C2- and C6- of the piperidine ring in compounds such as 6 and 8 to the N-protective group, and the likely existence of conformations that favor cyclization reactions, would limit the utility of such synthons in the preparation of the required heteroaromatic analogues of 1.
Scheme 2.
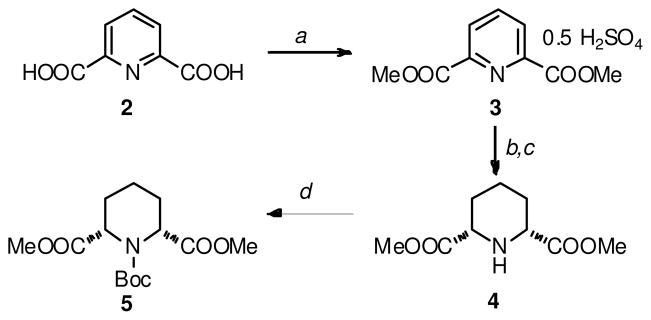
Reagents and Conditions for the synthesis of 5. (a) H2SO4, dimethoxypropane, MeOH, reflux, 4h, 100%; (b). H2, Pd-C, MeOH, 12h, bicarbonate workup, 88% de.;(c). crystallization from EtOAc, 70%, 100% de.; (d). Boc2O (10 equiv.), DMAP (50 mol%), 8h, 98%.
Scheme 3.

Reagents and conditions for the attempted synthesis of 3. (a) NaBH4-LiCl, DME-H2O (6:1), 50 °C, 12 h, 70%.; (b) TsCl, 2,6-lutidine, CH2Cl2, 8 h, 90%.; (c) 3-picoline (2 equiv.), LDA (2 equiv), THF, −78 °C to rt. (d). SOCl2, DMSO, CH2Cl2, −78 °C, 15 min. then Et3N, 15 min. (e) DMSO, DCC, CH2Cl2, 0 °C, 10 min. NOEs considered as proof of relative configuration are depicted as biheaded arrows.
These unsuccessful attempts at utilizing piperidine-based synthons prompted the delay the hydrogenation of the central core unit until the heteroaromatic appendages had been attached. Lee and Freudenberg have synthesized analogues of 1 through the condensation of 2,6-lutidine with various aromatic aldehydes, forming 2,6-distyryl derivatives, followed by N-methylation and catalytic hydrogenation.12 Since in our case, side-chain azaheteroaromatic moieties such as pyridyl and quinolyl, would be susceptible to N-methylation, the methiodide of 2,6-lutidine (11a) was employed as the starting point in the synthetic sequence. Condensation of 11a with pyridine-3-carboxaldehyde proceeded quantitatively to afford 12 as a bright yellow crystalline solid (Scheme 4).13 Attempts at hydrogenation of 12 revealed that the iodide counter-ion completely poisoned both Pd- and Pt-based catalysts. Therefore the corresponding tosylate 13, which underwent nonselective hydrogenation in methanol, was prepared, yielding a complex mixture resulting from reduction of the side-chain pyridine moiety in addition to the desired reduction of the central pyridinium and olefinic moieties. Addition of CHCl3 as an inhibitor caused a decrease in reaction rate, but there was no indication of improved selectivity with either Pd-C or PtO2 catalysts. The use of EtOAc as a solvent with Pd-C as the hydrogenation catalyst, however, led to the formation of 14 with negligible amounts of bi-products. These conditions could be extended to the 4-pyridyl pyridinium analogue 15, but a substantially longer reaction time was required. It was not possible to obtain a ‘general’ set of conditions for the synthesis of all of the target heteroaromatic analogues, as optimization or complete development of reaction conditions was necessary for each type of heterocyclic moiety (Table 1). Chemoselective hydrogenation of the furyl derivative 19 was not possible, since the furyl ring underwent preferential hydrogenation over the olefinic moieties. Also, hydrogenation of the thienyl analogue 20 was not possible due to strong catalyst poisoning by the divalent sulfur. This poisoning was insurmountable, even after addition of excess catalyst (500 wt%), use of various Pd and Pt catalysts, and increases in reaction temperature (oil bath, H2-balloon). Compounds 14, and 21–24 were converted to their hydrochloride or fumarate salts prior to pharmacological characterization. These salts were found to be freely soluble in water.
Scheme 4.

Synthesis of target heterocyclic analogs of 1.
Table 1.
Reagents and conditions for the synthesis of 14, and 21–24
| Het | Conditions-A | Yield | Conditions-B | Yield |
|---|---|---|---|---|
| 3-pyridyl | NaOH (2 equiv), H2O, 5 min. | 100% (13) | Pd-C, EtOAc, 4h, 50 psi | 43% (14)14 |
| 4-pyridyl | NaOH (2 equiv), H2O, 5 min. | 63% (15) | Pd-C, EtOAc, 24h, 50 psi | 80% (21)15 |
| 2-quinolyl | NaOH (2 equiv), 50% EtOH, 8h | 90% (16) | Pt sponge, 16 AcOH (2 equiv), EtOH, 12h, 30 psi | 35% (22)17 |
| 4-quinolyl | DIPEA, 50% EtOH, 40 °C, 12h | 47% (17) | Pd-C, AcOH (2 equiv), EtOH, 72h, 50 psi | 52% (23)18 |
| 3-indolyl | DIPEA, EtOH, reflux, 8h | 80% (18) | Pd-C, NaOH (2 equiv), EtOH, 72h, 50 psi | 18% (24)19 |
| 2-furyl | NaOH (0.5 equiv), 10% EtOH, 4h | 20% (19) | Unsuccessful selective reduction | NA |
| 2-thienyl | Ac2O, TsOH, DMAP, rt, 1h | 16% (20) | Unsurmountable catalyst poisoning | NA |
The current synthetic studies have been successful in affording 5 heteroaromatic analogues of lobelane, but have revealed a lack of a general practical synthetic methodology for the preparation of heteromeric lobelane analogues of different classes, especially with respect to the terminal hydrogenation procedure. Particularly lacking is a suitable catalyst that is resistant to the poisoning effects of analogues containing thienyl rings.
The ability of lobelane-derived analogues to bind to the [3H]DTBZ-binding site on VMAT2 may not be an accurate measure of their ability to inhibit VMAT2 function.8 In light of these findings, the above heteroaromatic analogues were examined for their ability to inhibit both the uptake of [3H]DA into synaptic vesicles and their affinity for the [3H]DTBZ-binding site on VMAT2 (Table 2). The 3-pyridinyl analogue 14 was about 30-fold less potent than lobelane in binding to the [3H]DTBZ binding site, while the 4-pyridinyl analogue 21 was marginally lower in binding affinity when compared to lobelane. The quinolyl analogues, 22 and 23, bound to the [3H]DTBZ binding site almost as avidly as lobelane, with the 2-quinolyl analogue, 22, possessing submicromolar affinity. The 3-indolyl analogue, 24, exhibited greatly diminished affinity for the [3H]DTBZ binding site. From these data, it would seem that the putative binding surface of these analogues in the [3H]DTBZ binding site is topologically well-defined with rigid structural requirements for ligand interaction. This bodes well for the future design of ligands that are selective for this binding site. The substantial inhibitory activity of the quinolyl analogs, 22 and 23, is interesting since we have previously shown that the corresponding naphthyl analogues also bind avidly to the [3H]DTBZ-binding site on VMAT2.6 Thus, this indicates that the quinolyl analogs interact with the binding site in a similar manner to the naphthyl analogs and the presence of the aza atom does not compromise the interaction with the binding site. The diminished activity of the 3-indolyl analogue 24 would may well be due to the H-bonding donor properties of the indolyl rings, which may interfere with binding.
Table 2.
Pharmacological activity at VMAT2 of analogues 14, 21–24 in comparison with 1
| Analogue | [3H]-DTBZ; Ki ± SEM (μM) | VMAT2 [3H]DA uptake Ki ± SEM (μM) |
|---|---|---|
| 1•0.5 H2SO4 | 0.97 ± 0.19 | 0.045 ± 0.002 |
| 14 | 33.3 ± 16.3 | 3.72 ± 0.32 |
| 21 | 5.87 ± 1.72 | 5.23 ± 3.03 |
| 22•HCl | 0.96 ± 0.37 | 0.067 ± 0.015 |
| 23•HCl | 2.64 ± 1.41 | 0.051 ± 0.005 |
| 24•C4H4O4 | 26.01 ± 2.88 | 2.13 ± 0.28 |
The pyridinyl analogues 14 and 21 were 100-fold lower in potency when compared to lobelane in inhibiting [3H]DA uptake into synaptic vesicles (Table 2), while the 2- and 4-quinolyl analogs 22 and 23, respectively, were equipotent with lobelane. The 3-indolyl analogue 24 also had 100-fold lower inhibitory potency when compared to lobelane in this assay. Thus, the potency of these analogues for inhibition of [3H]DA uptake into synaptic vesicles appears to depend largely on the overall stereo-character of the heteroaromatic moiety, rather than the specific location of charge or H-bond donor or acceptor elements.
There was no obvious relationship between the ability of analogues 14 and 21–24 to bind to the [3H]DTBZ binding site on VMAT2 and their ability to inhibit VMAT2 function. The lack of relationship is consistent with our earlier observations in the pyrrolidine series analogues of lobelane, and reinforces our earlier inference that affinity of lobelane-derived analogues for the [3H]DTBZ binding site does not necessarily predict their ability to inhibit VMAT2.
The isomeric quinolines 22 and 23 exhibit pharmacological profiles similar to those of lobelane with regards to their interaction with the binding site and ability to inhibit VMAT2. However, 22 and 23 have improved water solubility characteristics compared to lobelane. In particular, the trihydrochloride salt of 23 (termed ‘Quinlobelane’) has an aqueous solubility of ≥ 20 mg/mL in water when compared to 1, whose aqueous solubility is only 0.3 mg/mL. Thus, one of the primary objectives of identifying analogues of lobelane as VMAT2 inhibitors with improved water solubility characteristics has been met, and quinlobelane has been advanced to behavioral testing.
Acknowledgments
The authors would like to thank the National Institutes of Health, NIDA grant DA013519 for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Foster RH, Moench K, Lucille J, Clark HC. J Pharmacol. 1946;87:73. [PubMed] [Google Scholar]
- 2.Harrod SB, Dwoskin LP, Crooks PA, Klebaur JE, Bardo MT. J Pharmacol Exp Ther. 2001;298:172. [PubMed] [Google Scholar]
- 3.Dwoskin LP, Crooks PA. Biochem Pharmacol. 2002;63:89. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- 4.http://clinicaltrials.gov/ct2/show/NCT00439504?term=lobeline&rank=7
- 5.Flammia D, Dukat M, Damaj MI, Martin B, Glennon RA. J Med Chem. 1999;42:3726. doi: 10.1021/jm990286m. [DOI] [PubMed] [Google Scholar]
- 6.Miller DK, Crooks PA, Zheng G, Grinevich VP, Norrholm SD, Dwoskin LP. J Phar Exp Ther. 2004;310:1035. doi: 10.1124/jpet.104.068098. [DOI] [PubMed] [Google Scholar]
- 7.Zheng G, Dwoskin LP, Deaciuc AG, Norrholm SD, Crooks PA. J Med Chem. 2005;48:5551. doi: 10.1021/jm0501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng G, Dwoskin LP, Deaciuc AG, Crooks PA. Bioorg Med Chem Lett. 2008;18:6509. doi: 10.1016/j.bmcl.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nickell JR, Krishnamurthy S, Norrholm S, Deaciuc G, Zheng G, Crooks PA, Dwoskin LP. J Phar Exp Ther. 2009;332:612. doi: 10.1124/jpet.109.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vartak AP, Nickell JR, Chagkutip J, Dwoskin LP, Crooks PA. J Med Chem. 2009;52:7878. doi: 10.1021/jm900770h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chenevert R, Morin M. Tetrahedron: Asymmetry. 1996;7:2161. [Google Scholar]
- 12.Lee J, Freudenberg W. J Org Chem. 1944;9:537. [Google Scholar]
- 13.Fichera M, Fortuna CG, Giuseppe I, Musumarra G. Eur J Org Chem. 2002;1:145. [Google Scholar]
- 14.Characterization data for 14: Clear oil; Rf = 0.4 (CH2Cl2/EtOAc/Et3N, 1:1:0.2) 1H NMR (300 MHz, CDCl3) δ ppm 8.28–8.17 (m, 4H), 7.80–7.51 (m, 2H), 7.35 (t, J = 6.2 Hz, 2H), 2.86–2.50 (m, 6H), 2.17 (s, 3H), 1.62–1.41 (m, 10H); 13C NMR (75 MHz, CDCl3) δ ppm 149.2, 149.0, 139.5, 138.2, 128.3, 66.3, 42.8, 34.0, 32.8, 32.2, 22.9; EI-MS m/z = 309.1 (M)+, 203.1 (α-fragmentation).
- 15.Characterization data for 21: Clear oil; Rf = 0.7 (EtOAc/EtOH/Et3N, 10:1:0.5); 1H NMR (300 MHz, CDCl3) δ ppm 8.41 (d, J = 9.2 Hz, 4H), 7.49 (d, J = 9.2 Hz, 4H), 2.63 (t, J = 5.2 Hz, 4H), 2.20 (s, 3H), 2.34–2.07 (m, 2H), 1.71–1.39 (m, 10H); 13C NMR (75 MHz, CDCl3) δ ppm 154.1, 148.3, 123.0, 68.1, 41.5, 33.8, 33.5, 31.4, 22.0; EI-MS m/z 309.1 (M)+, 203.1 (α-fragmentation).
- 16.‘Pt sponge’ was generated by shaking PtO2 in EtOH under an atmosphere of H2 (30 psi) for 15 min. It was observed that the use of Pt-sponge generated in this manner, instead of co-activation with the substrate, resulted in a faster and cleaner reaction.
- 17.Characterization data for 22•HCl: crystalline solid; mp = 102–104 °C; Rf (free base) = 0.3 (toluene/EtOH/NH4OH, 10:1:1); 1H NMR (300 MHz, D2O) δ ppm 8.41 (d, J = 2.2 Hz, 2H), 7.98 (d, J = 5.3 Hz, 2H), 7.82–7.61 (m, 6H), 7.03 (d, J = 3.6 Hz, 2H), 2.81–2.42 (m, 4H), 2.22 (s, 3H), 2.31–2.17 (m, 2H), 1.65–1.40 (m, 10H); 13C NMR (75 MHz, D2O) δ ppm 161.8, 150.2, 137.0, 131.4, 130.1, 128.5, 128.4, 125.7, 123.9, 68.8, 42.2, 34.0, 33.1, 32.6, 21.2; EI-MS m/z 409.3 (M)+, 253.2 (α-fragmentation).
- 18.Characterization data for 23•HCl: crystalline solid; mp = 98–100 °C; Rf (free base) = 0.25 (toluene/EtOH/NH4OH, 10:1:1); 1H NMR (300 MHz, D2O) δ ppm 8.64 (d, 2H, J = 8.0 Hz), 8.22–8.19 (m, 2H), 8.02–7.91(m, 2H), 7.74–7.53 (m, 4H), 7.28 (s, br, 2H), 2.52–2.48 (m, 4H), 2.23 (s, 3H), 2.21–2.08 (m, 2H), 1.65–1.30 (m, 10H); 13C NMR (75 MHz, D2O) δ ppm 152.0, 150.8, 144.6, 131.1, 130.9, 128.6, 128.2, 124.0, 120.3, 70.1, 38.2, 33.7, 32.0, 29.3, 22.4; EI-MS m/z 409.3 (M)+, 253.2 (α-fragmentation).
- 19.Characterization data for 24•C4H4O4 white solid; mp = 80–82 °C, Rf (free base) = 0.5 (EtOAc/Et3N, 9:1); 1H NMR (300 MHz, D2O) δ ppm 7.64–7.04 (m, 8H), 7.20 (s, 2H), 6.63 (s, 2H, fumaric acid), 2.38–2.23 (m, 4H), 2.24 (s, 3H), 2.30–2.18 (m, 2H), 1.83–1.22 (m, 10H); 13C NMR (75 MHz, D2O) δ ppm 165.8 (COOH, fumaric acid), 138.1, 128.2, 127.7, 124.2, 124.0, 120.0, 118.4, 110.3, 109.5, 67.1, 41.1, 35.5, 32.1, 26.0, 21.8; EI-MS m/z 385.3 (M)+, 241.2 (α-fragmentation), 144.1, 116.1.