

Abstract
Ring size reduction of the central piperidine ring of lobelane yielded pyrrolidine analogs that showed marked inconsistencies in their ability to bind to the dihydrotetrabenazine (DTBZ) binding site on the vesicular monoamine transporter-2 (VMAT2) and their ability to inhibit VMAT2 function. The structure activity relationships indicate that structural modification within the pyrrolidine series resulted in analogs that interact with two different sites, i.e., the DTBZ binding site and an alternative site on VMAT2 to inhibit transporter function.
Keywords: VMAT2; 2,5-disubstituted pyrrolidines; dihydrotetrabenazine; methamphetamine abuse
Introduction
The vesicular monoamine transporter-2 (VMAT2) transports monoamines from the neuronal cytosol into the synaptic vesicle, a process that prevents metabolism by monoamine oxidase (MAO) and stores monoamines, such as dopamine (DA), for eventual release by exocytosis.1 Psychostimulants, such as methamphetamine (METH), inhibit the function of VMAT2, thereby increasing cytosolic levels of DA.2 METH also inhibits MAO and reverses the DA transporter (DAT), which together with the increased cytosolic levels of DA results in a substantial efflux of neurotransmitter into the synaptic cleft. Thus, the interaction of METH with VMAT2 is the first step in a series of events that leads ultimately to its pharmacological effects, including the psychological high which results from METH administration. The design and development of VMAT2- selective inhibitors is therefore a worthwhile pursuit toward the broader goal of developing efficacious therapeutics as a means to clinically intervene in the abuse of psychostimulants such as METH.3
The piperidine alkaloid and principal constituent of Lobelia inflata, lobeline (1), was found to inhibit the uptake of DA into synaptic vesicles.4 Since lobeline and its analogs competitively inhibit the binding of [3H]dihydrotetrabenazine (DTBZ), a known VMAT2 inhibitor, this binding site was identified as a target for the development of potential therapeutic agents for the treatment METH abuse. Thus, the DTBZ binding site on VMAT2 was utilized as a probe in preliminary screening of lobeline analogs for potency and selectivity for VMAT2 interaction. Large increases in potency were achieved through complete deoxygenation and reduction of the lobeline molecule, which resulted in the identification of a lead compound, cis-N-methyl-2,6-diphenethylpiperidine (2, lobelane).5,6,7 2, which is also a minor alkaloid of Lobelia inflata, was found to be more selective than 1 as a VMAT2 ligand, possessing little affinity for α4β2* nicotinic acetylcholine receptors (nAChRs), to which 1 binds with high potency.8 The cis-2,6-diethylarylpiperidine structural motif in 2 was therefore regarded as an initial pharmacophore on which to develop subsequent generations of VMAT2 inhibitors. Initial structure-activity studies on 2 were focused on the incorporation of substituents into the aromatic rings, variation in the number of methylenes between the central piperidine ring and the phenyl rings, and the positioning of the two phenethyl moieties at different sites around the central piperidine ring. Modest increases in affinity for the DTBZ binding site on VMAT2 were obtained through substitution of anisyl and naphthyl moieties for phenyl moieties in the phenethyl side chains,7 while departure from the 2,6-diphenethyl substitution pattern around the piperidine ring was found to be generally deleterious.9 A minimum of two methylene units in the alkane linker moiety was found to be essential for retention of binding affinity exhibited by 2 at the DTBZ binding site on VMAT2. Lastly, the trans-configured enantiomeric analogs of 2 (trans-2s, 4 and 5) exhibited diminished affinity for the DTBZ binding site, as did N-demethylated analogs, including nor-2 (3).
Design rationale
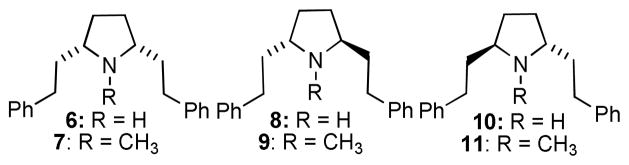
Available SAR suggests that the intramolecular distance between the two aromatic rings in 2, and the relative orientation of the phenethyl moieties, are determinants for affinity at the DTBZ binding site on VMAT2. To improve affinity, we investigated the effect of restricting the flexibility of the central piperidine ring to afford more conformationally-defined analogs. In this respect, the current study investigates the effect of replacing the central piperidine ring in 2 with a more conformationally restricted pyrrolidine ring. A series of six pyrrolidine analogs 6–11 were chosen as the initial target molecules. Compound 7 is a pyrrolidino analog of 2, and the trans-configured analogs 9 and 11 represent the pyrrolidino-analogs of trans-2s 4 and 5. Analogs 6, 8 and 10 were also included as analogs of nor-2 (3), to determine the importance of the N-methyl group in the SAR of these pyrrolidine analogs.
Results
Scheme 1 shows the synthesis of the initial 2,5-diphenethylpyrrolidine analogs. The absolute and relative stereochemistries were achieved utilizing Katritzky’s elegant benzotriazole-oxazolidine approach.10 Briefly, L- and D-phenylglycinol were both obtained through NaBH4 reduction of the corresponding phenylglycine methyl esters. A three-component condensation of L-phenylglycinol with benzotriazole and succinaldehyde afforded the key synthon 12 in useful quantities. Addition of a sixfold excess of phenethyl magnesium bromide to synthon 12 at room temperature led to a slightly exothermic reaction, affording a 2:1 mixture of the cis- and trans-configured diastereomers of 13 within a few hours; these diastereomers were separated by silica gel chromatography. The 2R,5S- and 2R,5R-diastereomers, 14 and 15, were hydrogenolyzed separately by catalytic-transfer hydrogenation with palladium hydroxide-over-carbon, employing ammonium formate as the hydrogen source in refluxing methanol. These conditions afforded quantitative conversion to the respective products, 6 and 8, within 20 minutes, as opposed to 12 h utilizing alternate conditions (i.e. H2/Pd-C).5 The resulting meso- and (+)-trans-2R,5R pyrrolidines 6 and 8 were then each N-methylated with paraformaldehyde and sodium triacetoxyborohydride/acetic acid in acetonitrile followed by treatment with HCl in diethyl ether to afford the meso isomer 7 and the (+)-trans-2R,5R isomer 9, respectively. Utilization of D-phenylglycinol in place of L-phenylglycinol in the above synthetic scheme provided synthon 16, which could be converted into 18 and 19. Hydrogenolysis of 19 followed by hydrochloride salt formation afforded 10 ((−)-trans-2S,5S), which in turn was N-methylated to 11 ((−)-trans-2S,5S). Note, compound 18 also forms the cis-meso isomer 6 upon hydrogenolysis, making further elaboration of this compound redundant.
Scheme 1. Synthesis of 2,5-diphenethylpyrrolidines.

Reagents and conditions. a. Ph-(CH2)n-MgBr (6 equiv.), THF, 0 °C to rt, 8 h; b. Silica gel chromatographic separation. c. HCI, ether d. NH4COO, Pd(OH)2/C, MeOH, reflux, 20 min. then conc. HCI; e. paraformaldehyde, Na(OAc)3BH, CH3CN, rt, 4h;
The pyrrolidine analogs in Scheme 1 were evaluated for affinity at the DTBZ binding site on VMAT2 (Table 1). When the 2,5-diphenethyl substituents are in the meso cis-configuration, overall binding affinity clearly decreases upon replacement of the piperidine ring with the pyrrolidine ring, since 7 is about 10-times less potent than 2 (2). When the phenethyl substituents are in the 2,5-trans configuration (analogs 9 and 11) the binding affinity does not appreciably change with azacyclic ring-size reduction when compared to the piperidine (2) counterparts 4 and 5, respectively. Interestingly, the (S,S)-trans nor-N-methyl analog 10 is about 10-fold more potent than its N-methyl derivative 11, whereas replacement of the N-methyl group in both the cis- and trans-analogs with an N-phenethanolyl substituent afforded no significant change in binding affinity, with analogs 14, 15, 18 and 19 possessing affinities comparable to the nor- and N-methyl pyrrolidine analogs.
Table 1.
Interaction of 2 and pyrrolidine analogs with VMAT2.
| Analog | [3H]DTBZ; Ki ± SEM (μM) | VMAT2 [3H]-DA Uptake Ki ± SEM (μM) | Ratio [3H]DTBZ binding/[3H]-DA uptake |
|---|---|---|---|
| Tetrabenazine | 0.013 ± 0.001 | 0.054 ± 0.015 | 0.24 |
| 1 | 2.76 ± 0.64 | 0.470 ± 0.045 | 5.87 |
| 2 | 0.97 ± 0.19 | 0.033 ± 0.041 | 29.4 |
| 3 | 2.31 ± 0.21 | 0.023 ± 0.004 | 100 |
| 4 | 5.32 ± 0.45 | 4.05 ± 0.612 | 1.31 |
| 5 | 6.46 ± 1.70 | 2.11 ± 0.50 | 3.06 |
| 6 | 3.06 ± 0.17 | 0.029 ± 0.007 | 106 |
| 7 | 8.80 ± 2.30 | 0.270 ± 0.035 | 32.6 |
| 8 | 3.40 ± 0.030 | 0.054 ± 0.017 | 63.0 |
| 9 | 3.97 ± 0.64 | 0.120 ± 0.015 | 33.1 |
| 10 | 1.33 ± 0.19 | 0.035 ± 0.006 | 38.0 |
| 11 | 14.45 ± 2.13 | 0.170 ± 0.030 | 85.0 |
| 14 | 4.34 ± 0.71 | 1.290 ± 0.240 | 3.36 |
| 15 | 5.05 ± 1.05 | 1.940 ± 0.470 | 2.60 |
| 18 | 2.75 ± 0.63 | 0.140 ± 0.049 | 19.6 |
| 19 | 5.61 ± 0.91 | 0.270 ± 0.053 | 20.7 |
| 22 | 1.64 ± 0.12 | 0.0093 ± 0.00075 | 176 |
| 23 | 2.46 ± 0.42 | 0.019 ± 0.00091 | 130 |
| 25 | 0.27 ± 0.0067 | 0.014 ± 0.0011 | 19.3 |
| 26 | 2.78 ± 0.63 | 0.12 ± 0.011 | 23.2 |
The ultimate goal of this SAR study was to identify chemical entities that would inhibit VMAT2 function. It is known that there are multiple binding sites on VMAT2 that may influence transporter function, i.e., a ketanserine binding site near the N-terminus, a TBZ binding site near the C-terminus, and two reserpine binding sites (a high affinity site and a low affinity site).11 In this respect, the affinity of a ligand toward the DTBZ binding site on VMAT2 may not necessarily be a sufficient indicator of the activity of the ligand in altering function of VMAT2. In light of these data and the possibility that other, as yet unidentified, binding sites on VMAT2 may exist, the ability of the current analogs to inhibit the uptake of [3H]-DA into isolated synaptic vesicles was also evaluated (Table 1).
While the affinities of the pyrrolidine ligands for the DTBZ binding site did not vary much with respect to structural change (Ki values were in the range of 0.3–14 μM), their ability to inhibit the uptake of [3H]-DA into synaptic vesicles varied greatly with subtle structural changes, with Ki values ranging from 9 nM to 1.94 μM. Ring-size reduction of the piperidine moiety in 2 afforded a 10-fold decrease in the potency to inhibit [3H]-DA uptake into vesicles when compared to 2 (meso cis- analogs 7 and 2) However, a similar ring-size reduction in nor-2 had no effect on inhibition of [3H]-DA uptake, since 3 and 6 were equipotent. The most striking difference in [3H]-DA uptake inhibition was observed between the trans-configured piperidine- and pyrrolidine-analogs. Thus, (S,S)-trans-2 (4) (Ki = 4.05 μM) and (R,R)-trans-2 (Ki = 2.11 μM) (5) are 33- and 12-fold, respectively, less potent than their corresponding pyrrolidine analogs 9 (Ki = 120 nM) and 11 (Ki = 170 nM), respectively. Within the pyrrolidine series, the presence of the N-methyl group is clearly deleterious toward potency of inhibition of [3H]-DA uptake, since the N-methyl pyrrolidines 7, 9 and 11 are all less potent than their nor-N-methyl counterparts 6, 8 and 10, with the effect of N-methyl substitution being most prominent in the cis-configured analogs 6 and 7. Also, within the N-phenethanolyl pyrrolidine series of analogs, the configuration of the pendant phenethanolyl stereocenter appears to be a factor contributing that contributes to the potency for [3H]-DA uptake inhibition, since the R-configured analogs 18 and 19 are about 10-fold more potent than the S-configured analogs 14 and 15.
A marked disparity exists in the abilities of the above series of analogs to bind to the DTBZ binding site on VMAT2 and to inhibit [3H]-DA uptake into vesicles, as can be seen in the ratios of the Ki values, as well as in the rank order of potency of the compounds in the two assays; this is evident for both the piperidine and pyrrolidine analogs (Table 1, right column). Compound 3 and its pyrrolidine analog 6 are both about 100-fold more potent in inhibiting [3H]-DA uptake into isolated synaptic vesicles than in binding to the DTBZ-binding site, while their N-methyl counterparts 2 and 7 are both about 30-times more potent in this regard. While the N-methyl trans-configured pyrrolidines 9 and 11 are both significantly more potent in inhibiting [3H]-DA uptake than in binding to the DTBZ binding site, their piperidine counterparts (i.e. the trans-2 enantiomers 4 and 5) are relatively weak [3H]-DA uptake inhibitors (Ki > 1 μM) and show relatively low potency in the DTBZ binding assay (Kis 5.32 μM and 6.46 μM, respectively). In the N-phenethanolyl series, the 2′R-analogs 18 and 19 exhibit significant differences in their ability to bind to the DTBZ binding site on VMAT2 (Ki 2.75 μM and 5.61 μM, respectively), and their ability to inhibit [3H]-DA uptake into vesicles (Ki 140 nM and 270 nM, respectively). Pyrrolidines 18 and 19 are more potent as [3H]-DA uptake inhibitors, compared to their corresponding 2′S-configured analogs 14 [Ki (binding) = 4.34 μM, Ki (uptake) = 1.29 μM)] and 15 [Ki (binding) = 5.05 μM, Ki (uptake) = 1.94 μM].
cis-2,5-Diphenethylpyrrolidine (6) was identified as the most potent pyrrolidine analog for the inhibition of VMAT2 function, and structural optimization of this lead molecule was further investigated. Thus, compound 22, in which the methylene linker moieties have been truncated to a single methylene unit, as well as compound 25, in which the methylene units have been increase from two to three were synthesized. The synthesis of pyrrolidine 22 was approached utilizing the method illustrated in Scheme 2. Treatment of 12 with benzyl magnesium chloride led to an inseparable ~3:1 mixture of diastereomers with the 2,5-cis-configured diastereomer 21 being the predominant isomer. Since diastereomer 21 was required for further elaboration, conditions were identified to increase the proportion of this diastereomer in the reaction product. During the synthesis of the 2,5-diphenethyl pyrrolidines, it was noted that the presence of MgBr2 in the reaction mixture (originating from the activation of Mg turnings by dibromoethane) resulted in diastereomeric ratios that favored the 2,5-cis diastereomers (14 and 18) over the 2,5-trans diastereomers (15 and 19). A solution of 12 was treated with progressively increasing equivalents of MgBr2 (generated in-situ from Mg and dibromoethane) prior to the addition of benzylmagnesium chloride. When 10 equiv. of MgBr2 had been utilized, the product mixture consisted solely of the free-base of 21 (the trans-configured diastereomer could not be detected by 1H NMR or by gas chromatography), however, this favorable exclusive formation of the desired diastereomer was achieved at the expense of a significantly lower yield (42%), as compared to the 80% yield obtained in the reaction without MgBr2. Hydrogenolysis of 21 led to 22, which was N-methylated and treated with HCl in diethylether to afford 23. The diastereomerically pure propyl homolog 25 and its N-methylated derivative 26 were prepared in a similar manner utilizing the aforementioned procedure employing phenpropyl magnesium bromide.
Scheme 2. Synthesis of second-generation pyrrolidine analogs.
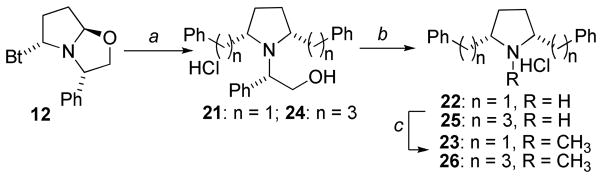
Reagents and conditions. a. R-MgX, MgBr2 (10 equiv), THF, rt. b. Pd(OH)2, NH4COO, CH3OH, c. Na(OAc)3BH, CH3CN, paraformaldehyde
The methylene truncated analogs 22 and 23 bound to the DTBZ-binding site of VMAT2 with affinities comparable to that of the parent pyrrolidines 6 and 7, whereas the methylene elongated nor-analog 25, bound with high affinity (Ki = 270 nM), making it one of the most potent 2-derived ligands known for this binding site. N-Methylation of 25 afforded 26, which was 10-times less potent than 25. Pyrrolidine 22 was the most potent [3H]-DA uptake inhibitor in this series of analogs, with a Ki of 9.3 nM. Its N-methylated derivative 23 was also a good uptake inhibitor (Ki = 19 nM). Pyrrolidine 25 was also a potent uptake inhibitor (Ki = 14 nM); its N-methylated analog 26 was about 10-times less potent.
As seen in Table 1, the disparity between [3H]-DTBZ binding and [3H]-DA uptake inhibition is more prominent for the methylene truncated pyrrolidines 22 and 23, which inhibit [3H]-DA uptake 176- and 129-fold more potently than they inhibit [3H]-DTBZ binding to VMAT2. The methylene elongated pyrrolidines 25 and 26 inhibit [3H]-DA uptake about 20-times more potently than they inhibit DTBZ binding, irrespective of their relative potencies in the individual assays.
Discussion
In summary, ring size reduction of the central piperidine ring of 2 yielded pyrrolidine analogs that showed marked disparities in their ability to bind to the [3H]-DTBZ binding site on VMAT2 and their ability to inhibit VMAT2 function. Unlike tetrabenazine, the compounds in this series of pyrrolidine analogs of 2 were more potent inhibiting VMAT2 function than they were inhibiting the binding of [3H]-DTBZ to VMAT2. The SAR indicate that structural modification within the pyrrolidine series results in analogs that likely interact with two different sites on VMAT2, i.e., the DTBZ binding site, and perhaps an alternative site on VMAT2, to inhibit transporter function. While the affinity of the pyrrolidine analogs toward the DTBZ binding site is not significantly sensitive to structural changes, the potency for [3H]-DA uptake inhibition is sensitive to subtle structural changes, such as alteration in methylene linker length, N-substitution, and stereochemistry in the case of the N-phenethanolyl analogs. Such sensitivity suggests that the putative binding site on VMAT2 whose interaction with the pyrrolidine analogs leads to functional inhibition, is topographically distinct from the DTBZ ligand binding site. It should be noted that compound 22, which has the greatest potency to inhibit [3H]-DA uptake into synaptic vesicles, can be considered a structural dimer of methamphetamine (Figure 3), which is itself known to inhibit VMAT2 function.12 In this respect, 2 (2) could also be considered to be a “dimeric” methamphetamine analog; however, this compound does not itself support self-administration, but rather decreases methamphetamine self-administration in animal models.13
Figure 3.
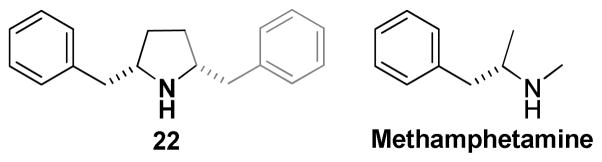
Pyrrolidine 22 may be regarded structurally a ‘dimer’ of methamphetamine.
The results of this study are expected to have a significant impact on the future design of lobelane-derived VMAT2 inhibitors. Due caution needs to be exercised in utilizing the SAR from [3H]-DTBZ-binding assays for the design of lobelane-derived VMAT2 inhibitors, since such data may not be functionally relevant. The pyrrolidine ring has been identified as a valid replacement for the piperidine ring in 2, and nor-analogs (i.e. N-demethylated homologs of 2) appear to be optimal for both binding to the DTBZ binding site on VMAT2 and inhibition of VMAT2 function. A potent VMAT2 ligand, 25, and a potent water-soluble inhibitor of VMAT2 function (22) were also identified during this study, which appear to be suitable for further development and advancement into behavioral studies.
Experimental section
(2′S)-2-[(2R,5S)-2,5-Di-(2-phenethyl)-tetrahydro-1H-1-pyrrolyl]-2-phenylethan-1-ol (14) and (2′S)-2-[(2R,5R)-2,5-di-(2-phenethyl)-tetrahydro-1H-1-pyrrolyl]-2-phenylethan-1-ol (15)
To 306 mg (1 mmol) of 12 suspended in 20 mL of THF was added a solution of phenethylmagnesium bromide in THF (6 equiv.) over a period of 10 minutes at 0 °C. The solution was warmed to ambient temperature over 8 h, 10 mL of 2N NaOH were added, and the biphasic mixture was extracted with diethyl ether (3 × 100 mL). The etherial layers were combined, diluted with 50 mL of CH2Cl2, dried with anhydrous Na2SO4 and evaporated under reduced pressure to afford a yellow oil. The oil was dissolved in 20 mL of dry diethyl ether through which HCl gas had been bubbled with stirring for 10 minutes. The supernatant was decanted and the resulting gummy sediment was triturated with diethyl ether (5 × 50 mL). Aqueous NaOH solution (2N, 20 mL) was then added, and the suspension extracted with diethyl ether (2 × 100 mL). The organic layers were washed with brine, dried, and the oily residue was applied to a silica gel column (10 g). The column was eluted with 15:1 (hexanes/diethyl ether) to afford pure fractions of compounds 14 (240 mg, 60 %) and 15 (60 mg, 15%) as colorless oils. 14 Rf = 0.4 (8:1, hexanes/EtOAc), 1H NMR (300 MHz, CDCl3) δ ppm 7.45–7.03 (m, 15H, Ar), 4.02–3.83 (m, 2H), 3.78–3.60 (m, 1H), 3.35–3.20 (br, s, 1H), 3.15–2.95 (m, 2H), 2.80–2.45 (m, 4H), 1.95–1.20 (m, 10H). 13C NMR (75 MHz, CDCl3) δ ppm 143.7, 137.2, 129.1, 128.5, 128.5, 127.9, 125.9, 64.8, 63.6, 61.9, 57.6, 38.1, 36.4, 29.8, 29.2, 28.9. [α]D20 −13.2° (c 1.0, CH2Cl2); Anal. C28 H33NO (C,H,N) 15 Rf = 0.2 (8:1, hexanes/EtOAc) 1H NMR (300 MHz, CDCl3) δ ppm 7.50–6.98 (m, 15H), 4.30–3.92 (m, 2H), 3.84–3.79 (m, 1H), 3.32–3.16 (br, s, 1H), 3.17–2.92 (m, 2H), 2.73–2.56 (m, 4H), 2.03–1.20 (m, 10H) 13C NMR (75 MHz, CDCl3) δ ppm 146.3, 138.2, 125.4, 127.9, 128.8, 126.1, 125.4, 122.1, 66.2, 61.6, 57.6, 38.1, 29.2, 28.9. [α]D20 −2.0° (c 1.0, CH2Cl2). Anal. C28 H33NO (C,H,N).
(2′R)-2-(2S,5R)-2,5-Di-(2-phenethyl)-tetrahydro-1H-1-pyrrolyl-2-phenylethan-1-ol (18)
Compound 18 was synthesized from 16, and was the major diastereomer (55% yield). 1H and 13C NMR spectra were identical to 3; [α]D23 +12.1° (c 1.0, CH2Cl2). Anal. C28 H33NO (C,H,N).
(2′R)-2-(2S,5S)-2,5-Di(2-phenethyl)-tetrahydro-1H-1-pyrrolyl-2-phenylethan-1-ol (19)
19 was synthesized from 16, as the minor diastereomer (20% yield). 1H and 13C NMR spectra were identical to 4; [α]D20 +2.2° (c 1.0, CH2Cl2). Anal. C28 H33NO (C,H,N).
cis-2,5-Di-(2-phenethyl)-pyrrolidine hydrochloride (6)
To a solution of 14 (100 mg, 0.25 mmol) in MeOH (5 mL) was added Pd(OH)2-C (10 mg, 10 wt%) and ammonium formate (100 mg, 100 wt%). The mixture was refluxed for 20 min before being diluted with CH2Cl2 (30 mL) and filtered through celite. The filtrate was treated with 1 mL conc. HCl and evaporated to a crusty white solid under reduced pressure. The solid was triturated with diethyl ether, and precipitated from a mixture of CH2Cl2 and hexanes by addition of hexanes, to yield 6 (60 mg, 77%) as a white solid, mp = 155 °C, 1H NMR (300 MHz, CDCl3) δ ppm 10.22 (br, s, 1H), 9.03 (br, s, 1H), 7.32–7.08 (m, 10H), 3.62–3.43 (apparent s, br, 2H), 2.63–2.47 (m, 4H), 1.83–1.62 (m, 8H). 13C NMR (75 MHz, CDCl3) δ ppm 141.7, 128.6, 128.6, 126.2, 60.4, 41.1, 35.8, 29.0. Anal. C20H26ClN (C,H,N).
(2R,5R)-Di-(2-phenethyl)-pyrrolidine hydrochloride (8)
Compound 15 (100 mg, 0.25 mmol) was prepared as described for the preparation of 6, to yield 38 mg (48%) of 8 as a white solid, mp = 95 °C (deliquescent), 1H NMR (300 MHz, CDCl3) δ ppm 9.80 (br, s, 1H), 9.72 (br, s, 1H), 7.40–7.00 (m, 10H), 3.52–3.30 (s, br, 2H), 2.40–1.99 (m, 4H), 1.70–1.53 (m, 8H). 13C NMR (75 MHz, CDCl3) δ ppm 140.0, 129.6, 128.5, 126.1, 58.3, 43.8, 33.0, 29.4. [α]D20 = +7.5° (c = 1.0, CH2Cl2); Anal. C20H26ClN (C,H,N).
(2S,5S)-di-(2-phenethyl)-pyrrolidine hydrochloride (10)
Compound 10 was synthesized from 19 in a manner similar to that described for 8 (73% yield). Spectral characteristics were identical to those of 8, [α]D20 = −8.1° (c = 1.0, CH2Cl2); Anal. C20H26ClN (C,H,N).
1-Methyl-cis-2,5-di-(2-phenethyl)-pyrrolidine hydrochloride (7)
To a solution of 6 (50 mg, 0.14 mmol) in CH3CN (5 mL) and acetic acid (1 mL) was added NaBH4 (50 mg, excess) and paraformaldehyde (50 mg, excess). The initial suspension afforded a clear solution upon stirring for 12 h. The solution was then treated with conc. HCl (0.5 mL), stirred for 2 h, poured into 50% aqueous NaOH (10 mL) and extracted with CH2Cl2 (6 × 20 mL). The CH2Cl2 extracts were combined, dried (anhydrous MgSO4), filtered and the solvent evaporated to yield an oil that was treated with conc. HCl (1 mL) and azeotroped twice with toluene. Trituration of the residue with diethyl ether afforded a white solid 7 (43 mg, 83%), mp = 94 °C (deliquescent); 1H NMR (300 MHz, CDCl3) δ ppm 9.48 (br, s, 1H), 7.25–7.02 (m, 10H), 3.73–3.48 (m, 2H), 2.63–2.47 (m, 4H), 2.34 (s, 3H), 1.74–1.38 (m, 8H); 13C NMR (75 MHz, CDCl3) δ ppm 144.8, 127.2, 127.1, 126.5, 65.2, 48.3, 38.3, 32.2, 30.1. Anal. C21H28ClN•0.5 H2O (C,H,N).
1-Methyl-(2R,5R)-di-(2-phenethyl)-pyrrolidine hydrochloride (9)
Compound 8 was subjected to identical conditions to those described for the preparation of 7 to yield 30 mg (58%) of 9 as a white solid, mp = 113 °C; 1H NMR (300 MHz, CDCl3) δ ppm 9.90 (br, s, 1H), 7.30–6.99 (m, 10H), 3.84–3.65 (m, 2H), 2.50–2.23 (m, 4H), 2.10 (s, 3H), 1.66–1.20 (m, 8H); 13C NMR (75 MHz, CDCl3) δ ppm 144.3, 127.06, 127.00, 126.9, 63.1, 45.3, 37.2, 31.6, 31.1. [α]D20 = +46.2° (c = 1.5, MeOH); Anal. C21H28ClN•H2O (C,H,N).
1-Methyl-(2S,5S)-di-(2-phenethyl)-pyrrolidine hydrochloride (11)
Compound 10 was subjected to conditions similar to those described for the preparation of 9. Spectral characteristics were identical to those of 7; [α]D20 = −48.7° (c = 1.0, MeOH). Anal. C21H28ClN (C,H,N).
cis-2,5-Di(2-benzyl)-pyrrolidine hydrochloride (22)
To a solution of 21 (200 mg, 0.50 mmol) in MeOH (5 mL) was added Pd(OH)2-C (10 mg, 10 wt%) and ammonium formate (100 mg, 50 wt%). The mixture was refluxed for 20 min., diluted with CH2Cl2 (60 mL), and filtered through celite. The filtrate was treated with 5 mL conc. HCl and evaporated to a crusty white solid under reduced pressure. The solid was triturated with diethyl ether, and precipitated from a mixture of CH2Cl2 and hexanes by addition of hexanes, to yield 22 (108 mg, 74%) as a white solid, mp = 173 °C. 1H NMR (300 MHz, CDCl3) δ ppm 9.4 (s, 1H), 9.2 (br, s, 1H), 7.45–7.00 (m, 10H), 4.00–3.64 (s, br, 2H), 2.48–2.14 (m, 4H), 1.70–1.52 (m, 6H). 13C NMR (75 MHz, CDCl3) δ ppm 139.0, 126.3, 124.6, 120.8, 58.4, 37.1, 34.3, 33.1; Anal. C18H22ClN (C,H,N).
1-Methyl-cis-2,5-di(2-benzyl)-pyrrolidine hydrochloride (23)
To a solution of 22 (100 mg, 0.32 mmol) in MeOH (3 mL) was added NaCNBH3 (50 mg, excess) and paraformaldehyde (50 mg, excess). The initial suspension became a clear solution upon stirring for 6h. The solution was diluted with conc. HCl (10 mL) and stirred for 2 h, during which time all of the cyanoborohydride and hydrogen cyanide was assumed to be decomposed. The mixture was poured into 20% aq. NaOH (10 mL) and extracted with CH2Cl2 (100 mL). Drying and evaporation of the organic layer yielded an oil that was treated with conc. HCl (5 mL) and co-evaporated with toluene. Trituration of the residue afforded a white solid 22 (80 mg, 81%). mp = 126 °C (deliquescent); 1H NMR (300 MHz, CDCl3) δ ppm 9.98 (br, s, 1H), 7.33–7.00 (m, 10H), 3.63–3.53 (m, 2H), 2.46–2.23 (m, 4H), 2.42 (s, 3H), 1.51–1.22 (m, 6H); 13C NMR (75 MHz, CDCl3) 139.1, 128.9, 127.4, 125.1, 65.2, 48.3, 39.0, 30.1. Anal. C19H24ClN•H2O (C,H,N).
Cis-2,5-di(3-phenylpropyl)-pyrrolidine hydrochloride (25)
To compound 24 (100 mg, 0.21 mmol) was added Pd(OH)2-on-Carbon (10 wt%), MeOH (10 mL) and ammonium formate (500 mg). After refluxing for 2h, the solution was poured into CH2Cl2 (50 mL), filtered, and evaporated to obtain a white solid (70 mg, 94%). 1H NMR (300 MHz, CDCl3) δ ppm 10.31 (s, 1H), 9.03 (s, 1H), 7.27–7.13 (m, 15H, Ar), 3.50 (s, 1H), 2.62–2.14 (m, 4H), 2.12–1.68 (m, 14H). 13C NMR (75 MHz, CDCl3) δ ppm 140.2, 129.7, 127.2, 126.2, 65.66, 37.38, 29.5, 28.2, 27.85; Anal. C22H30ClN (C,H,N).
1-Methyl-cis-2,5-di(3-phenylpropyl)-pyrrolidine hydrochloride (26)
To a solution of 25 (20 mg, 0.08 mmol) in MeOH (3 mL) was added NaCNBH3 (20 mg, excess) and paraformaldehyde (20 mg, excess). The initial suspension became a clear solution upon stirring for 12h. The solution was diluted with conc. HCl (1 mL) and stirred for 2 h, during which time all of the cyanoborohydride and hydrogen cyanide was assumed to be decomposed. The mixture was then poured into 20% aq. NaOH (3 mL) and extracted with CH2Cl2 (10 mL). Drying and evaporation of the organic layer yielded an oil that was treated with conc. HCl (1 mL) and coevaporated with toluene. Trituration of the residue afforded a white solid (18 mg, 87%), mp = 78 °C. 1H NMR (300 MHz, CDCl3) δ ppm 9.48 (s, 1H), 7.45–7.03 (m, 10H), 3.47–3.18 (m, 2H), 2.58–2.47 (m, 4H), 2.19 (s, 3H), 1.91–1.36 (m, 6H); 13C NMR (75 MHz, CDCl3) δ ppm 140.6, 128.2, 128.13, 128.02, 64.1, 37.6, 36.4, 29.5, 29.2, 28.3; Anal. C23H32ClN•H2O (C,H,N).
Preparation of rat brain synaptic vesicles
Fresh whole brain (excluding cerebellum and brain stem) was homogenized in 20 vol of ice-cold 0.32 M sucrose with a glass homogenizer (7 strokes of a Teflon pestle, clearance = 0.003 in). Homogenates were centrifuged at 1000 g for 12 min at 4 °C. Resulting supernatants (S1) were centrifuged at 22,000 g for 10 min. Resulting pellets (P2), containing the synaptosomes, were resuspended in 18 mL ice-cold Milli-Q water for 5 min with 7 strokes of the Teflon pestle homogenizer. Osmolarity was restored by immediate addition of 2 mL of 25 mM HEPES and 100 mM K2-tartrate buffer (pH 7.5). Samples were centrifuged at 20,000 g for 20 min. MgSO4 (final concentration, 1 mM) was added to the resulting supernatants (S3). Final centrifugations were performed at 100,000 g for 45 min. Pellets (P4) were resuspended immediately in ice-cold buffer (see below) providing ~15 μg protein/100 μL.
[3H]DTBZ binding assay
100 μL of vesicles suspension was incubated in assay buffer (in mM: 25 HEPES, 100 K2-tartrate, 5 MgSO4, 0.1 EDTA and 0.05 EGTA, pH 7.5, 25 °C) in the presence of 5 nM [3H]DTBZ and 1 nM–1 mM lobelane analogue (final concentrations) for 30 min at room temperature. Nonspecific binding was determined in the presence of 10 μM Ro4-12084. Assays were performed in duplicate using Unifilter-96 96-well GF/B filter plates (presoaked in 0.5% polyethylenimine) and terminated by harvesting using the FilterMate harvester. After washing 5 times with 350 μL of the ice-cold wash buffer (in mM: 25 HEPES, 100 K2-tartrate, 5 MgSO4 and 10 NaCl, pH 7.5), filter plates were dried, bottoms-sealed and each well filled with 40 μL Packard’s MicroScint 20 cocktail. Bound [3H]DTBZ was measured using a Packard TopCount NXT scintillation counter and a Packard Windows NT-based operating system.
[3H]-DA uptake inhibition assay
Inhibition of [3H]-DA uptake was conducted in a preparation of isolated synaptic vesicles.14 Briefly, rat striata were homogenized with 10 strokes of a Teflon pestle homogenizer (clearance ~ 0.003″) in 14 ml of 0.32 M sucrose solution. Homogenate was centrifuged (2,000 g for 10 min at 4° C), and the resulting supernatant was centrifuged again (10,000 g for 30 min at 4°C). The pellet was resuspended in 2 ml of 0.32 M sucrose solution and subjected to osmotic shock by adding 7 ml of ice-cold water to the preparation, followed by the immediate restoration of osmolarity by adding 900 μl of 0.25M HEPES buffer and 900 μl of 1.0 M potassium tartrate solution. Samples were centrifuged (20,000 g for 20 min at 4° C), and the resulting supernatant was centrifuged again (55,000g for 1 hr at 4° C), followed by the addition of 100 μl of 10 mM MgSO4, 100 μl of 0.25 M HEPES and 100 μl of 1.0 M potassium tartrate solution prior to the final centrifugation (100,000 g for 45 min at 4° C). The final pellet was resuspended in 2.4 ml of assay buffer (25 mM HEPES, 100 mM potassium tartrate, 50 μM EGTA, 100 μM EDTA, 1.7 mM ascorbic acid, 2 mM ATP-Mg2+, pH 7.4). Aliquots of the vesicular suspension (100 μl) were added to tubes containing assay buffer, various concentrations of inhibitor (0.1 nM – 10 mM) and 0.1 μM [3H]-DA in a final volume of 500 μl. Nonspecific uptake was determined in the presence of Ro4-1284 (10 μM). Reactions were terminated by filtration, and radioactivity retained by the filters was determined by liquid scintillation spectroscopy (Liquid scintillation analyzer; PerkinElmer Life and Analytical Sciences, Boston, MA).
Supplementary Material
Figure 1.

Structures of 1, 2 and synthetic analogs.
Figure 2.
Structures of initial pyrrolidine analogs of 2 and nor-2
Acknowledgments
The authors thank Agripina G. Deaciuc for technical assistance with the pharmacological assays, and the National Institutes of Health, NIDA Grant DA013519 for financial support.
Abbreviations
- VMAT2
Vesicular MonoAmine Transporter 2
- MAO
MonoAmine Oxidase
- DA
DopAmine
- METH
METHamphetamine
- DTBZ
DihydroTetraBenaZine
- nAChRs
nicotinic ACetylcHoline Receptors
Footnotes
Special Footnote. The authors would like to express profound gratitude to the ACS Division of Medicinal Chemistry for its relentless efforts toward fostering research and training in the discipline of Medicinal Chemistry over the past 100 years.
Supporting Information Available. Combustion analysis data for all compounds, and preparation and characterization data for 21 and 24 can be found in the supporting information. This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Eiden LE. The vesicular neurotransmitter transporters: current perspectives and future prospects. The FASEB Journal. 2000;14:2396–2400. doi: 10.1096/fj.00-0817rev. [DOI] [PubMed] [Google Scholar]
- 2.Ascher JA, Cole JO, Colin JN, Feighner RM, Ferris RM, Fibiger HC, Golden RN, Martin P, Potter WZ, Richelson E, Sulser F. Bupropion: a review of its mechanism of antidepressant activity. J Clin Psychiatry. 1995;56:395–401. [PubMed] [Google Scholar]
- 3.Zheng G, Dwoskin LP, Crooks PA. Vesicular monoamine transporter 2: role as a novel target for drug development. The AAPS Journal. 2006;8:E682–E692. doi: 10.1208/aapsj080478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dwoskin LP, Crooks PA. A novel mechanism of action and potential use for lobeline as a treatment for psychostimulant abuse. Biochem Pharmacol. 2002;63:89–98. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- 5.Terry L, Williamson R, Gattu M, Beach JW, McCurdy CR, Sparks JA, Pauly JR. Lobeline and structurally simplified analogs exhibit differential agonist activity and sensitivity to antagonist blockade when compared to nicotine. Neuropharmacology. 1998;37:93–102. doi: 10.1016/s0028-3908(97)00142-1. [DOI] [PubMed] [Google Scholar]
- 6.Flammia D, Dukat M, Damaj MI, Martin B, Glennon RA. Lobeline: Structure-Activity Investigation of Nicotinic Acetylcholinergic Receptor Binding. J Med Chem. 1999;42:3726–3731. doi: 10.1021/jm990286m. [DOI] [PubMed] [Google Scholar]
- 7.Zheng G, Dwoskin LP, Deaciuc AG, Norrholm SD, Crooks PA. Defunctionalized lobeline analogues: structure-affinity of novel ligands for the vesicular monoamine transporter. J Med Chem. 2005;48:5551–5560. doi: 10.1021/jm0501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Court JA, Perry EK, Spurden D, Lloyd S, Gillespie JI, Whiting P, Barlow R. Comparison of the binding of nicotinic agonists to receptors from human and rat cerebral cortex and from chick brain (α4β2) transfected into mouse fibroblasts with ion channel activity. Brain Res. 1994;667:118–122. doi: 10.1016/0006-8993(94)91721-3. [DOI] [PubMed] [Google Scholar]
- 9.Zheng G, Dwoskin LP, Deaciuc AG, Crooks PA. Synthesis and evaluation of a series of homologues of lobelane at the vesicular monoamine transporter-2. Bioorg Med Chem Lett. 2008;18:6509–6512. doi: 10.1016/j.bmcl.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katritzky AR, Cui X, Baozhen Y, Steel PJ. Asymmetric syntheses of 2-substituted and 2,5-disubstituted pyrrolidines from (3S,5R,7aR)-5-(benzotriazol-1-yl)-3-phenyl[2,1-b]oxazolopyrrolidine. J Org Chem. 1999;64:1979–1985. doi: 10.1021/jo9821426. [DOI] [PubMed] [Google Scholar]
- 11.Gopalakrishnan A, Sievert M, Ruoho AE. Identification of the substrate binding region of vesicular monoamine transporter-2 (VMAT-2) using iodoaminoflisopolol as a novel photoprobe. Mol Pharmacol. 2007;72:1567–1575. doi: 10.1124/mol.107.034439. [DOI] [PubMed] [Google Scholar]
- 12.Ugarte YV, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE. Methamphetamine rapidly decreases mouse vesicular dopamine uptake: role of hyperthermia and dopamine D2 receptors. Eur J Pharmacol. 2003;472:165–171. doi: 10.1016/s0014-2999(03)01911-3. [DOI] [PubMed] [Google Scholar]
- 13.Neugebauer NM, Harrod SB, Stairs DJ, Crooks PA, Dwoskin LP, Bardo MT. Lobelane decreases methamphetamine self-administration in rats. Eur J Pharmacol. 2007;571:33–38. doi: 10.1016/j.ejphar.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. Lobeline and nicotine evoked [3H] overflow from rat striatal slices preloaded with [3H]dopamine: differential inhibition of synaptosomal and vesicular [3H]dopamine uptake. J Pharmacol Exp Ther. 1997;280:1432–1444. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.