

Abstract
Phosphocitrate (PC) inhibited meniscal calcification and the development of calcium crystal-associated osteoarthritis (OA) in Hartley guinea pigs. However, the mechanisms remain elusive. This study sought to examine the biological activities of PC in the absence of calcium crystals and test the hypothesis that PC is potentially a meniscal protective agent. We found that PC downregulated the expression of many genes classified in cell proliferation, ossification, prostaglandin metabolic process, and wound healing, including bloom syndrome RecQ helicase-like, cell division cycle 7 homolog, cell division cycle 25 homolog C, ankylosis progressive homolog, prostaglandin-endoperoxide synthases-1/cyclooxygenase-1, and plasminogen activator urokinase receptor. In contrast, PC stimulated the expression of many genes classified in fibroblast growth factor receptor signaling pathway, collagen fibril organization, and extracellular structure organization, including fibroblast growth factor 7, collagen type I, alpha 1, and collagen type XI, alpha 1. Consistent with its effect on the expression of genes classified in cell proliferation, collagen fibril organization, and ossification, PC inhibited the proliferation of OA meniscal cells and meniscal cell-mediated calcification while stimulating the production of collagens. These findings indicate that PC is potentially a meniscal-protective agent and a disease-modifying drug for arthritis associated with severe meniscal degeneration.
1. Introduction
Osteoarthritis (OA) is one of the most prevalent causes of disability in the aging population and has enormous economic and social consequences. However, existing nonsurgical treatment options only provide symptomatic relief but have no effect on the progression of the underlying disease or cartilage degeneration. The lack of progress in the development of disease-modifying drugs for OA therapy is largely due to our limited understanding of the pathogenesis of OA and our insufficient knowledge about the molecular targets for OA therapy.
Knee OA is not merely an articular cartilage disease, but a disease of the whole joint. An important local factor is the structural integrity of the menisci. In recent years, there has been a dramatic advance in our understanding of the integral role of the menisci for knee function and the consequences of meniscal abnormality on the development of OA. Studies have found that meniscal degeneration is a general feature of knee OA and contributes to joint space narrowing [1, 2]. Meniscal lesions at baseline were more common in knees that developed OA than in the knees that did not develop OA during a 30-month follow-up period [3]. OA meniscal cells displayed a distinct gene expression profile different from normal meniscal cells [4]. These observations indicate that the meniscus is not a passive bystander in the disease process of knee OA [5, 6].
Basic calcium phosphate crystal and calcium pyrophosphate dihydrate crystal are the two most common articular calcium crystals. The presence of these crystals in OA articular cartilage and synovial fluid is well recognized. These crystals are also present in knee menisci of patients with end-stage OA [7, 8]. Studies found that these crystals stimulate cell mitogenesis, cell endocytotic activity, and the production of matrix metalloproteinases (MMPs) and inflammatory cytokines including interleukin-1 (IL-1) and prostaglandin-endoperoxide synthase 2/cyclooxygenase-2 (PTGS2/Cox-2) [9–11]. However, there is still controversy as to whether these crystals are causative factors, factors that exacerbate OA, or simply bystanders.
Phosphocitrate (PC) is a naturally occurring compound originally identified in rat liver mitochondrial extracts and crab hepatopancreas [12]. Moro et al. and Romanello et al. suggested that PC could be formed in vivo through cytosolic phosphorylation of citric acid [13, 14], which explains why it is nontoxic. Since its original identification, PC has been shown to be a powerful calcification inhibitor [15, 16]. Tew et al. speculated that PC prevented CaPO4 precipitation in cells or cellular compartments containing high concentration of Ca2+ and PO4 [12]. PC prevented soft tissue calcification in vivo and did not produce any significant toxic side effect in rats when administered through intraperitoneal injections in doses up to 150 μmol/kg/day [17]. In cell cultures, PC inhibits calcium crystal-induced mitogenesis, expression of MMPs, and crystal-induced cell death [18–20]. In the Hartley guinea pig model of crystal-associated OA, PC inhibited meniscal calcification and reduced the severity of cartilage degeneration [21]. These observations provide support for the notion that crystals may play an important role in the development of OA and that calcification inhibitors are potentially disease-modifying drugs for crystal-associated OA therapy. However, two bisphosphonates, which are potent calcification inhibitors, failed to inhibit the development of OA in animal models of OA [22, 23], raising doubts as to whether calcification inhibitors are disease-modifying drugs for crystal-associated OA as well as the exact role of calcium crystals in the development of OA. In this study, we sought to examine the biological activities of PC in the absence of calcium crystals and test the hypothesis that PC has unique crystal-independent biological activities which may be responsible, at least in part, for its disease-modifying activity on OA and that PC is potentially a meniscal protective agent.
2. Materials and Methods
Dulbecco's Modified Eagle Medium, StemPro chondrogenesis differentiation medium, fetal bovine serum, Hank's balanced salt solution, and stock antibiotic and antimycotic mixture were products of Invitrogen (Carlsbad, CA, USA). PC was synthesized according to the procedure described [24]. All other chemicals are purchased from Sigma (St. Louis, MO, USA).
2.1. Cells
OA meniscal cells were prepared from menisci derived from patients with end-stage OA. Briefly, the medial menisci derived from patients with end-stage OA were processed to remove fatty and synovial tissues, minced into small pieces, and cultured in 100 mm plates at 37°C in medium containing 0.5% antibiotic/antimycotic solution and 10% serum. Every three or four days, the culture medium was changed. When the cells reached 70% confluence, they were passaged and maintained in medium containing 10% serum. Human foreskin fibroblasts were obtained from American Type Culture Collection (CRL-2429, Manassas, VA, USA). OA meniscal cells prepared from three OA patients were used in this study. OA menisci were collected with the approval of the authors' Institutional Review Board from OA patients undergoing knee joint replacement surgery. The need for informed consent was waived because those menisci were surgical waste, and no private patient information was collected.
2.2. Cell Culture and RNA Extraction
OA meniscal cells derived from three OA patients were harvested from cell culture plates and mixed and replated in four 100 mm cell culture plates at 90% confluence. On the second day, medium containing 1% serum was added. Twenty-four hours later, the medium in two plates was replaced with medium containing 1% serum and PC (1 mM), and the medium in the other two plates was replaced with medium containing 1% serum without PC. Twenty-four hours later, total RNA was extracted from these cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and purified using Oligotex kit (Qiagen, Valencia, CA, USA). We repeated the experiment twice. Microarray was performed using these RNA samples (total six RNA samples).
2.3. Microarray
RNA samples extracted from three independent experiments were used for microarray analysis experiments. Briefly, double-stranded DNA was synthesized using SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen, San Diego, CA, USA). The DNA product was purified using GeneChip Sample Cleanup Module (Affymetrix, Santa Clara, CA, USA). cRNA was synthesized and biotin-labeled using BioArray high yield RNA transcript labeling kit (Enzo Life Sciences, Farmingdale, NY, USA). The cRNA product was purified using GeneChip Sample Cleanup Module and subsequently chemically fragmented. The fragmented and biotinylated cRNA was hybridized to HG-U133_Plus_2 GeneChip using Affymetrix Fluidics Station 400 (Affymetrix, Santa Clara, CA, USA). The fluorescent signals were quantified during two scans by Agilent Gene Array Scanner G2500A (Agilent Technologies, Palo Alto, CA, USA) and GeneChip operating Software (Affymetrix, Santa Clara, CA, USA). Genesifter (VizX Labs, Seattle, WA, USA) was used for the analysis of differential gene expression and gene ontology.
2.4. Real-Time RT-PCR
After microarray analyses, we mixed the RNA samples extracted from PC-treated OA meniscal cells (PC-treated RNA sample) and the RNA samples extracted from untreated OA meniscal cells (untreated RNA sample) and performed RT-PCR experiments. Briefly, cDNA was synthesized using TaqMan Reverse Transcription Reagents (Applied Biosystems, University Park, IL, USA) using the RNA samples described. Quantification of relative transcript levels for selected genes and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was performed using ABI7000 Real-Time PCR system (Applied Biosystems, University Park, IL, USA). TaqMan Gene Expression assay (Applied Biosystems, University Park, IL, USA) was used. CDNA samples were amplified with an initial Taq DNA polymerase activation step at 95°C for 10 minutes, followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing at 60°C for one minute. Fold change was calculated, and the expression level of the genes to be examined was normalized to the expression level of GAPDH. RT-PCR experiment was performed in triplicates using the same RNA sample.
2.5. Cell Proliferation
OA meniscal cells (2 × 104) were plated in six well cluster plates and cultured in medium containing 10% serum in the presence of increasing amounts of PC (triplicates). Medium was changed every three days until the OA meniscal cells in the wells without PC reached 85% confluence. Cells were then harvested and cell numbers were determined using a hemocytometer. This experiment was repeated 3 times using OA meniscal cells derived from different patients. This proliferation assay was also performed using human primary foreskin fibroblasts.
2.6. Micromass Culture and Histology
OA meniscal cells were harvested from several 100 mm culture plates and suspended in medium containing 10% serum. For preparing a micromass, a droplet of the cell suspension containing 6 × 106 cells was placed in a well of a 24-well plate. After placing all droplets, the plate was incubated for 4 hours at 37°C in a tissue culture incubator. These micromasses were then fed with StemPro chondrogenesis differentiation medium with PC (1 mM) or without PC every three days throughout the experiment for 14 days. Each well was then rinsed twice with 500 μL of Hank's balanced salt solution, and two drops of eosin were added to these wells. Five minutes later, eosin was aspirated off and the micromasses were transferred to a strip of filter paper which sat on the top of an ethanol-soaked sponge within a plastic cassette. The cassettes sat in a 10% formalin solution for one hour. These micromasses then underwent routine paraffin embedding. Sections were cut at 5 μm thick and stained with picrosirius red for collagens, alcian blue for proteoglycans, and alizarin red for calcium deposits.
2.7. Cell-Mediated Calcification in Monolayer Culture
OA meniscal cells were plated in twenty-four well plates at 90% confluence. The next day, medium was replaced with StemPro chondrogenesis differentiation medium containing 1 mM adenosine-5′-triphosphate (ATP) in the absence or presence of PC (1 mM). The cells were cultured for 14 days and fed with StemPro chondrogenesis differentiation medium containing ATP every three or four days throughout the experiment. At the end of the experimental period, media were removed. Calcification was examined using alizarin red.
2.8. Statistical Analysis
For cell proliferation, data are expressed as the mean ± SD, and the difference between two groups was analyzed using Student's t-test. For real-time RT-PCR, experiment was repeated in triplicates. The difference between two experimental groups was analyzed using Student's t test. In all cases, P values less than 0.01 were considered significant. Statistical analysis was performed using the SAS software, version 9.3.
3. Results
3.1. Effect of PC on Gene Expression
We performed three microarray experiments (PC-treated RNA sample I and untreated RNA sample I; PC-treated RNA sample II and untreated RNA sample II; PC-treated RNA sample III and untreated RNA sample III) as described. The results of the three microarray experiments were quite similar. The results of the first microarray experiment showed that of the more than 50,000 transcripts, 2445 transcripts displayed significant differential expression (more than 1.6-fold changes) in the PC-treated OA meniscal cells compared with the untreated OA meniscal cells. A total of 1795 transcripts displayed decreased expression and 650 transcripts displayed increased expression. The genes that fell into specific biological processes previously implicated in OA, or suspected to have a role in OA are listed in Tables 1 and 2.
Table 1.
Differentially expressed genes in PC-treated via untreated OA meniscal cells.
| Biological process |
Gene name |
Gene ID | Differential expression* |
Description |
|---|---|---|---|---|
| Cell proliferation | BLM | NM_000057 | −7.41 | Bloom syndrome, RecQ helicase-like |
| NDP | NM_000266 | −4.68 | Norrie disease (pseudoglioma) | |
| HELLS | AF155827 | −4.46 | Helicase, lymphoid specific | |
| E2F7 | AI341146 | −4.12 | E2F transcription factor 7 | |
| CDC7 | NM_003503 | −4.11 | Cell division cycle 7 homolog (S. cerevisiae) | |
| CDCA7 | AY029179 | −2.71 | Cell division cycle-associated 7 | |
| CDC25C | NM_001790 | −2.36 | Cell division cycle 25 homolog C (S. pombe) | |
| BRCA1 | AF005068 | −3.60 | Breast cancer 1, early onset | |
| BRCA2 | X95152 | −3.30 | Breast cancer 2, early onset | |
| PRKRA | AA279462 | −3.34 | PKinase, interferon-indu double-stranded RNA-dependent activator | |
| HHIP | AK098525 | −3.28 | Hedgehog interacting protein | |
| CHEK1 | AA224205 | −3.11 | CHK1 checkpoint homolog (S. pombe) | |
| PTPRK | AU145587 | −3.08 | Protein tyrosine phosphatase, receptor type, K | |
| GINS1 | NM_021067 | −3.07 | GINS complex subunit 1 (Psf1 homolog) | |
| TCF19 | BC002493 | −2.93 | Transcription factor 19 | |
| MKI67 | AU147044 | −2.92 | Antigen identified by monoclonal antibody Ki-67 | |
| PDS5B | AK026889 | −2.88 | PDS5, regulator of cohesion maintenance, homolog B | |
| UHRF1 | AK025578 | −2.77 | Ubiquitin-like with PHD and ring finger domains 1 | |
| AURKB | AB011446 | −2.64 | Aurora kinase B | |
| MKI67 | AU132185 | −2.64 | Antigen identified by monoclonal antibody Ki-67 | |
| FIGNL1 | NM_022116 | −2.62 | Fidgetin-like 1 | |
| KIF15 | NM_020242 | −2.60 | Kinesin family member 15 | |
| LRP6 | NM_002336 | −2.55 | Low-density lipoprotein receptor-related protein 6 | |
| ANXA1 | AU155094 | −2.47 | Annexin A1 | |
| DDX11 | U33833 | −2.47 | DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 11 | |
| FANCA | AW083279 | −2.46 | Fanconi anemia, complementation group A | |
| SPRY2 | NM_005842 | −2.43 | Sprouty homolog 2 (Drosophila) | |
| RECQL4 | NM_004260 | −2.43 | RecQ protein-like 4 | |
| NTN1 | BF591483 | −2.42 | Netrin 1 | |
| ADRB2 | NM_000024 | −2.41 | Adrenergic, beta-2, receptor, surface | |
| CUL4A | AU155661 | −2.40 | Cullin 4A | |
| DLC1 | NM_024767 | −2.39 | Deleted in liver cancer 1 | |
| STIL | NM_003035 | −2.39 | SCL/TAL1 interrupting locus | |
| CHEK1 | NM_001274 | −2.37 | CHK1 checkpoint homolog (S. pombe) | |
| TIMELESS | NM_003920 | −2.37 | Timeless homolog (Drosophila) | |
| SMAD4 | AL832789 | −2.23 | SMAD family member 4 | |
| SMAD1 | NM_015583 | −2.19 | SMAD family member 1 | |
| PCNA | NM_002592 | −2.18 | Proliferating cell nuclear antigen | |
| PRKCD | NM_006254 | −2.17 | Protein kinase C, delta | |
| RBBP4 | AI972451 | −2.16 | Retinoblastoma binding protein 4 | |
| PTGS1 | NM_000962 | −2.13 | Prostaglandin-endoperoxide synthase 1 | |
| ASPM | NM_018123 | −2.13 | Abnormal spindle (asp) homolog, microcephaly associated | |
| GLMN | AA814383 | −2.11 | Glomulin, FKBP associated protein | |
| NASP | NM_002482 | −2.11 | Nuclear autoantigenic sperm protein (histone binding) | |
| CCNA2 | NM_001237 | −2.09 | Cyclin A2 | |
| TBX2 | AW173045 | −2.08 | T-box 2 | |
| KIF2C | U63743 | −2.08 | Kinesin family member 2C | |
| PDCD1LG2 | AF329193 | −2.06 | Programmed cell death 1 ligand 2 | |
| BUB1B | NM_001211 | −2.05 | Budding uninhibited by benzimidazoles 1 homolog beta (yeast) | |
| POLA1 | NM_016937 | −2.04 | Polymerase (DNA directed), alpha 1, catalytic subunit | |
| TACC3 | NM_006342 | −2.02 | Transforming, acidic coiled-coil containing protein 3 | |
| CDK2 | M68520 | −2.01 | Cyclin-dependent kinase 2 | |
| DLX5 | NM_005221 | 2.99 | Distal-less homeobox 5 | |
| VCAM1 | NM_001078 | 2.88 | Vascular cell adhesion molecule 1 | |
| ADORA1 | NM_000674 | 2.48 | Adenosine A1 receptor | |
| TNFRSF9 | NM_001561 | 2.38 | Tumor necrosis factor receptor superfamily, member 9 | |
| FGF9 | NM_002010 | 2.38 | Fibroblast growth factor 9 (glia-activating factor) | |
| FABP3 | NM_004102 | 2.24 | Fatty acid binding protein 3, muscle, and heart | |
| FGFR2 | M87771 | 2.20 | Fibroblast growth factor receptor 2 | |
| BAMBI | NM_012342 | 2.13 | BMP and activin membrane-bound inhibitor homolog | |
| CD24 | L33930 | 2.09 | CD24 molecule | |
| HSF1 | AI393937 | 2.07 | Heat shock transcription factor 1 | |
|
| ||||
| Ossification | COL13A1 | M33653 | −3.39 | Collagen, type XIII, alpha 1 |
| SATB2 | AK025127 | −3.13 | SATB homeobox 2 | |
| ADRB2 | NM_000024 | −2.41 | Adrenergic, beta-2, receptor, surface | |
| SMAD1 | NM_015583 | −2.19 | SMAD family member 1 | |
| ENPP1 | BF591996 | −2.02 | Ectonucleotide pyrophosphatase/phosphodiesterase 1 | |
| BMPR1B | AA935461 | −1.93 | Bone morphogenetic protein receptor, type IB | |
| NAB1 | AF045452 | −1.87 | NGFI-A-binding protein 1 (EGR1-binding protein 1) | |
| GNAS | AA810695 | −1.86 | GNAS complex locus | |
| FGF18 | BC006245 | −1.73 | Fibroblast growth factor 18 | |
| TNFRSF11A | AW026379 | −1.67 | Tumor necrosis factor receptor superfamily, member 11a | |
| EGFR | K03193 | −1.61 | Epidermal growth factor receptor | |
| PLA2G4A | M68874 | −1.61 | Phospholipase A2, group IVA (cytosolic, calcium dependent) | |
| ANKH | T99215 | −1.60 | Ankylosis, progressive homolog (mouse) | |
| DLX5 | NM_005221 | 2.99 | Distal-less homeobox 5 | |
| FGF9 | NM_002010 | 2.38 | Fibroblast growth factor 9 (glia-activating factor) | |
| IGFBP5 | R73554 | 1.78 | Insulin-like growth factor-binding protein 5 | |
| GABBR1 | N45228 | 1.72 | Gamma-aminobutyric acid (GABA) B receptor, 1 | |
| MMP14 | NM_004995 | 1.72 | Matrix metallopeptidase 14 (membrane inserted) | |
| TUFT1 | NM_020127 | 1.66 | Tuftelin 1 | |
| COL1A1 | AI743621 | 1.62 | Collagen, type I, alpha 1 | |
| MGP | NM_000900 | 1.61 | Matrix Gla protein | |
| SMAD3 | BF971416 | 1.60 | SMAD family member 3 | |
|
| ||||
| BMP signaling pathway | UBE2D3 | AI239832 | −2.44 | Ubiquitin-conjugating enzyme E2D 3 (UBC4/5 homolog, yeast) |
| ZFYVE16 | BC032227 | −2.43 | Zinc finger, FYVE domain containing 16 | |
| SMAD4 | AL832789 | −2.23 | SMAD family member 4 | |
| SMAD1 | NM_015583 | −2.19 | SMAD family member 1 | |
| BMPR1B | AA935461 | −1.93 | Bone morphogenetic protein receptor, type IB | |
| HIPK2 | AW300045 | −1.91 | Homeodomain interacting protein kinase 2 | |
| MSX1 | AI421295 | −1.84 | Msh homeobox 1 | |
| GREM2 | NM_022469 | −1.73 | Gremlin 2, cysteine knot superfamily, homolog (Xenopus laevis) | |
|
| ||||
| Prostaglandin metabolic process | PTGS1 | NM_000962 | −2.13 | Prostaglandin-endoperoxide synthase 1 |
| AKR1C2 | BF508244 | −1.91 | Aldo-keto reductase family 1, member C2 | |
| PLA2G4A | M68874 | −1.61 | Phospholipase A2, group IVA (cytosolic, calcium dependent) | |
| PTGS2 | NM_000963 | −1.33 | Prostaglandin-endoperoxide synthase 2 | |
| PTGIS | D38145 | 1.79 | Prostaglandin I2 (prostacyclin) synthase | |
| PDPN | AW590196 | 1.61 | Podoplanin | |
|
| ||||
| Response to wounding | THBD | AW119113 | −4.24 | Thrombomodulin |
| PLAT | NM_000930 | −3.12 | Plasminogen activator, tissue | |
| TFPI | J03225 | −2.66 | Tissue factor pathway inhibitor | |
| TFPI2 | AL574096 | −2.38 | Tissue factor pathway inhibitor 2 | |
| SDC1 | NM_002997 | −2.38 | Syndecan 1 | |
| SMAD1 | NM_015583 | −2.19 | SMAD family member 1 | |
| PLAUR | AY029180 | −1.94 | Plasminogen activator, urokinase receptor | |
| TMPRSS6 | AI912086 | 1.96 | Transmembrane protease, serine 6 | |
| JUB | NM_032876 | 1.92 | Jub, ajuba homolog (Xenopus laevis) | |
*Negative number indicates decreased expression and positive number indicates elevated expression (fold change) in PC-treated OA meniscal cells compared with untreated OA meniscal cells.
Table 2.
Differentially expressed genes in PC-treated via untreated OA meniscal cells.
| Biological process |
Gene name |
Gene ID | Differential expression* |
Description |
|---|---|---|---|---|
| FGF receptor signaling pathway | FGF9 | NM_002010 | 2.38 | Fibroblast growth factor 9 (glia-activating factor) |
| FGFR2 | M87771 | 2.20 | Fibroblast growth factor receptor 2 | |
| FGF7 | NM_002009 | 1.83 | Fibroblast growth factor 7 (keratinocyte growth factor) | |
| NDST1 | AL526632 | 1.71 | N-deacetylase/N-sulfotransferase (heparan glucosaminyl) 1 | |
| THBS1 | AI812030 | 1.68 | Thrombospondin 1 | |
| HHIP | AK098525 | −3.28 | Hedgehog interacting protein | |
| FGF18 | BC006245 | −1.73 | Fibroblast growth factor 18 | |
|
| ||||
| Collagen fibril organization | COL14A1 | M64108 | 2.06 | Collagen, type XIV, alpha 1 |
| COL11A1 | J04177 | 1.82 | Collagen, type XI, alpha 1 | |
| COL1A1 | AI743621 | 1.62 | Collagen, type I, alpha 1 | |
| SERPINH1 | NM_004353 | 1.66 | Serpin peptidase inhibitor, clade H, member 1 | |
| DPT | AI146848 | 1.65 | Dermatopontin | |
| TRAM2 | BC028121 | 1.60 | Translocation-associated membrane protein 2 | |
| COL5A1 | AI983428 | 1.52 | Collagen, type V, alpha 1 | |
| COL5A2 | NM_000393 | 1.44 | Collagen, type V, alpha 2 | |
|
| ||||
| Extracellular structure organization | CACNA1A | NM_023035 | 2.55 | Calcium channel, voltage dependent, P/Q type, alpha 1A subunit |
| COL14A1 | M64108 | 2.06 | Collagen, type XIV, alpha 1 | |
| ECM2 | NM_001393 | 2.04 | Extracellular matrix protein 2, female organ, and adipocyte specific | |
| MYO6 | AA877789 | 1.98 | Myosin VI | |
| TMPRSS6 | AI912086 | 1.96 | Transmembrane protease, serine 6 | |
| COL11A1 | J04177 | 1.82 | Collagen, type XI, alpha 1 | |
| CRISPLD2 | AL136861 | 1.74 | Cysteine-rich secretory protein LCCL domain containing 2 | |
| HSD17B12 | BC012536 | 1.68 | Hydroxysteroid (17-beta) dehydrogenase 12 | |
| SERPINH1 | NM_004353 | 1.66 | Serpin peptidase inhibitor, clade H, member 1 | |
| DCN | AI336924 | 1.65 | Decorin | |
| DPT | AI146848 | 1.65 | Dermatopontin | |
| COL1A1 | AI743621 | 1.62 | Collagen, type I, alpha 1 | |
| APLP1 | U48437 | 1.61 | Amyloid beta (A4) precursor-like protein 1 | |
| NFASC | AI821777 | −1.99 | Neurofascin homolog (chicken) | |
| MMP9 | NM_004994 | −1.91 | Matrix metallopeptidase 9 | |
|
| ||||
| Inflammatory response | ADORA1 | NM_000674 | 2.48 | Adenosine A1 receptor |
| CD24 | L33930 | 2.09 | CD24 molecule | |
| BCL6 | S67779 | 1.98 | B-cell CLL/lymphoma 6 | |
| NGF | NM_002506 | 1.84 | Nerve growth factor (beta polypeptide) | |
| CFD | NM_001928 | 1.82 | Complement factor D (adipsin) | |
| ITGB2 | NM_000211 | 1.80 | Integrin, beta 2 | |
| CFI | BC020718 | 1.74 | Complement factor I | |
| NDST1 | AL526632 | 1.71 | N-deacetylase/N-sulfotransferase (heparan glucosaminyl) 1 | |
| IL17C | AF152099 | 1.70 | Interleukin 17C | |
| THBS1 | AI812030 | 1.68 | Thrombospondin 1 | |
| SERPINA3 | NM_001085 | 1.67 | Serpin peptidase inhibitor, clade A, member 3 | |
| AGTR1 | NM_004835 | 1.65 | Angiotensin II receptor, type 1 | |
| TFRC | N76327 | 1.64 | Transferrin receptor (p90, CD71) | |
| PDPN | AW590196 | 1.61 | Podoplanin | |
| MASP1 | AI274095 | 1.61 | Mannan-binding lectin serine peptidase 1 | |
| NFKBIZ | BE646573 | −2.58 | NFKb inhibitor, zeta | |
| ANXA1 | AU155094 | −2.47 | Annexin A1 | |
| ADRB2 | NM_000024 | −2.41 | Adrenergic, beta-2, receptor, surface | |
| SMAD1 | NM_015583 | −2.19 | SMAD family member 1 | |
| CXCL6 | NM_002993 | −2.19 | Chemokine (C-X-C motif) ligand 6 | |
| C2 | NM_000063 | −2.18 | Complement component 2 | |
| BMPR1B | AA935461 | −1.93 | Bone morphogenetic protein receptor, type IB | |
| TNFRSF1B | NM_001066 | −1.74 | Tumor necrosis factor receptor superfamily, member 1B | |
| EDNRA | NM_001957 | −1.67 | Endothelin receptor type A | |
| PLA2G4A | M68874 | −1.61 | Phospholipase A2, group IVA (cytosolic, calcium dependent) | |
| GPR68 | AI805006 | −1.60 | G protein-coupled receptor 68 | |
*Negative number indicates decreased expression and positive number indicates elevated expression (fold change) in PC-treated OA meniscal cells compared with untreated OA meniscal cells.
As shown in Table 1, the expression of numerous genes classified in cell proliferation was downregulated by PC. Of the 62 differentially expressed genes (more than 2-fold changes) classified in cell proliferation, the expression of 52 genes, including bloom syndrome recq helicase-like (BLM; −7.41-fold change), lymphoid-specific helicase (Hells; −4.46-fold change), cell division cycle 7 homolog (CDC7; −4.11-fold change), CDC 25 homolog C (CDC25C; −2.36-fold change), and cyclin E2 (CCNE2, −3.51-fold change), was downregulated by PC. This specific downregulatory effect of PC on the expression of genes classified in cell proliferation suggests that PC may inhibit the proliferation of OA meniscal cells.
The expressions of many genes classified in ossification and bone morphogenetic protein (BMP) signaling pathway were also downregulated by PC (Table 1). Of the 22 differentially expressed genes classified in ossification, the expression of 13 genes, including collagen, type XIII, alpha 1 (COL13A1; −3.39-fold change), special AT-rich sequence-binding protein (SATB2; −3.13), mothers against DPP homolog 1/SMAD family member 1 (SMAD1; −2.19), ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1; −2.02-fold change), and ankylosis progressive homolog (ANKH; −1.60-fold change), was downregulated by PC. The expression of 9 genes, including insulin-like growth factor-binding protein 5 (IGFBP5; 1.78-fold change) and matrix Gla protein (MGP; 1.61-fold change), was upregulated by PC. Of the 8 differentially expressed genes classified in BMP signaling pathway, the expression of all 8 genes, including BMP receptor, type IB (BMPR1B; −1.93-fold change), was downregulated by PC.
In addition, the expression of several genes classified in prostaglandin metabolic process and respond to wounding was downregulated by PC (Table 1). Of the 5 differentially expressed genes classified in prostaglandin metabolic process, the expression of 3 genes, including prostaglandin-endoperoxide synthase 1/cyclooxygenase-1 (PTGS1/Cox-1; −2.13-fold change), phospholipase A2, group IVA (PLA2G4A; −1.61-fold change), was downregulated by PC. Of the 9 differentially expressed genes classified in respond to wounding, the expression of 7 genes, including thrombomodulin (THBD; −4.24-fold change), plasminogen activator, tissue (PLAT, −3.12-fold change) and plasminogen activator, urokinase receptor (PLAUR; −1.94-fold change), was downregulated by PC.
Many genes that were upregulated by PC fell into the biological processes of fibroblast growth factor (FGF) receptor signaling pathway, collagen fibril organization, and extracellular structure organization (Table 2). Of the 7 differentially expressed genes classified in FGF receptor signaling pathway, the expression of 5 genes, including FGF7 (1.83-fold change), FGF9 (2.38-fold change), and FGF receptor 2 (FGFR2; 2.20 fold change), was upregulated by PC. Of the 6 differentially expressed genes classified in collagen fibril organization, the expression of all 6 genes, including collagen type I, alpha 1 (COL1A1; 1.62-fold change), collagen type XI, alpha 1 (COL11A1; 1.82-fold change), and collagen type XIV, alpha 1 (COL14A1; 2.06-fold change), was upregulated by PC. Further examination of the microarray data indicated that the expression of collagen type V, alpha 1 (COL5A1; 1.52-fold change) and collagen type V, alpha 2 (COL5A2; 1.44-fold change) was also upregulated by PC although the changes were less than 1.6-fold (Table 2). Of the 15 genes differentially expressed genes classified in extracellular structure organization, the expression of 13-genes, including extracellular matrix protein 2 (ECM2; 2.04-fold change), was upregulated by PC. Only 2 genes, including MMP9 (−1.91-fold change), were downregulated by PC (Table 2).
Finally, of the 26 genes classified in inflammatory response, the expression of 15 genes, including adenosine A1 receptor (ADORA1; 2.48-fold change), CD24 molecule (CD24; 2.09-fold change), and B-cell CLL/lymphoma 6 (BCL6; 1.98-fold change), was upregulated by PC. The expression of 11 genes, including NF-kappaB inhibitor zeta (NFKBIZ; −2.58-fold change), adrenergic, beta-2, receptor (ADRB2; −2.41-fold change), and chemokine (C-X-C motif) ligand 6 (CXCL6; −2.19-fold change), was downregulated by PC (Table 2).
3.2. Real-Time RT-PCR
Real-time RT-PCR was used to confirm expression of selected genes. The results are listed in Table 3. As shown, the differential expression of the genes examined was confirmed by real-time RT-PCR (P < 0.01).
Table 3.
Differential expression confirmed by real-time RT-PCR.
| Gene name | Gene ID | Differential expression microarray |
Differential expression* RT-PCR |
Differential expression** RT-PCR |
|---|---|---|---|---|
| BLM | NM_000057 | −7.41 | −5.54 | −6.54 |
| HELLS | AF155827 | −4.46 | −4.17 | −4.87 |
| CDC25C | NM_001790 | −2.36 | −2.18 | −2.92 |
| CDC6 | NM_001254 | −2.00 | −2.20 | −3.41 |
| CCNE2 | NM_004702 | −3.51 | −2.94 | −3.46 |
| CCNA2 | A1346350 | −2.00 | −2.34 | −1.78 |
| ANKH | T99215 | −1.60 | −1.42 | −1.86 |
| PTGS1 | NM_000962 | −2.13 | −2.43 | −2.98 |
| THBD | AW119113 | −4.24 | −4.51 | −3.79 |
| FGF7 | NM_002009 | 1.83 | 1.96 | 2.32 |
| FGF9 | NM_002010 | 2.38 | 2.47 | 2.70 |
| COL11A1 | J04177 | 1.82 | 1.96 | 2.01 |
| ECM2 | NM_001393 | 2.04 | 2.71 | 2.15 |
| IGFBP5 | R73554 | 1.78 | 1.95 | 2.34 |
| CD24 | L33930 | 2.09 | 2.32 | 3.10 |
*RT-PCR was performed using the RNA samples that were used for microarray analyses. **RT-PCR was performed using RNA samples from another experiment.
3.3. PC Inhibited Proliferation of OA Meniscal Cells
The specific downregulatory effect of PC on the expression of genes classified in cell proliferation suggests that PC may inhibit the proliferation of OA meniscal cells. To test this, we cultured OA meniscal cells in the absence and presence of PC for 9 days and then determined the cell number using a hemocytometer. Indeed, we found that PC inhibited the proliferation of OA meniscal cells (Figure 1(a), right bar group). There were 55% fewer OA meniscal cells in the PC-treated wells than that in the untreated wells (P < 0.01). In contrast, PC had no effect on the proliferation of human foreskin fibroblasts (Figure 1(a), left bar group). For comparison, we also examined the effect of disodium salt of ethane-1-hydroxy-1, 1-bisphosphonic acid (EHDP), which is a bisphosphonate, together with PC on the proliferation of OA meniscal cells. As shown in Figure 1(b), both PC and EHDP inhibited the proliferation of OA meniscal cells in a dose-dependent manner. In addition, the morphology of PC-treated OA meniscal cells and untreated OA meniscal cells was similar (not shown), indicating that the reduction in cell number was not due to cellular toxicity of PC.
Figure 1.
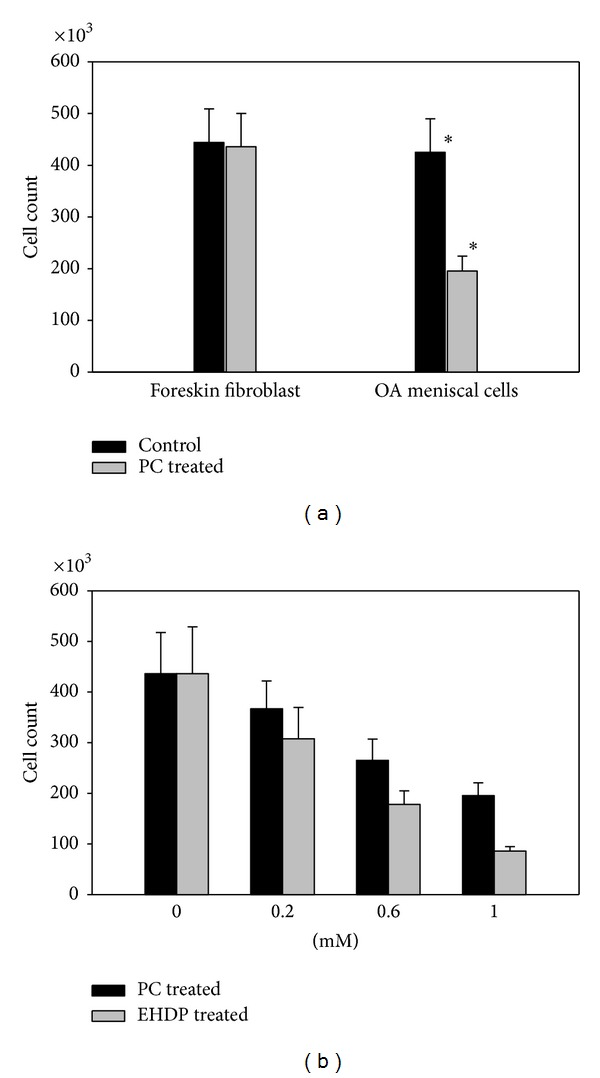
PC inhibited the proliferation of OA meniscal cells. OA meniscal cells and human foreskin fibroblast were plated in six-well cluster plates and cultured in the presence or absence of PC. (a) There were about 55% fewer OA meniscal cells in the PC-treated (1 mM PC) wells than that in the untreated wells (the right bar group; P = 0.0032). In contract, PC had no effect on the proliferation of human foreskin fibroblast (the left group bars). (b) PC and EHDP inhibited the proliferation of OA meniscal cells in a dose-dependent manner.
3.4. PC Stimulated Production of Collagens
The upregulatory effect of PC on the expression of genes classified in collagen fibril organization, including COL1A1 (1.62-fold change), COL11A1 (1.82-fold change), COL14A1 (2.06-fold change), COL5A1 (1.52-fold change), and COL5A2 (1.44-fold change), suggests that PC may stimulate the production of collagens by the OA meniscal cells. To examine this, we prepared micromasses of OA meniscal cells (triplicates) and examined the production of collagens using picrosirius red staining. The results confirmed that PC stimulated the production of collagens by OA meniscal cells. Representative images of picrosirius red staining are shown in Figure 2. As shown, the picrosirius red staining in the PC-treated micromasses (Figure 2(b)) was stronger than that in the untreated micromasses (Figure 2(a)). These micromasses were also stained with alcian blue. Results indicated that PC had no detectable effect of the production of proteoglycans (not shown).
Figure 2.
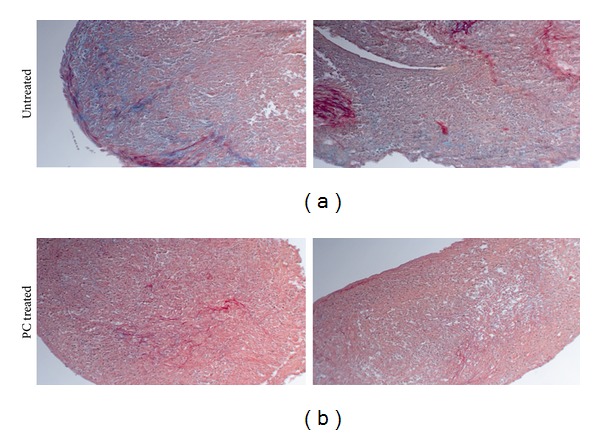
PC stimulated the production of collagens. Micromasses of OA meniscal cells were cultured in the absence of PC ((a); magnification 10x) or the presence of 1 mM PC ((b); magnification 10x) for 14 days. These micromasses were then processed and stained with picrosirius red. Note the much stronger picrosirius red staining in the PC-treated micromass than that in the untreated micromass.
3.5. PC Inhibited Meniscal Cell-Mediated Calcification
These micromasses were also stained with alizarin red for calcium deposits. Representative images of alizarin red staining are shown in Figure 3. As shown, many calcium deposits were detected in the micromasses of OA meniscal cells cultured in the absence of PC (Figure 3(a)), but no calcium deposits were detected in the micromasses cultured in the presence of PC (Figure 3(b)). PC treatment (1 mM) abolished OA meniscal cell-mediated calcium deposition in micromass culture completely.
Figure 3.
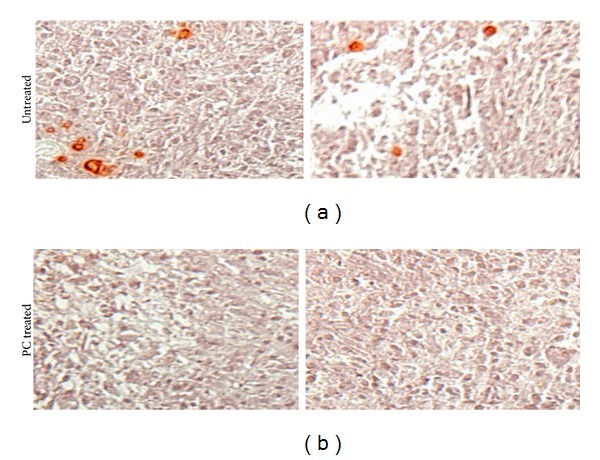
PC inhibited OA meniscal cells-mediated calcification in micromass culture. Micromasses of OA meniscal cells were stained with alizarin red. In the absence of PC, calcium deposits were detected ((a); magnification 10x). In the presence of 1 mM PC, calcium deposits were abolished ((b); magnification 10x).
The inhibitory effect of PC on OA meniscal cell-mediated calcification was also examined using monolayer culture. As shown in Figure 4, calcium deposits were detected in the monolayer culture of OA meniscal cells (Figure 4(a), but no calcium deposits were detected in the monolayer culture of OA meniscal cells cultured in the presence of PC (Figure 4(b)).
Figure 4.
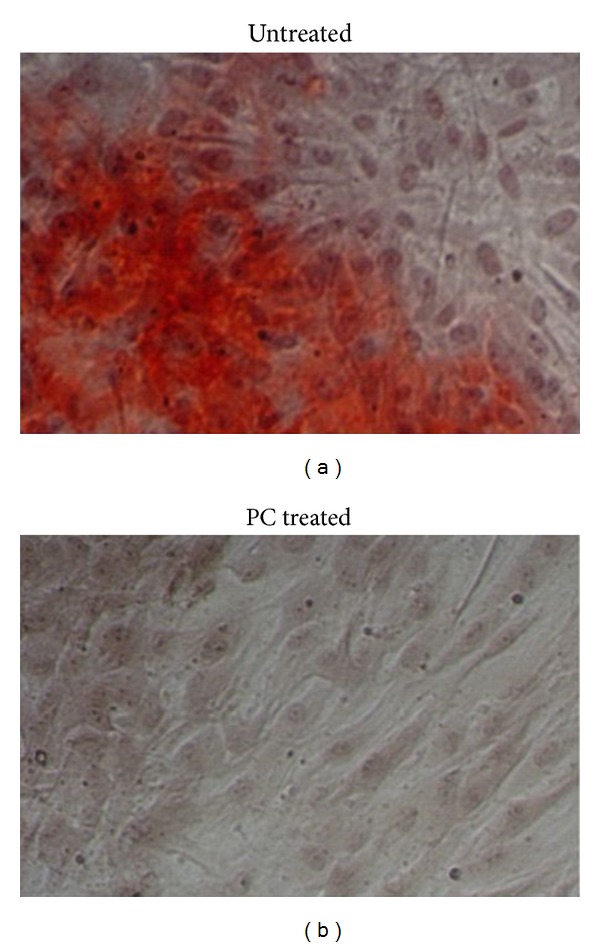
PC inhibited OA meniscal cells-mediated calcification in monolayer culture. In the absence of PC, calcium deposits were formed ((a); magnification 20x). In the presence of 1 mM PC, no calcium deposits were formed ((b); magnification 20x).
4. Discussion
Increased number and size of cell clusters are the hallmark histological feature of OA articular cartilage [25, 26]. Cell clusters are activated cells and represent an important source of pathological mediator. The number of proliferating chondrocytes increases during OA progression [26, 27]. Cell clusters are also detected in OA menisci [28]. These observations indicate that abnormal cell activation and proliferation may play a role in the development and/or progression of OA. In this study, we demonstrated that PC downregulated the expression of numerous genes associated with cell proliferation. Consistent with this regulatory effect, PC inhibited the proliferation of OA meniscal cells. Although the exact implication of this effect on the development of OA is unclear at present, these findings suggest that PC may inhibit the activation of OA meniscal cells or inhibit the formation of cell clusters.
Studies consistently reported apoptotic cells in cell clusters [29, 30]. One of the mechanisms of apoptosis is abnormal hypertrophic differentiation of chondrocytes and subsequent pathological calcification. There is abundant evidence indicating the expression of hypertrophic makers, including collagen X, osteocalcin, and MMP13, within the cell clusters [31–33]. In addition, staining of OA cartilage sections showed colocalization of calcium deposits and clusters containing apoptotic cells adjacent to the calcium deposits [34]. Mineralization of extracellular matrix surrounding cell clusters was also detected in OA menisci [28]. These observations indicate that abnormal cell activation, terminal differentiation, pathological calcification, and apoptosis may be mutually linked disease processes in OA. In this study, we demonstrated that PC inhibited the expression of numerous genes associated with cell proliferation, differentiation, and pathological calcification, including CDC7 (−4.11-fold change), CDC25C (−2.36-fold change), COL13A1 (−3.39-fold change), SATB2 (−3.13-fold-change), SMAD1 (−2.19-fold change), ENPP1 (−2.02-fold change), and ANKH (−1.60-fold change). Consistent with this regulatory activity, PC inhibited the proliferation of OA meniscal cells and abolished meniscal cells-mediated calcification. These findings suggest that PC may exert its disease-modifying activity in part by targeting the genes associated with cell proliferation, differentiation, and pathological calcification.
Several previous observations provide support for this potential mechanism. For example, studies found that COL13 was increased in OA articular cartilage and that transgenic mice overexpressing COL13 had abnormally high bone mineral density [35, 36]. The expression of ENPP1 and ANKH was elevated in OA meniscal cells, menisci, or articular cartilage [4, 34, 37]. SMAD1 induced terminal differentiation of chondrocytes and promoted calcification [38]. SATB2 stimulated osteogenic differentiation of adult stem cells [39]. These previous findings demonstrated that elevated expressions of COL13, ENPP1, ANKH, SMAD1, and SATB2 in OA cartilage or OA menisci were associated with terminal differentiation and biomineralization. The downregulatory effect of PC on these genes indicates that PC may reverse the processes of terminal differentiation and biomineralization.
Early studies demonstrated that MGP was a calcification inhibitor and that inhibition of IGFBP5 breakdown reduced articular cartilage loss in an experimental OA [40, 41]. In this study, we demonstrated that PC upregulated the expression of MGP (1.61-fold change) and IGFBP5 (1.78-fold change). This upregulatory effect of PC may lead to elevated production of MGP and IGFBP5.
FGF signaling pathway plays an important role in the regulation of osteogenesis and chondrogenesis. Studies found that FGF18 induced chondrocyte hypertrophy and mineralization [42, 43], while FGF9 inhibited terminal differentiation of calvaria-derived cells and mineralization [44, 45]. FGF7 was a potent inhibitor of phosphate transport [46]. In this study, we found that PC downregulated the expression of FGF18 (−1.73-fold change), while it upregulated the expression of FGF9 (2.38-fold change) and FGF7 (1.83-fold change). These regulatory effects indicate that PC may inhibit OA meniscal cell differentiation and pathological calcification in part by modulating the FGF signaling pathway. Consistently, studies have demonstrated that constitutive expression of thrombospondin 1 (THBS1), a protein involved in FGF signaling pathway, inhibited biomineralization and that THBS1 gene therapy suppressed the progression of arthritis in a rat model of OA [47, 48]. Another study demonstrated that the absence of signaling through FGF receptor 3 (FGFR3) leads to premature cartilage degeneration and early arthritis [49]. These previous findings indicate a key role of FGF signaling pathway in the development of healthy articular cartilage. The specific upregulatory activity of PC on THBS1 (1.68-fold change) and FGF9 (a preferred ligand for FGFR3) indicates that PC may exert its disease-modifying activity on OA in part by modulating the FGF signaling pathway.
Severe loss of collagen occurs in OA menisci [50]. Disease-modifying drugs targeting meniscal degeneration for OA therapy should inhibit meniscal collagen loss. In this study, we demonstrated that PC stimulated the production of collagen by OA meniscal cells. This finding suggests that PC is not only potentially a disease-modifying drug for OA therapy but also potentially a meniscal protective agent.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used to treat arthritis. Traditional NSAIDs inhibit both PTGS1/Cox-1 and PTGS2/Cox-2, while new generation of NSAIDs selectively inhibits PTGS2/Cox-2. In this study, we demonstrated that PC downregulated the expression of PTGS1/Cox-1 (Table 1, −2.13-fold change). Further examination indicated that PC also downregulated the expression of PTGS2/Cox-2 (Table 1, −1.33-fold change). Moreover, PC downregulated the expression of PLA2G4A (−1.61-fold change), PLAUR (−1.94-fold change), and MMP9 (−1.91-fold change), all of which have been previously implicated in the pathogenesis of arthritis [51–53]. The findings presented in this study indicate that PC is potentially an anti-inflammation agent.
Interestingly, many genes classified in the inflammatory response were upregulated by PC, including adenosine A1 receptor ADORA1 (2.48-fold change), CD24 (2.09-fold change), and BCL6 (1.98-fold change). It is worth noting that previous studies demonstrated that the activation of adenosine receptors reduced inflammation and joint destruction in a rat adjuvant-induced arthritis, CD24 repressed tissue damage-induced immune response, and that BCL6 inhibited the expression of chemokines and attenuated allergic airway inflammation [54–56]. These previous findings indicate that ADORA1, CD24, and BCL6 are potentially anti-inflammation molecules. The upregulation of these genes by PC is consistent with the notion that PC is potentially an anti-inflammation agent [57].
The genes downregulated by PC included NFKBIZ (−2.58-fold change), ADRB2 (−2.41-fold change), and CXCL6 (−2.19-fold change). Interestingly, studies demonstrated that NFKBIZ mediated IL-6 production, ADRB2 antagonists reduced the severity of arthritis, and that anti-CXCL16 antibody inhibited infiltration of inflammatory cells and arthritis [58–60]. These previous findings indicate that NFKBIZ, ADRB2, and CXCL6 are proinflammation molecules. The downregulatory effect of PC on these genes is consistent with the notion that PC is potentially an anti-inflammation agent [57].
5. Conclusions
These specific biological activities of PC are intrinsic properties of PC and are not dependent on the presence of calcium crystals. PC is potentially an anti-inflammation and meniscal protective agent. PC is not only potentially a disease-modifying drug for crystal-associated OA but also potentially a disease-modifying drug for arthritis associated with severe meniscal degeneration. The findings presented in this study provide further support for the development of PC, and/or its analogues, as disease-modifying drugs for OA therapy.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
This study is supported in part by an NC Biotech Center Grant, a Charlotte-Mecklenburg Education and Research Foundation Grant, and a Mecklenburg County Medical Society Smith Arthritis Fund Grant (to Yubo Sun). This study was performed at Carolinas Medical Center, Charlotte, NC, USA. The authors would like to thank Natalia Zinchenko for her help with histological examinations of micromasses.
References
- 1.Bennett LD, Buckland-Wright JC. Meniscal and articular cartilage changes in knee osteoarthritis: a cross-sectional double-contrast macroradiograpahic study. Rheumatology. 2002;41(8):917–923. doi: 10.1093/rheumatology/41.8.917. [DOI] [PubMed] [Google Scholar]
- 2.Hunter DJ, Zhang YQ, Tu X, et al. Change in joint space width: hyaline articular cartilage loss or alteration in meniscus? Arthritis and Rheumatism. 2006;54(8):2488–2495. doi: 10.1002/art.22016. [DOI] [PubMed] [Google Scholar]
- 3.Englund M, Guermazi A, Roemer FW, et al. Meniscal tear in knees without surgery and the development of radiographic osteoarthritis among middle-aged and elderly persons: the multicenter osteoarthritis study. Arthritis and Rheumatism. 2009;60(3):831–839. doi: 10.1002/art.24383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Y, Mauerhan DR, Honeycutt PR, et al. Analysis of meniscal degeneration and meniscal gene expression. BMC Musculoskeletal Disorders. 2010;11(article 19) doi: 10.1186/1471-2474-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Y, Mauerhan DR. Meniscal calcification, pathogenesis and implications. Current Opinion in Rheumatology. 2012;24(2):152–157. doi: 10.1097/BOR.0b013e32834e90c1. [DOI] [PubMed] [Google Scholar]
- 6.Englund M, Guermazi A, Lohmander LS. The meniscus in knee osteoarthritis. Rheumatic Disease Clinics of North America. 2009;35(3):579–590. doi: 10.1016/j.rdc.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Mauerhan DR, Honeycutt PR, et al. Calcium deposition in osteoarthritic meniscus and meniscal cell culture. Arthritis Research and Therapy. 2010;12(2, article R56) doi: 10.1186/ar2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pauli C, Grogan SP, Patil S, et al. Macroscopic and histopathologic analysis of human knee menisci in aging and osteoarthritis. Osteoarthritis and Cartilage. 2011;19(9):1132–1141. doi: 10.1016/j.joca.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy GM, Mitchell PG, Cheung HS. The mitogenic response to stimulation with basic calcium phosphate crystals is accompanied by induction and secretion of collagenase in human fibroblasts. Arthritis and Rheumatism. 1991;34(8):1021–1030. doi: 10.1002/art.1780340812. [DOI] [PubMed] [Google Scholar]
- 10.Sun Y, Zeng XR, Wenger L, Cheung HS. Basic calcium phosphate crystals stimulate the endocytotic activity of cells—inhibition by anti-calcification agents. Biochemical and Biophysical Research Communications. 2003;312(4):1053–1059. doi: 10.1016/j.bbrc.2003.11.048. [DOI] [PubMed] [Google Scholar]
- 11.Morgan MP, Whelan LC, Sallis JD, McCarthy CJ, Fitzgerald DJ, McCarthy GM. Basic calcium phosphate crystal-induced prostaglandin E2 production in human fibroblasts: role of cyclooxygenase 1, cyclooxygenase 2, and interleukin-1β . Arthritis and Rheumatism. 2004;50(5):1642–1649. doi: 10.1002/art.20223. [DOI] [PubMed] [Google Scholar]
- 12.Tew WP, Malis CD, Howard JE, Lehninger AL. Phosphocitrate inhibits mitochondrial and cytosolic accumulation of calcium in kidney cells in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(9):5528–5532. doi: 10.1073/pnas.78.9.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moro L, Stagni N, Luxich E, Sallis JD, De Bernard B. Evidence in vitro for an enzymatic synthesis of phosphocitrate. Biochemical and Biophysical Research Communications. 1990;170(1):251–258. doi: 10.1016/0006-291x(90)91267-v. [DOI] [PubMed] [Google Scholar]
- 14.Romanello M, Michalsky M, Stagni N, Moro L, Favretto D, Traldi P. High-performance liquid chromatographic and mass spectrometric identification of phosphocitrate synthesized by human kidney homogenate. Journal of Chromatography. 1993;616(2):167–173. doi: 10.1016/0378-4347(93)80383-f. [DOI] [PubMed] [Google Scholar]
- 15.Williams G, Sallis JD. Structure-activity relationship of inhibitors of hydroxyapatite formation. Biochemical Journal. 1979;184(1):181–184. doi: 10.1042/bj1840181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams G, Sallis JD. Structural factors influencing the ability of compounds to inhibit hydroxyapatite formation. Calcified Tissue International. 1982;34(2):169–177. doi: 10.1007/BF02411229. [DOI] [PubMed] [Google Scholar]
- 17.Shankar R, Crowden S, Sallis JD. Phosphocitrate and its analogue N-sulpho-2-amino tricarballylate inhibit aortic calcification. Atherosclerosis. 1984;52(2):191–198. doi: 10.1016/0021-9150(84)90117-5. [DOI] [PubMed] [Google Scholar]
- 18.Nair D, Misra RP, Sallis JD, Cheung HS. Phosphocitrate inhibits a basic calcium phosphate and calcium pyrophosphate dihydrate crystal-induced mitogen-activated protein kinase cascade signal transduction pathway. Journal of Biological Chemistry. 1997;272(30):18920–18925. doi: 10.1074/jbc.272.30.18920. [DOI] [PubMed] [Google Scholar]
- 19.Cheung HS, Sallis JD, Struve JA. Specific inhibition of basic calcium phosphate and calcium pyrophosphate crystal-induction of metalloproteinase synthesis by phosphocitrate. Biochimica et Biophysica Acta. 1996;1315(2):105–111. doi: 10.1016/0925-4439(95)00106-9. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Reuben P, Wenger L, Sallis JD, Demadis KD, Cheung HS. Inhibition of calcium phosphate-DNA coprecipitates induced cell death by phosphocitrates. Frontiers in Bioscience. 2005;10:803–808. doi: 10.2741/1574. [DOI] [PubMed] [Google Scholar]
- 21.Cheung HS, Sallis JD, Demadis KD, Wierzbicki A. Phosphocitrate blocks calcification-induced articular joint degeneration in a guinea pig model. Arthritis and Rheumatism. 2006;54(8):2452–2461. doi: 10.1002/art.22017. [DOI] [PubMed] [Google Scholar]
- 22.Ding M, Danielsen CC, Hvid I. The effects of bone remodeling inhibition by alendronate on three-dimensional microarchitecture of subchondral bone tissues in guinea pig primary osteoarthrosis. Calcified Tissue International. 2008;82(1):77–86. doi: 10.1007/s00223-007-9093-2. [DOI] [PubMed] [Google Scholar]
- 23.Walton M. The effects of long-term administration of ethane-1-hydroxy-1, 1-diphosphonate on osteoarthrosis and heterotopic ossification in the mouse knee joint. Clinical Orthopaedics and Related Research. 1981;155:218–223. [PubMed] [Google Scholar]
- 24.Turhanen PA, Demadis KD, Peräniemi S, Vepsäläinen JJ. A novel strategy for the preparation of naturally occurring phosphocitrate and its partially esterified derivatives. Journal of Organic Chemistry. 2007;72(4):1468–1471. doi: 10.1021/jo061709c. [DOI] [PubMed] [Google Scholar]
- 25.Mankin HJ, Lippiello L. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. Journal of Bone and Joint Surgery A. 1970;52(3):424–434. [PubMed] [Google Scholar]
- 26.Lotz MK, Otsuki S, Grogan SP, Sah R, Terkeltaub R, D’Lima D. Cartilage cell clusters. Arthritis and Rheumatism. 2010;62(8):2206–2218. doi: 10.1002/art.27528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfander D, Körtje D, Weseloh G, Swoboda B. Cell proliferation in osteoarthritic human cartilage. Zeitschrift fur Orthopadie und Ihre Grenzgebiete. 2001;139(5):375–381. doi: 10.1055/s-2001-17977. [DOI] [PubMed] [Google Scholar]
- 28.Hellio Le Graverand MP, Sciore P, Eggerer J, et al. Formation and phenotype of cell clusters in osteoarthritic meniscus. Arthritis & Rheumatism. 2001;44(8):1808–1818. doi: 10.1002/1529-0131(200108)44:8<1808::AID-ART318>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto S, Ochs RL, Komiya S, Lotz M. Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis & Rheumatism. 1998;41(9):1632–1638. doi: 10.1002/1529-0131(199809)41:9<1632::AID-ART14>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.Blanco FJ, Guitian R, Vazquez-Martul E, de Toro FJ, Galdo F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis & Rheumatism. 1998;41(2):284–289. doi: 10.1002/1529-0131(199802)41:2<284::AID-ART12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 31.Von der Mark K, Kirsch T, Nerlich A, et al. Type X collagen synthesis in human osteoarthritic cartilage: indication of chondrocyte hypertrophy. Arthritis and Rheumatism. 1992;35(7):806–811. doi: 10.1002/art.1780350715. [DOI] [PubMed] [Google Scholar]
- 32.Pullig O, Weseloh G, Ronneberger DL, Käkönen SM, Swoboda B. Chondrocyte differentiation in human osteoarthritis: expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcified Tissue International. 2000;67(3):230–240. doi: 10.1007/s002230001108. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Manner PA, Horner A, Shum L, Tuan RS, Nuckolls GH. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis and Cartilage. 2004;12(12):963–973. doi: 10.1016/j.joca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Johnson K, Hashimoto S, Lotz M, Pritzker K, Goding J, Terkeltaub R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis & Rheumatism. 2001;44(5):1071–1081. doi: 10.1002/1529-0131(200105)44:5<1071::AID-ANR187>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Karlsson C, Dehne T, Lindahl A, et al. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis and Cartilage. 2010;18(4):581–592. doi: 10.1016/j.joca.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Ylönen R, Kyrönlahti T, Sund M, et al. Type XIII collagen strongly affects bone formation in transgenic mice. Journal of Bone and Mineral Research. 2005;20(8):1381–1393. doi: 10.1359/JBMR.050319. [DOI] [PubMed] [Google Scholar]
- 37.Ijiri K, Zerbini LF, Peng H, et al. Differential expression of GADD45β in normal and osteoarthritic cartilage: potential role in homeostasis of articular chondrocytes. Arthritis and Rheumatism. 2008;58(7):2075–2087. doi: 10.1002/art.23504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hellingman CA, Davidson ENB, Koevoet W, et al. Smad signaling determines chondrogenic differentiation of bone-marrow-derived mesenchymal stem cells: inhibition of Smad1/5/8P prevents terminal differentiation and calcification. Tissue Engineering A. 2011;17(7-8):1157–1167. doi: 10.1089/ten.TEA.2010.0043. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Tu Q, Grosschedl R, et al. Roles of SATB2 in osteogenic differentiation and bone regeneration. Tissue Engineering A. 2011;17(13-14):1767–1776. doi: 10.1089/ten.tea.2010.0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oyoung J, Liao Y, Xiao Y, et al. Matrix gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. Journal of the American Chemical Society. 2011;133(45):18406–18412. doi: 10.1021/ja207628k. [DOI] [PubMed] [Google Scholar]
- 41.Clemmons DR, Busby WH, Jr., Garmong A, et al. Inhibition of insulin-like growth factor binding protein 5 proteolysis in articular cartilage and joint fluid results in enhanced concentrations of insulin-like growth factor 1 and is associated with improved osteoarthritis. Arthritis and Rheumatism. 2002;46(3):694–703. doi: 10.1002/art.10222. [DOI] [PubMed] [Google Scholar]
- 42.Liu Z, Lavine KJ, Hung IH, Ornitz DM. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Developmental Biology. 2007;302(1):80–91. doi: 10.1016/j.ydbio.2006.08.071. [DOI] [PubMed] [Google Scholar]
- 43.Zalutskaya AA, Cox MK, Demay MB. Phosphate regulates embryonic endochondral bone development. Journal of Cellular Biochemistry. 2009;108(3):668–674. doi: 10.1002/jcb.22302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weksler NB, Lunstrum GP, Reid ES, Horton WA. Differential effects of fibroblast growth factor (FGF) 9 and FGF2 on proliferation, differentiation and terminal differentiation of chondrocytic cells in vitro. Biochemical Journal. 1999;342(part 3):677–682. [PMC free article] [PubMed] [Google Scholar]
- 45.Fakhry A, Ratisoontorn C, Vedhachalam C, et al. Effects of FGF-2/-9 in calvarial bone cell cultures: differentiation stage-dependent mitogenic effect, inverse regulation of BMP-2 and noggin, and enhancement of osteogenic potential. Bone. 2005;36(2):254–266. doi: 10.1016/j.bone.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. Journal of Clinical Endocrinology and Metabolism. 2005;90(2):1012–1020. doi: 10.1210/jc.2004-0357. [DOI] [PubMed] [Google Scholar]
- 47.Ueno A, Miwa Y, Miyoshi K, et al. Constitutive expression of thrombospondin 1 in MC3T3-E1 steoblastic cells inhibits mineralization. Journal of Cellular Physiology. 2006;209(2):322–332. doi: 10.1002/jcp.20735. [DOI] [PubMed] [Google Scholar]
- 48.Hsieh JL, Shen PC, Shiau AL, et al. Intraarticular gene transfer of thrombospondin-1 suppresses the disease progression of experimental osteoarthritis. Journal of Orthopaedic Research. 2010;28(10):1300–1306. doi: 10.1002/jor.21134. [DOI] [PubMed] [Google Scholar]
- 49.Valverde-Franco G, Binette JS, Li W, et al. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Human Molecular Genetics. 2006;15(11):1783–1792. doi: 10.1093/hmg/ddl100. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y, Mauerhan DR, Kneisl JS, et al. Histological examination of collagen and proteoglycan changes in osteoarthritic menisci. Open Rheumatology Journal. 2012;6:24–32. doi: 10.2174/1874312901206010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leistad L, Feuerherm AJ, Faxvaag A, Johansen B. Multiple phospholipase A2 enzymes participate in the inflammatory process in osteoarthritic cartilage. Scandinavian Journal of Rheumatology. 2011;40(4):308–316. doi: 10.3109/03009742.2010.547872. [DOI] [PubMed] [Google Scholar]
- 52.Tai N, Kuwabara K, Kobayashi M, et al. Cytosolic phospholipase A2 alpha inhibitor, pyrroxyphene, displays anti-arthritic and anti-bone destructive action in a murine arthritis model. Inflammation Research. 2010;59(1):53–62. doi: 10.1007/s00011-009-0069-8. [DOI] [PubMed] [Google Scholar]
- 53.Chu SC, Yang SF, Lue KH, Hsieh YS, Hsiao TY, Lu KH. Urokinase-type plasminogen activator, receptor, and inhibitor correlating with gelatinase-B (MMP-9) contribute to inflammation in gouty arthritis of the knee. Journal of Rheumatology. 2006;33(2):311–317. [PubMed] [Google Scholar]
- 54.Boyle DL, Moore J, Yang L, Sorkin LS, Firestein GS. Spinal adenosine receptor activation inhibits inflammation and joint destruction in rat adjuvant-induced arthritis. Arthritis and Rheumatism. 2002;46(11):3076–3082. doi: 10.1002/art.10595. [DOI] [PubMed] [Google Scholar]
- 55.Chen GY, Tang J, Zheng P, Liu Y. CD24 and siglec-10 selectively repress tissue damage—induced immune responses. Science. 2009;323(5922):1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seto T, Yoshitake M, Ogasawara T, et al. Bcl6 in pulmonary epithelium coordinately controls the expression of the CC-type chemokine genes and attenuates allergic airway inflammation. Clinical and Experimental Allergy. 2011;41(11):1568–1578. doi: 10.1111/j.1365-2222.2011.03836.x. [DOI] [PubMed] [Google Scholar]
- 57.Sun Y, Mauerhan DR, Franklin AM, Norton J, Hanley EN, Jr., Gruber HE. Phosphocitrate is potentially a disease-modifying drug for noncrystal-associated osteoarthritis. BioMed Research International. 2013;2013:11 pages. doi: 10.1155/2013/326267.326267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seshadri S, Kannan Y, Mitra S, Parker-Barnes J, Wewers MD. MAIL regulates human monocyte IL-6 production. Journal of Immunology. 2009;183(8):5358–5368. doi: 10.4049/jimmunol.0802736. [DOI] [PubMed] [Google Scholar]
- 59.Levine JD, Coderre TJ, Helms C, Basbaum AI. β2-Adrenergic mechanisms in experimental arthritis. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(12):4553–4556. doi: 10.1073/pnas.85.12.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis and Rheumatism. 2005;52(10):3004–3014. doi: 10.1002/art.21301. [DOI] [PubMed] [Google Scholar]