

Abstract
Insertional translocations (ITs) are rare events that require at least three breaks in the chromosomes involved and thus qualify as complex chromosomal rearrangements (CCR). In the current study, we identified 40 ITs from approximately 18,000 clinical cases (1:500) using array-comparative genomic hybridization (aCGH) in conjunction with fluorescence in situ hybridization (FISH) confirmation of the aCGH findings, and parental follow-up studies. Both submicroscopic and microscopically visible IT events were detected. They were divided into three major categories: (1) simple intrachromosomal and interchromosomal IT resulting in pure segmental trisomy, (2) complex IT involving more than one abnormality, (3) deletion inherited from a parent with a balanced IT resulting in pure segmental monosomy. Of the cases in which follow-up parental studies were available, over half showed inheritance from an apparently unaffected parent carrying the same unbalanced rearrangement detected in the propositi, thus decreasing the likelihood that these IT events are clinically relevant. Nevertheless, we identified six cases in which small submicroscopic events were detected involving known disease-associated genes/genomic segments and are likely to be pathogenic. We recommend that copy number gains detected by clinical aCGH analysis should be confirmed using FISH analysis whenever possible in order to determine the physical location of the duplicated segment. We hypothesize that the increased use of aCGH in the clinic will demonstrate that IT occurs more frequently than previously considered but can identify genomic rearrangements with unclear clinical significance.
Keywords: array-CGH, genomic rearrangement, chromosome rearrangement, insertion, submicroscopic, FISH, segmental aneusomy
INTRODUCTION
Segmental aneusomies generally distribute into six categories; deletion, duplication (direct or inverted), triplication, terminal translocation, insertional translocation (IT) (intrachromosomal and interchromosomal), and other rare complex rearrangements. Rearrangements involving multiple breaks are hypothesized to occur less frequently. IT events require at least three breakage events and therefore are likely to be rare compared to other rearrangements that require only one (e.g., deletion) or two (e.g., duplication, terminal translocation) breaks. A review of microscopically visible IT estimated the incidence to be 1:80,000 live births [Van Hemel and Eussen, 2000]. However, the occurrence of submicroscopic IT is just now being elucidated with the increased genomic resolution afforded by clinical implementation of array-comparative genomic hybridization (aCGH) [de Ravel et al., 2005; Shinawi et al., 2008; Bi et al., 2009; Carvalho et al., 2009]. Interestingly, the increased detection of IT events has reinforced the need to confirm copy number gains using methodologies such as fluorescence in situ hybridization (FISH) to determine the genomic position or location of the additional material.
IT events can affect gene function and consequently result in an abnormal phenotype in several ways: (1) The duplicated segment may contain one or more genes resulting in increased gene expression. (2) The duplication can disrupt a gene and result in either loss or gain of function. (3) Genes located at the insertion site may be deleted, duplicated, or disrupted. (4) Deletion or duplication in the flanking region of a gene may result in abnormal gene expression due to position effects.
We have identified 40 cases with IT wherein a copy number variation (CNV) was initially detected by aCGH analysis, and the insertion event was subsequently uncovered during confirmatory FISH or parental studies. The cases presented here include both microscopically visible and submicroscopic events. The diverse groups of cases include: (1) simple intrachromosomal or inter-chromosomal IT, (2) complex IT involving more than one abnormality, and (3) deletion inherited from a parent with a balanced IT.
Our experience with IT cases detected using aCGH analysis suggests that these types of rearrangements occur up to 160X more frequently than previously recognized (1:500 versus 1:80,000). One would presume that a microscopically visible genomic imbalance can result in an abnormal phenotype. This hypothesis is supported by reports of inheritance of IT from phenotypically abnormal parents thus allowing clear clinical correlation of the finding [Arens et al., 2004; Kolomietz et al., 2006; Liang et al., 2006].
However, the higher resolution achieved using aCGH analysis relative to karyotype analysis makes interpreting the clinical significance of smaller submicroscopic IT challenging. When submicroscopic IT segments involve dosage-sensitive genes, the clinical relevance of the rearrangement is more obvious [Bi et al., 2009]. Clinical correlation becomes more challenging when the unbalanced submicroscopic IT event is inherited from an apparently phenotypically “normal” parent. We present cases in which the parent and child share the same genomic aberration but the parent has no clear phenotype. We discuss the challenges to genetic counseling and clinical interpretation that these CNVs might present.
SUBJECTS AND METHODS
Patient Samples
All patient samples were referred to the Medical Genetics Laboratories at Baylor College of Medicine for clinical aCGH analysis. We identified a total of 40 patients with IT in approximately 18,000 unrelated patients referred from July 2005 to January 2009.
DNA was extracted from whole blood using the Puregene DNA extraction kit (Gentra, Minneapolis, MN) according to the manufacturer’s instructions. For additional studies, we obtained informed consent approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine.
BAC Microarrays
The aCGH analysis on CMA V5 BAC was conducted on a clinically available microarray containing: 853 BAC and PAC clones designed to interrogate genomic regions of 75 known genomic disorders, all 41 subtelomeric regions, and 43 pericentromeric regions; manufactured by Baylor College of Medicine [Cheung et al., 2005; Lu et al., 2007].
CMA V6 BAC microarray (Baylor College of Medicine) included 1,472 BAC and PAC clones covering approximately 10 Mb of the 41 subtelomeric regions, 43 pericentromeric regions as well as ~150 genomic disorders with minimum backbone coverage of every chromosome at the 650-band level of cytogenetic resolution [Ou et al., 2008a].
Clinical Oligonucleotide Microarrays
CMA V6 OLIGO microarrays are custom-targeted oligonucleotide arrays manufactured by Agilent Technologies (Santa Clara, CA) using approximately 44,000 oligonucleotides to emulate the CMA V6 BAC arrays [Ou et al., 2008a].
CMA V7 OLIGO microarrays (Agilent Technologies) contain approximately 105,000 oligonucleotides covering the human genome at an average resolution of 30 kb with increased coverage at known disease loci. Probes were selected to target over 270 genetic syndromes, 41 unique subtelomeric regions, all 43 unique peri-centromeric regions, and the mitochondrial genome. Some cases that were initially evaluated using the BAC microarrays were reevaluated on CMA V7 OLIGO arrays to further delineate the size of the CNV.
High-Resolution Microarray
The high-resolution Human Genome CGH Microarray Kit 244A (Agilent Technologies) contains approximately 244,000 oligonucleotides with an 8.9 kb overall median probe spacing and a 7.4 kb probe spacing in RefSeq genes. This array was used with consent to reexamine DNA from the patients to further refine the identified genomic gains and losses.
aCGH Analysis
The procedures for probe labeling and hybridization of our BAC arrays were reported previously [Cheung et al., 2005]. The procedures for DNA digestion, labeling, and hybridization for the oligonucleotide arrays were performed according to the manufacturer’s instructions with some modifications [Ou et al., 2008a]. The slides were scanned into image files using a GenePix Model 4000B microarray scanner (Molecular Devices, Sunnyvale, CA) or an Agilent G2565 laser scanner. BAC array data were quantified as previously described [Cheung et al., 2005; Lu et al., 2007]. Microarray image files of oligonucleotide arrays were quantified using Agilent Feature extraction software (v9.0) and imported into the Agilent CGH Analytics program (244K microarrays) or into our in-house analysis package for copy number analysis (CMA microarrays), as described previously [Ou et al., 2008a].
Chromosome and FISH Analyses
Confirmatory chromosome and FISH analyses were performed on peripheral blood lymphocytes using standard procedures following detection of copy number changes observed by aCGH analysis. FISH analyses with cloned fragments of DNA (typically BAC clones) were performed using standard procedures. Briefly, the BAC clone of interest was grown in TB media with 20 μg/ml chloramphenicol. DNA was extracted from the BAC clones (Eppendorf Plasmid Mini Prep kit, Hamburg, Germany) and directly labeled with fluorochrome conjugated-dUTP by nick translation (Vysis, Downer Grove, IL) according to the manufacturer’s instructions. Digital FISH images were captured using a Power Macintosh G3 System and MacProbe version 4.4 software (Applied Imaging, San Jose, CA).
RESULTS
We identified 40 cases with IT out of approximately 18,000 patients (~1 in 500) referred for clinical aCGH analysis using CMA V5, V6, or V7. Although the CNVs were originally detected by aCGH analysis, the IT events were identified during FISH confirmation of the aCGH results or follow-up parental studies. The results from the parental studies are presented when available.
The 40 IT cases fall into three groups: simple IT (Table I), complex IT (Table II), and deletions resulting from parental balanced IT (Table III). All the tables include patient information (gender, age at the time of testing, indication for referral, and previous chromosome studies) and the results of the clinical testing performed at BCM, including CMA version(s), the location of the inserted fragment with genomic coordinates, the insertion location, FISH clones used for confirmation, the size of the inserted fragment, and parental origin when available. The size of the IT fragments ranged from 0.097 to 30 Mb. There was no apparent preferential involvement of any individual chromosome (Fig. 1). Interestingly, four insertions occurred into chromosome 3p13.
TABLE I.
Simple Insertional Translocation
| Case | Sex | Age | Indication | Karyotype | CMA versions | CNV fragment IT site | CNV in size (Mb) | Confirmation | Parental origin |
|---|---|---|---|---|---|---|---|---|---|
| Intrachromosomal IT | |||||||||
| 1 | F | 1 m | CHD | NPb | V7.4 OLIGO | 7p15.1p14.1 (28559836–38185226)×3 7q31.32 |
9.62–10.16 | der(7)ins(7) (q31.32p14.1p15.1) (RP11-100A8++) | Pat (U) |
| 2 | M | 5 | Speech delay Self-injurious behavior | 46,XY | V7.2 OLIGO | Xq13.1 (71278278–71488432)×2 Xp21 |
0.21–0.284 | ish der(X)ins(X) (p21q13.1q13.1) (RP11-1145H7++) | Mat (U) |
| Interchromosomal IT | |||||||||
| 3 | M | 3 | NP | NPb | V6, V.7.2 OLIGO | 1p21.3p21.1 (93623096–104408284)×3 3q23 |
10.78–10.90 | ish der(3)ins(3;1) (q23;p21.1p21.3) (RP11-465K1+) | NP |
| 4 | M | 2½ | Seizures, DD | NPb | V6 OLIGO | 1q32.1q32.2(192363276–209052559)×3 5q14 |
16.69 | ish der(5)ins(5;1) (q14.2;q32.1q32.2) (RP11-279E18+) | NP |
| 5 | M | 7 m | MCA | 46,XYb | V6, V7.2 OLIGO | 2q23.2q31.1 (147651413–177914914)×3 6q16.2 |
30.26–30.33 | ish der(6)ins(6;2) (q16.2;q23.3q31.1) (RP11-2I8+) | Mat (B) |
| 6 | M | 2 | Abn karyotype | 46,XY,der(2)t (2:11) | V7.1 OLIGO | 11q22.1q24.2 (100029790–126660054)×3 2p21 |
26.62–26.71 | ish der(2)ins(2;11) (p21?;q22.1q24.2) (RP11-21G19+) | Mat (B) |
| 7 | M | 7 | Family history of MR | NPb | V7.4 OLIGO | 5q13.2q14.2 (70831688–81775089)×3 6p12.2 |
10.94–13.20 | ish der(6)ins(6;5) (p12.2;q13.2q14) (RP11-107N7+, RP11-428C6+) | NP |
| 8 | M | 1 | DD | 46,XY,der(1) ins(1;?) (q32.3;?) | V6 OLIGO | 20q12q13.31 (38195616–58827827)×3 1q32.3 |
20.6 | ish der(1)ins(1;20) (q32.3;q12q13.13) (RP11-298O6+) | Mat (B) |
| 9 | F | 7 | MCA | NP | V7.1 OLIGO | 1q21.1 (145085612–145518428)×3 7q35 |
0.433–4.048 | ish der(7)ins(7;1) (q35?;q21.1q21.1) (RP11-337C18+, RP11-533N14+) | Mat (U) |
| 10 | M | 4 | Mild DD/MR, ASD | 46,XY | V6.5, V7.2 OLIGO | 2q37.3 (240617514–240721504)×3 13q34 |
1.236 | ish der(13)ins(13;2) (q24;q27.3q27.3) (RP11-463B12+) | NAP |
| 11 | M | 6 | DF, DD, hypotonia, craniosynostosis | 46,XY | V6, V7.2 OLIGO | 4q27 (121334064–121647533)×3 9p12 |
0.313–0.408 | ish der(9)ins(9;4)(p12;q27q27) (RP11-679C8+) | NP |
| 12 | M | 3 | Severe DD MR | NP | V6.5 OLIGO | 10p15.3(1375145–1450405)×3 8q? |
0.075–0.99 | ish der(8)ins(8;10) (q?;p15.3p15.3) (RP11-398B16+) | Mat (U) |
| 13a | F | 8½ | DD | NP | V5 BAC | 17p13.3 (1098132–1449087)×3 13q12 |
0.35 | ish der(13)ins(13;17) (q12;p13.3p13.3) (RP11-818O24+) | NAP |
| 14 | M | 5 | DD, ASD | NP | V6.5 OLIGO | 22q13.33 (49346185–49512543)×3 11p13 |
0.166 | ish der(11)ins(11;22)(p?13;q13.33q13.33)(n66c4+) | NAP |
| 15a | M | 3½ | DD | NP | V5 BAC | Xq28(152508915–152994739)×3 Yp11.3 |
0.519 | ish der(Y)ins(Y;X) (p11.3;q28q28) (RP11-244I10+) | dn |
| 16 | M | 4½ | ASD | 46,XY | V6.1 BAC, V7.2 OLIGO | 15q26.3(99714596–99805974)×3 Xp21 |
0.091–0.284 | ish der(X)ins(X;15) (p21;q26.3q26.3) (RP11-497M17+) | NP |
| 17 | M | 1 | Radical hypoplasia | NP | V7.2 OLIGO | 11q14.1 (177825189–78225427)×3 Yq12 |
0.4–0.488 | ish der(Y)ins(Y;11) (q12;q14.1q14.1) (RP11-109P16+) | Pat (U) |
| 18 | M | 1 m | DF | 46,XY | V6, V7.2 OLIGO | 8q23.1(109099731–109414979)×3 Yp11.2 |
0.315–0.370 | ish der(Y)ins(Y;8) (p11.2;q23.1q23.1) (RP11-80D8+) | NP |
| IT involving similar chromosomal segments | |||||||||
| 19 | F | 4 | Mod. DD/MR | 46,XX | V5 BAC, V7.2 OLIGO | 6p25.3(468355–593492)×3 3p13 |
0.125–0.720 | ish der(3)ins(6;3) (p25.3;p13p13) (RP1-20B11+) | Mat (U) |
| 20 | M | 8.5 | Learning disability | 46,XY | V6, V7.2 OLIGO | 6p25.3(468355–720370)×3 3p13 |
0.252–0.720 | ish der(3)ins(6;3) (p25.3;p13p13) (RP1-20B11+) | NP |
| 21 | M | 2 m | DF | 46,XY | V5 BAC, V7.2 OLIGO | 6p25.3(468355–582886)×3 3p13 |
0.115–0.593 | ish der(3)ins(6;3) (p25.3;p13p13) (RP1-20B11+) | Mat (U) |
| 22 | F | 18 | Cleft lip/palate, hydronephrosis | NP | V.7 OLIGO | 6p25.3(244440–593492)×3 3p13 |
0.349–0.701 | ish der(3)ins(6;3) (p25.3;p13p13) (RP1-20B11+) | Mat (U) |
| IT to the short arm acrocentric chromosomes | |||||||||
| 23 | M | 6½ | Abn karyotype | der(22) | V.7.2 OLIGO | 10p15.3p11.23 (214559–30674526)×3 22p12 |
30.46–30.87 | der(22)ins(22;10) (p12;p11.23p15.3) | NP |
| 24 | M | 9 | NP | NP | V6.5 OLIGO | 5q35.1q35.3
(172041555–180601312)×3 22p11.2 |
8.56–9.751 | ish der(22)ins(22;5) (p11.2;q35.1q35.3) (RP11-99N22+) | NP |
| 25a | M | 5 m | FTT | 46,XY | V7.2 OLIGO | 17q25.1.q25.3 (70570936–78638511)×3 22p12 |
8.068–8.124 | ish der(22)ins(22;17)(p12;q25.1q25.3) (RP11-16C1+, RP11-936I5+) | NAP |
| 26 | M | 9 m | DD and FTT | NP | V7.1 OLIGO | 11p12(36521870–3762764)×3 22p12 |
1.105–1.181 | ish der(22)ins(22;11) (p12?;p12p12) (RP11-65C9+) | Mat (U) |
| 27 | M | 3 m | MCA | 46,XY,add(15) (p11.2) | V5 BAC, V6.3 OLIGO | 15q25.1q25.3(79832451–100070968)×3 15p12 |
20.239 | ish der(15)ins(15)(p12q25.1q25.3) (RP11-121E15++, RP11-437B10++) | NP |
| 28 | M | 4 m | NP | NP | V6, V7.2 OLIGO | 18q22.2(65659843–66052052)×3 15p12 |
0.392–0.453 | ish der(15)ins(15;18) (p12;q22.2q22.2) (RP11-704G7+) | NP |
NP, not provided; CHD, congenital heart defect; m, months; ASD, autistic spectrum disorder; Mat, maternal; DD, developmental delay; Pat, paternal; MCA, multiple congenital anomalies; U, unbalanced in parent; Abn, abnormal; B, balanced in parent; MR, mental retardation; NAP, mother’s test is normal, father not tested; DF, dysmorphic features; dn, de novo; FTT, failure to thrive.
Published case report.
Retrospective analysis of G-banded chromosomes determined that the abnormality was microscopically visible.
TABLE II.
Complex Insertional Translocation
| Case | Sex | Age | Indication | Karyotype | CMA versions | CNV fragment IT site | CNV size (Mb) | Confirmation | Parental origin |
|---|---|---|---|---|---|---|---|---|---|
| 29 | M | 33 | Moderate DD/MR, DF | 46,XY,der(1) (p36.1) | V6 BAC, V7.2 OLIGO | 1q31.1q31.2
(187916046–190322755)×3 1p36.1 1q32.1(203196581–204185740)×3 1p36.1? |
2.407–2.846 0.989–1.018 |
ish der(1)ins(1)(p36q31.1q31.1) (RP11-445K1+) ish der(1)ins(1)(p36q32.1q32.1) (RP11-379J10+) |
NP |
| 30 | M | 1½ | DF, food allergy | 46,XY,dup(9) (q21.1q22.2)dn | V6 OLIGO | 9q31.3q33.1 (112610768–120263701)×3 9q13 |
7.65–15.48 | ish der(9)ins(9)(q13q31.3q33.1) (RP11-35N6++, RP11-18B16+++, RP11-451E16++) | dn |
| 31 | F | 1 m | DF | NPb | V7.1 OLIGO | 7p22.3p22.2 (1109663–3956207)×3 8q12.1q12.3 (58038717–65409558)×1 |
2.847–2.904 7.371–7.436 |
ish der(8)del(8)(q12.1q12.3)
(RP11-33I11−) ins(8;7)(q12.1;p22.2p22.3) (RP11-6A1+) |
Mat (B) |
| 32a | M | 2 m | MCA | 46,XY,der (13)pat | V5 BAC | 14q22.3q23.3
(55774576–67571522)×3 13q21(62339014–66728435)×1 |
11.79 4.38 |
der(13)ins(13;14) (q21.3;q22.3q23.3) del(13)(q21.31q21.32) |
Pat (U) |
| 33 | M | 8 | DD/MR, DF, MCA | 46,XYb | V6 BAC, V7.2 OLIGO | Xq28(153176364–154533675)×2 6q25(151157788–159568296)×1 |
1.357–1.606 8.41–8.61 |
ish der(6)del(6)(q25.3q25.3)
(RP11-115G17−) ins(6;X)(q25.3;q28q28)(RP11–211L10+) |
dn |
| 34 | F | 1 m | CHD | 46,XX | V7.2 OLIGO | 5p15.33(774848–1306298)×3 Xq28(153009381–153106702)×3 |
0.531–0.640 0.097 |
ish der(X)ins(X;5)(q28;p15.3p15.3) (RP11-43F13+) | NP |
| 35 | M | 1½ | NP | NP | V6, 7.2 OLIGO | 9q34.3(138497871–138575754)×3 Xp22.3(RP13-167H21)×2 |
0.078–0.080 0.1 |
ish der(X)ins(X;9)(p22.3;q34.3q34.3) (RP11-611D20+)dup(X)(p22.3p22.3) (RP13-167H21+) | Mat (U) |
| 36 | F | 12 | Severe DD/MR, DF | 46,XX | V5 BAC 244K | 16p13.3(486776–660154)×3 Xp22.3(RP13-391G2)×3 |
0.173 0.1 |
ish der(X)ins(X;16)(p22.3;p13.3p13.3) (RP11-64L12+)dup(X) (p22.3p22.3)(RP13-391G2+) | dn |
NP, not provided; m, months; U, unbalanced in parent; DD, developmental delay; B, balanced in parent; MR, mental retardation; dn, de novo; DF, dysmorphic features; Mat, maternal; MCA, multiple congenital anomalies; Pat, paternal; CHD, congenital heart defect.
Published case report.
Karyotype analysis was reportedly normal; retrospective analysis of G-banded chromosomes by our laboratory determined that the abnormality was microscopically visible.
TABLE III.
Deletion Associated With Parental Balanced Insertion
| Case | Sex | Age | Indication | Karyotype | CMA versions | CNV fragment IT site | CNV in size (Mb) | Confirmation | Parental origin |
|---|---|---|---|---|---|---|---|---|---|
| 37 | M | 5 m | Growth delay | NPb | V6.2; V.7.2 OLIGO | 7p15.1(25981198–30568448)×1 9p24(mat) |
4.587–4.632 | ish der(7)del(7)(p15.1p15.3) ins(9;7)(p24;p15.1p15.3) (RP11-112E16−)mat |
Mat (B) |
| 38a | M | 7 | DD | 46,XYb | V5 BAC | 7q31(111.4–120.5 Mb)×1 10q24.3(mat) |
9.1 | ish der(7)del(7)(q31.1q31.31) ins(10;7)(q24.3;q31.1q31.31) (RP11-12L9−)mat |
Mat (B) |
| 39 | F | 13 | DD | 46,XXb | V7.1 OLIGO | 12p12.3p11.23
(17779542–26586202)×1 12q13(mat) |
8.807–8.918 | ish der(12)del(12) (p12.3p12.3) ins(12)(q13;p12.3p12.3) (RP11-459D22−)mat |
Mat (B) |
| 40a | F | 7½ | DD | 46,XX | V6 OLIGO | 21q22.12(33833315–35646946)×1 11q13(pat) |
1.8 | ish der(21)del(21)(q22.12q22.12) ins(11;21)(q13.5;q22.12q22.12) (RP11-17O20−)pat |
Pat (B) |
m, Months; NP, not provided; Mat, maternal; Pat, paternal; B, balanced in parent; DD, developmental delay.
Published case report.
Retrospective analysis of G-banded chromosomes by our laboratory determined that the abnormality was microscopically visible.
FIG. 1.

Summary karyogram. An ideogram of each chromosome at an 850-band level resolution (http://www.peds.ufl.edu/divisions/genetics/teaching/chromosomes.htm) is shown with the insertion site on the left indicated with a triangle and the CNV fragment on the right indicated by the horizontal line. The size of the line represents the relative size of the CNV (not drawn to scale). Red represents copy number loss (deletion), green represents copy number gain, and black represents no copy number change (“copy neutral”). Parent of origin is provided when available (U, unbalanced in the parent; B, balanced in the parent; dn, de novo).
Simple Insertional Translocation
We define simple IT as a single copy number gain due to the insertion of a genomic segment into another location resulting in pure segmental trisomy. When the insertion occurs on a homologous chromosome it is referred to as an intrachromosomal event, when it inserts into a non-homologous chromosome it is an interchromosomal event. For the purposes of this publication, we subdivided the simple IT events into the following categories: (1) intrachromosomal, (2) interchromosomal, (3) involving similar chromosomal regions, and (4) involving the short arm of acrocentric chromosomes (Table I).
Intrachromosomal IT
We identified 2 of the 40 cases (5%) in which a simple, apparently three-break IT event occurred within the same homologous chromosome (Table I). The inserted segment was translocated from one arm of the chromosome to the other arm in both cases. One case (Case 1) was determined to be microscopically visible. Parental studies were available for both cases.
Case 1 was a newborn male referred for aCGH testing with an indication of cardiac anomalies and pulmonary insufficiency (Table I). The aCGH results on the Chromosomal Microarray Analysis version 7 array (CMA V7) revealed an ~9.6 Mb gain in copy number of the chromosome 7p15.1p14.1 segment (Fig. 2A). FISH confirmation of the aCGH results established that this segment from the short arm of chromosome 7 had inserted into the long arm of chromosome 7 and partial karyotype analysis of G-banded chromosomes determined that the insertion occurred at band 7q31.32 (Fig. 2A). This region of chromosome 7 (genomic coordinates 28559836–38185226) contains 47 annotated RefSeq genes (UCSC Browser), including the T-BOX 20 (TBX20) gene, which has been implicated in atrial septal defect (ASD) seen in patients with congenital heart disease [Kirk et al., 2007]. However, parental FISH analysis revealed that the same unbalanced insertion was also present in the father (data not shown). No additional information was provided regarding the medical history for the father.
FIG. 2.

Simple intrachromosomal and interchromosomal insertional translocations. A: Case 1: The aCGH summary table is on top. To the left is the aCGH result showing the oligonucleotide probes specific for chromosome 7 next to the chromosome 7 ideogram. The probes detecting the gain in copy number are shown in green and circled. To the right is the FISH result showing insertion of chromosome 7p14 material (probe RP11-100A8 in red) into 7q. The control probe is RP11-61L9 labeled in green. A partial karyotype of chromosome 7 (G-banded) is shown below. The arrow indicates the derivative chromosome 7 with the box showing the location of the insertion. B: Case 2: The aCGH summary table is on top. On the bottom is the FISH result showing insertion of chromosome Xq material (probe RP11-1145H7 in red) into Xp. The control probe is RP11-383C12 labeled in green. The inset panel is the same X chromosome FISH image shown in inverse DAPI. C: Case 4: The aCGH summary table is on top. To the left is the aCGH result showing the oligonucleotide probes specific for chromosome 1 next to the chromosome 1 ideogram. The probes detecting the gain in copy number are shown in red and circled. To the right is the FISH result showing insertion of chromosome 1q32 material (probe RP11-279E18 in red) into chromosome 5q. The control probe is RP11-339I2 labeled in green. A partial karyotype of chromosome 5 (G-banded) is shown below. The arrow indicates the derivative chromosome 5 with the box showing the location of the insertion. D: Case 25: The aCGH summary table is on top. On the bottom is the inverse DAPI FISH image showing insertion of chromosome 17q material (probes RP11-497H17 in red and RP11-16C1 in green) into chromosome 22p. E: Case 26: The aCGH summary table is on top. On the bottom is the FISH result showing insertion of chromosome 11p material (probe RP11-65C9 in red) into chromosome 22p. The control probe is RP11-709C9 labeled in green. The inset panel is the same chromosome 22 FISH image shown in inverse DAPI.
Case 2 was a 5-year-old male with speech delay and self-injurious behavior at the time of referral. The CMA V7 detected an ~0.21 Mb gain of chromosome Xq13.1 involving six annotated RefSeq genes (Fig. 2B). FISH confirmation revealed that this segment of chromosome X is inserted into the short arm of chromosome X at band Xp21 (Fig. 2B). Parental FISH analysis with the same clone revealed that the same unbalanced insertion was present in the mother (data not shown). No additional information was provided and no further testing was performed on the mother. Although it was recommended that additional male maternal relatives be tested to help clarify the clinical significance of this finding, no additional family members have been tested to date.
Interchromosomal IT
The most common class of IT involves interchromosomal events in which a segment from one chromosome inserts into another non-homologous chromosome. This class of IT also results in pure segmental trisomy for the inserted genomic segment. Although we identified a total of 26 cases in this category, we subdivided cases with apparently similar chromosomal segments (N =4) and IT involving the short arm of acrocentric chromosomes (N =6).
Of the remaining 16 simple interchromosomal IT cases, six (37.5%) were microscopically visible (Table I). In addition, 10 of the 16 cases had follow-up studies on at least one parent. Three cases (5, 6, and 8) were inherited from a mother carrying the balanced form of the rearrangement, three (Cases 9, 12, and 17) were inherited from a parent carrying the same unbalanced rearrangement, one (Case 15) was de novo, and three (Cases 10, 13, and 14) were not present in the parent that was available for testing. Of the six cases with no parental follow-up studies, three had large microscopically visible insertions and therefore were interpreted as pathogenic (Cases 3, 4, and 7), while the remaining three were submicroscopic with unclear clinical significance (Cases 11, 16, and 18).
Case 4 was a 2½-year-old male referred for evaluation due to a history of seizures with unknown etiology and developmental delay. His neurological examination revealed mild global hypotonia. The family history was significant for the mother’s sister with mild developmental delay of unknown etiology and consistent with independent living. aCGH using CMA V6 revealed an ~16 Mb copy number gain of chromosome 1q32.1q32.2 material (Fig. 2C). FISH and partial karyotype analyses revealed that this segment of chromosome 1 is inserted into chromosome 5 at band 5q14.2 (Fig. 2C). Although no follow-up information is available to date, the rearrangement observed in this propositus is likely pathogenic given the size of the duplicated segment.
Interchromosomal IT involving similar chromosomal regions
Of the 26 interchromosomal events identified, four unrelated cases involved the same bands on chromosomes 3 and 6 (Table I). All four cases (19–22) showed a copy number gain of chromosome 6p25 with overlapping genomic coordinates 468355–582886 detected by aCGH analysis. FISH confirmation using probe RP1-20B11 revealed an insertion of chromosome 6p25 material into chromosome 3p13 in all four cases. The sizes of the duplications ranged from a minimum of 125 to 349 kb, and the same unbalanced insertion was present in the mother in three of four cases (parental follow-up studies were not available for Case 20).
Case 20 was an 8½-year-old male who was evaluated for global developmental delay and an abnormal EEG. He carried a diagnosis of static encephalopathy. He was noted to be delayed at 1 year of age with no words, and no speech was developed until 4 years of age. An EEG revealed right temporal spikes during sleep but with no accompanying clinical seizures. A brain MRI performed at 8 years of age was normal. There were no significant dysmorphic features observed and the neurological exam was normal. aCGH revealed that an ~250 kb segment from the distal subtelomeric region of chromosome 6p was inserted into chromosome 3 at band 3p13 (Fig. 3A). Parental studies were not available.
FIG. 3.

Interchromosomal insertions involving similar regions of chromosome 6p material into chromosome 3p. FISH analysis was performed using probe RP1-20B11 (chromosome 6). A: Case 20: To the left is the genome view of the aCGH results with the copy number gain indicated by the red circle. The aCGH summary table is to the right. On the bottom right is the FISH result. The control probe is RP11-239K6 labeled in green. The inset panel is the FISH image shown in inverse DAPI. B: Case 22: The aCGH summary table is on top. On the bottom is the inverse DAPI FISH image for the propositus (left) and the mother (right). The control probe is RP11-69L16 labeled in green.
Case 22 was an 18-year-old female who sought genetic counseling for her reproductive risk in the future. She was referred for aCGH testing with multiple congenital anomalies including: cleft lip and palate, syndactyly between the third and fourth digits, and bilateral hydronephrosis, which were surgically repaired. Family history indicated that her parents were fourth cousins once removed. aCGH and FISH confirmation revealed that an ~350 kb segment from the distal subtelomeric region of chromosome 6p was inserted into chromosome 3 at band 3p13 (Fig. 3B, left image). The same unbalanced insertion was also present in her mother who has a history of premature colorectal cancer but was otherwise normal (Fig. 3B, right image).
The first megabase of chromosome 6 sequence annotated on the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway) is identified as a region of many structural variations in the normal population (Fig. 4). There are 167 “normal” individuals with gains and losses within this region in the Toronto Database of Genomic Variants (http://projects.tcag.ca/variation/?source=hg18). Although there are four annotated RefSeq genes within this interval (DUSP22, IRF4, EXOC2, and HUS1B), there is no apparent clinical correlation. The above case reports combined with the CNV database suggest that these rearrangements are not likely to be related to the phenotypic findings in these patients.
FIG. 4.

Schematic representation of chromosome 6p25 genomic structure. The ideogram for chromosome 6 is shown on top. The red box indicates the region of interest. The enlarged view of the first 1 Mb of chromosome 6p25 is shown below. The blue bar indicates the location of the FISH probe. RefSeq genes are indicated with arrows. The orange bars indicate regions of segmental duplication (Seg Dup). The green bars indicate regions of copy number polymorphisms (CNP).
IT to the short arm of acrocentric chromosomes
Rearrangements involving the short arm of acrocentric chromosomes often appear cryptic, especially when present in an unbalanced form. We identified six cases with insertions into the short arms of chromosome 15 or 22 (Table I). Two cases (23 and 27) were submitted to further characterize a known karyotype abnormality; one case (25) had a reportedly normal karyotype, although the array showed the inserted material was greater than 8 Mb in size; and karyotype information was not provided in three cases (24, 26, and 28).
Case 25 was a 5-month-old male referred for aCGH and chromosome analyses to evaluate his failure to thrive. He presented with dysmorphic features including; microcephaly, large anterior fontanelle, downslanting palpebral fissures, triangular appearance of the face, mild micrognathia, and ears that were borderline low set and posteriorly rotated. The family history was non-contributory, with two healthy siblings. A brain MRI at 4 months of age showed microcephaly with mild-to-moderate cerebral global volume loss as well as minimal cerebellar volume loss. The temporal horns had a ballooned configuration raising concerns for communicating hydrocephalus. The aCGH analysis (CMA V7) showed an ~8 Mb gain of chromosome 17q25 material (Fig. 2D). FISH confirmation revealed that the additional material was inserted into the short arm of chromosome 22 (Fig. 2D). However, the chromosome analysis performed concurrently was normal (data not shown). Only the maternal sample was available for testing and FISH analysis using the same probe was normal (data not shown).
Case 26 was a 9-month-old male referred for aCGH testing with developmental delay, failure to thrive, and cyclic episodes of vomiting and neutropenia. The aCGH analysis performed on CMA V7 showed a gain of 1.1 Mb in size of chromosome 11p12 material (Fig. 2E). FISH analysis showed the extra copy of chromosome 11p material inserted into the short arm of chromosome 22 (Fig. 2E). This same unbalanced insertion was also present in the mother (data not shown).
There are three annotated RefSeq genes, RAG1, RAG2, and C11orf74, within the minimum interval (genomic coordinates chr11: 36521870–37627264). Mutations in RAG1 and RAG2 resulting in decreased activity of these two proteins cause Omenn syndrome (OMIM 603554), which is characterized by reticuloendotheliosis with eosinophilia, severe combined immunodeficiency, and in some instances failure to thrive [Schwarz et al., 1996; Villa et al., 1998]. Although duplication of the RAG1 and RAG2 genes has not been previously reported in association with a clinical phenotype, the presence of cyclic neutropenia in this patient may suggest B cell lineage involvement, and makes RAG1 and RAG2 good candidates for the neutropenia seen in this patient. However, since the mother also carries the same unbalanced insertion, the clinical significance of this rearrangement remains uncertain.
Complex Insertional Translocation
Complex ITs are defined as insertions that are generated by more than three breakage and joining events. We identified eight cases with complex ITs (Table II). Two cases (29 and 30) were intra-chromosomal, whereas six were interchromosomal (Cases 31–36).
Case 29 was a 33-year-old male referred for aCGH analysis to characterize the derivative chromosome 1 identified by chromosome analysis. He presented with cognitive delays (IQ scores in the 60s), but otherwise was noted to be verbal and social, and able to live independently. He had a history of seizures and fine motor control abnormalities. He currently receives some supportive services and is under his mother’s guardianship. Previous chromosome analysis identified a derivative chromosome 1 with a rearrangement involving the 1p36.1 region (Table II). aCGH analysis (CMA V7) revealed a gain in copy number in the long arm of chromosome 1 at band 1q31.1q31.2 encompassing 2.4 Mb and a gain in copy number in the long arm of chromosome 1 at band 1q32.1 spanning 0.98 Mb, both of which were shown by FISH analysis to be inserted to the short arm of chromosome 1, most likely at band 1p36.1 (Fig. 5A). An interval of 16.8 Mb between the two segments on the long arm of chromosome 1 showed no evidence of copy number change. FISH analysis revealed that this segment remained on the long arm of chromosome 1 (data not shown). Parental samples were not available. It is unclear whether this rearrangement arose as independent or related events.
FIG. 5.

Complex insertional translocations. A: Case 29: The aCGH summary table is on top. To the left is the aCGH result showing the oligonucleotide probes specific for chromosome 1 next to the chromosome 1 ideogram. The probes detecting the gain in copy number are shown in green and circled. To the right is the FISH result showing insertion of chromosome 1q32 material (probe RP11-379J10 in red) into chromosome 1p36. The control probe is RP5-888M10 labeled in green, which maps to chromosome 1p36.22. The inset panel is the FISH image shown in inverse DAPI. B: Case 33: The aCGH summary table is on top. On the bottom is the inverse DAPI FISH image showing insertion of chromosome 6q material (probe RP11-211L10 in red) into chromosome Xq. C: Case 34: The aCGH summary table is on top. On the bottom is the FISH image showing insertion of chromosome 5p material (probe RP11-43F13 in green) into chromosome Xq. The X chromosome probes are RP11-119A22 labeled in red and the centromere probe in green (Vysis).
Three of the eight complex IT cases were insertions with deletions near the insertion location (Table II; Cases 31–33). Case 33 was an 8-year-old male at the time of referral with an indication of developmental delay, mental retardation, dysmorphic features, and multiple congenital anomalies. Chromosome studies were reportedly normal. The aCGH analysis on V6 and V7 showed a loss in copy number on chromosome 6q25 of greater than 8 Mb in size; as well as a gain in copy number on chromosome Xq28 of ~1.5 Mb in size (Fig. 5B). FISH analysis showed that the duplicated segment from chromosome Xq28 is inserted into the long arm of the deleted chromosome 6 at band 6q25 (Fig. 5B). It is possible that the deletion observed on chromosome 6 was the result of the insertion event. Follow-up parental FISH studies revealed that this rearrangement was a de novo event (data not shown).
Three cases were insertions with duplications near the insertion location (Table II; Cases 34–36). Case 34 was a newborn female with patent ductus arteriosus and transient tachypnea when referred for aCGH testing. Chromosomal microarray analysis revealed a gain in copy number in the chromosome 5p15.33 region of ~0.531 Mb as well as a gain in copy number in the chromosome Xq28 region spanning ~100 kb (Fig. 5C). The gain on Xq28 encompasses the first two exons of the MECP2 gene. FISH analysis revealed that the third copy of the chromosome 5p15.33 segment is inserted into one X chromosome at band Xq28 (Fig. 5C). At the level of resolution of FISH analysis, we were unable to determine whether the segment from 5p15.33 was inserted into the same region where MECP2 is located and whether the duplication on chromosome Xq28 was a direct result of the insertion event. No follow-up information is available to date.
Deletion Resulting From Parental Balanced Insertion
There were four cases that required parental studies to provide the correct diagnosis and recurrence risk for the family. The aCGH analysis on all four cases revealed what appeared to be a simple interstitial deletion ranging in size from 1.8 to 9.1 Mb, which were all confirmed by FISH analysis (Table III, Cases 37–40). Parental FISH studies were recommended to determine whether the observed abnormalities were de novo or inherited from a parental rearrangement. Balanced insertions were present in the mother in three cases (37–39) and in the father in one (Case 40). Interestingly, Cases 38 and 39 had previous normal karyotype analyses; however, both the abnormalities detected were large enough (~9 Mb in size) to be microscopically visible. A detailed description of Case 38 was previously published [Lennon et al., 2007].
The deletion observed in Case 40 was submicroscopic (~1.8 Mb in size). The aCGH results and a brief clinical description were previously published [Shinawi et al., 2008]. More detailed information is presented here. The patient was a 7½-year-old female referred for aCGH testing. She had been extensively evaluated for chronic thrombocytopenia, severe failure to thrive, developmental delay, behavioral problems, mild dysmorphic features, and autistic features. The family history was significant for a paternal cousin with failure to thrive. In addition, the patient’s father and four siblings had familial short stature but otherwise were healthy. The patient’s mother had one previous first trimester miscarriage.
At 19 months of age, a brain MRI showed moderate diffuse sulcal widening with delayed myelination and a mildly dysgenetic rostrum of the corpus callosum. Her blood count showed macrocytic anemia and thrombocytopenia. The megakaryocytes were present in all stages of development. The patient continued to display severe failure to thrive despite intensive nutritional therapy. Two months later, the anemia resolved but the thrombocytopenia and macrocytosis persisted. Chromosome analysis performed at this time was reportedly normal.
The results of the aCGH analysis showed a loss of chromosome 21q22.12 material including the RUNX1 gene (Fig. 6A), which was confirmed by FISH analysis (Fig. 6B, bottom left) [Shinawi et al., 2008]. Surprisingly, parental FISH studies revealed that the father carried a balanced insertion of chromosome 21q22.12 material into chromosome 11 at band 11q13 (data not shown). Therefore, the proposita inherited the derivative chromosome 21 from the father. Testing of other family members revealed that one of her brothers also carried the balanced insertion (Fig. 6B, bottom right).
FIG. 6.
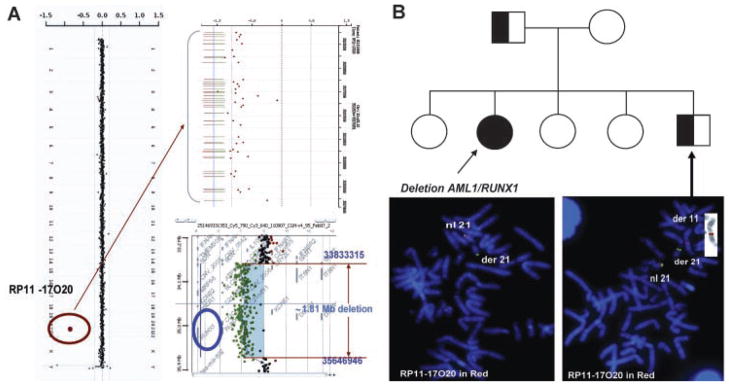
Deletion resulting from a balanced translocation in a parent (Case 40). A: To the left is the genome view of the aCGH results with the copy number gain indicated by the red circle. The top right is a zoomed in view of the individual oligonucleotide probes detecting the deletion. The bottom right is the deleted region detected on the high-density 244K Agilent array. B: A pedigree of the family. The propositus is indicated by the filled in circle. The father and brother carry the balanced insertional translocation (half-filled squares). Below the pedigree is the FISH image showing the deletion in the propositus (left) and the balanced insertion in the brother (right). The FISH probes are RP11-17O20 in red, which maps to chromosome 21p22.12, and the chromosome 21 telomeric probe (Vysis) in green. The inset panel is the inverse DAPI FISH image of the derivative chromosome 11 in the brother.
DISCUSSION
ITs are rarely seen on routine chromosome analysis. Two reports estimate the frequency to be approximately 1:10,000 patients karyotyped [Fryns et al., 1984; Van Hemel and Eussen, 2000]. Using a combination of aCGH and FISH analyses, we identified 40 IT cases out of approximately 18,000 cases (1:500) referred for molecular chromosomal testing (aCGH), a figure 20-fold greater than the established literature. This dramatic increase in detection rate likely reflects the high-resolution genome analysis afforded by aCGH technology compared to conventional cytogenetic studies.
In our cohort of cases, there were 9 female and 31 male patients. In addition, of the 23 cases with follow-up parental studies, only 4 were paternal in origin while 15 cases were maternal in origin, and four were de novo. Interestingly, in 11 of the 19 (58%) inherited cases (1, 2, 9, 12, 17, 19, 21, 22, 26, 32, and 35), a parent had the same unbalanced rearrangement seen in the propositus. When only the submicroscopic IT cases are examined (21 of 40 =53%) the number of cases where a parent carries the same unbalanced IT increases to 69% (9/13) (Table IV). This observation supports the contention that unbalanced ITs that are inherited may have little phenotypic consequences. However, the possibility that the parent and child do not carry the identical CNV is typically not ruled out. Since the parents are often studied using FISH analysis, the actual size of the CNV detected in the parent is frequently unresolved. Although it is assumed that the parent and child have the same aberration, there is now evidence in the literature that indicates this may not be true for terminal deletions [South et al., 2008].
TABLE IV.
Submicroscopic IT
| Inherited from parent w/balanced IT | 1 | |
| Maternal | 0 | |
| Paternal | 1 | |
| Inherited from parent w/unbalanced IT | 9 | |
| Maternal | 8 | |
| Paternal | 1 | |
| De novo | 2 | |
| Pending parental studies | 9 | |
| Total | 21 |
There were five cases (9, 13, 14, 15, and 40) of small, submicroscopic IT events involving known disease genes/regions that were interpreted as causative for the observed phenotype. For example, Case 9 is a female referred for aCGH testing with an indication of multiple congenital anomalies. The CMA V7 aCGH analysis detected a gain on chromosome 1q21.1 at genomic coordinates 145085612–145518428 (Table I). FISH analysis revealed that the duplicated 1q21.1 segment is inserted into the long arm of chromosome 7 (data not shown). This region of chromosome 1 has recently been associated with microcephaly when deleted and macrocephaly when duplicated as well as developmental delay and congenital anomalies [Brunetti-Pierri et al., 2008; Mefford et al., 2008]. The head circumference of our patient was not provided and, as with many of the patients published in the literature, the rearrangement was inherited from a parent (Table I).
Case 14 is a male referred for aCGH testing with an indication of developmental delay and autistic spectrum disorder (Table I). The aCGH and confirmatory FISH analysis revealed that a third copy of the SHANK3 gene inserted into chromosome 11p13 (data not shown). Deletions of, and mutations in, this gene have been associated with language and speech delay, and autistic spectrum disorder [Durand et al., 2007]. Therefore, although it is likely that the dosage imbalance resulting from duplication of the SHANK3 gene is responsible for the observed phenotype in this patient, there are no publications to our knowledge of pure segmental trisomy for the SHANK3 gene alone. Similarly, clinical correlations were made for Case 13 showing pure trisomy for the YWHAE gene within the Miller–Dieker region on chromosome 17p13.3 and brain abnormalities [Bi et al., 2009], and for Case 40 with pure monosomy for the RUNX1 gene region and thrombocytopenia [Shinawi et al., 2008].
There are, however, other examples of CNVs involving known disease genes where the clinical significance is less clear. Cases 15 and 34 both involved segmental aneusomy of the MECP2 gene. Duplication of this gene in males results in Lubs syndrome (OMIM 300260) characterized by developmental delay, hypotonia, limited/absent speech, and dysmorphic features and is likely the cause of the phenotype observed in Case 15. However, the clinical consequence of this duplication in females (as seen in Case 34) may be more subtle and is likely to be dependent on patterns of X-chromosome inactivation (XCI). Reports in the literature describe MECP2 gene duplication inheritance from apparently phenotypically normal mothers; however, these women have extremely skewed XCI [Meins et al., 2005; Van Esch et al., 2005; del Gaudio et al., 2006]. More recently, Ramocki et al. [2009] reported results of formal clinical evaluations of MECP2 gene duplication carriers and suggested that MECP2 duplication in females may result in abnormal neuropsychiatric phenotypes even in the presence of skewed XCI.
In addition, some IT events will likely end up categorized as clinically benign such as in Cases 19–22. All four of these cases have apparently similar IT events involving the same region of chromosome 6p25 inserted into chromosome 3p13. The region on chromosome 6 from genomic coordinates 1–1000000 was shown to contain common CNVs seen in the normal population (Fig. 4 and the Database of Genomic Variants). We also have cases in which both the propositus and an apparently phenotypically normal parent carry a duplication of the same region on chromosome 6p25, further supporting the hypothesis that this region may be polymorphic and clinically benign. Interestingly, although there are many annotated segmental duplications within this region, none show homology to chromosome 3p13. Although these insertions appear recurrent at the level of cytogenetic bands, further molecular studies are needed to explore the mechanistic underpinnings. Regardless of the mechanism that resulted in the IT event, the phenotypes observed in Cases 19–22 currently cannot be causally linked to the rearrangements.
Although the incidence of IT at the chromosomal level is much lower, it is more likely that these events were causative due to the large size of the DNA fragments involved in the insertion events. For example, Case 32 was a 2-month-old male when referred for aCGH testing with an indication of multiple congenital anomalies. aCGH, FISH, and partial karyotype analyses revealed a deletion of chromosome 13q21 (~4.38 Mb in size) with an apparent insertion of chromosome 14q22.3q23.3 (~11.79 Mb in size) in its place, suggesting that the deletion on chromosome 13 was a result of the insertion event [Ou et al., 2008b]. Interestingly, analysis of the parental blood samples by FISH revealed that the father carried the same unbalanced complex rearrangement [Ou et al., 2008b]. Furthermore, there are family studies reported in the literature of microscopically visible insertions resulting in partial trisomy and/or monosomy that are inherited from a parent carrying the same unbalanced IT [Kolomietz et al., 2006; Liang et al., 2006] or the balanced form of the insertion [Arens et al., 2004; Fogu et al., 2007]. However, depending on the size and location of the insertion, it remains challenging to discern whether some de novo events are pathogenic, and likewise, whether some inherited events are clinically benign.
Despite the fact that IT was detected more frequently using aCGH, the clinical relevance of some of these findings is often unclear, especially when the CNV does not involve disease-associated genomic segments. In the past several years, many groups have studied normal populations to identify structural variants in the human population, the results of which are available in the Database of Genomic Variants web page (http://projects.tcag.ca/variation/). It has been estimated that there are ~100 CNVs/per individual and new CNVs are identified each year [Feuk et al., 2006]. Our current study illustrates that insertion events are also likely to be more common than previously recognized. Regardless of size, when a CNV is inherited from a seemingly normal parent, it is less likely to be interpreted as pathogenic. However, additional confounding factors, such as (1) parent of origin effects, (2) unmasking of recessive alleles, (3) decreased penetrance, and (4) possible differences in CNV size, must be considered when counseling these families.
CONCLUSIONS
There is no question that the emergence of aCGH in the clinic has revolutionized the way segmental aneusomies are detected. Developments in array platforms and bioinformatic tools have rapidly improved our ability to detect CNVs with resolutions less than 1 kb leading to observations of CNVs even in apparently “balanced” structural rearrangements [Rosenberg et al., 2005; De Gregori et al., 2007; Simovich et al., 2007; Baptista et al., 2008]. However, in spite of these amazing new technologies, a major roadblock remains unchanged. When a copy number gain is detected, it is impossible to determine where in the genome the additional material resides using the array data alone. FISH analysis is the preferred method to identify both the location and the copy number of the interrogated genomic segment. The finding that insertion events (whether pathogenic or benign) occur more frequently than previously predicted reestablishes the need to confirm the location of a copy number gains detected by aCGH analysis, especially in the clinical setting.
The importance of follow-up parental studies is also illustrated in the current study. Four of the 40 IT cases (10%) were deletions resulting from inheritance of a derivative chromosome present in the balanced state in the parent (Table III). In studying the propositus alone, one would assume that the interstitial deletion was a sporadic event with a low recurrence rate. All four cases presented in this category are likely to be pathogenic. Since the results of the parental studies demonstrated that the deletions were inherited from a parental balanced rearrangement, the recurrence risk and genetic counseling for these families were dramatically altered. It is likely that interstitial deletions resulting from parental rearrangements are under-ascertained given that follow-up studies of both biological parents are frequently unavailable.
Molecular cytogenetics as a field must proceed with caution as the density and resolution of microarrays used for clinical diagnoses rapidly increase. This will inevitably raise the number of CNVs detected per individual, thus further complicating clinical correlation of aCGH findings. Nevertheless, the potential for new discoveries will also increase, ultimately expanding our understanding of genomic disorders.
Acknowledgments
The authors thank the staff in the Medical Genetics Laboratories and, in particular, Caroline Borgan for her assistance.
References
- Arens YH, Engelen JJ, Govaerts LC, van Ravenswaay CM, Loneus WH, van Lent-Albrechts JC, van der Blij-Philipsen M, Hamers AJ, Schrander-Stumpel CT. Familial insertion (3;5)(q25.3;q22.1q31.3) with deletion or duplication of chromosome region 5q22.1–5q31.3 in ten unbalanced carriers. Am J Med Genet Part A. 2004;130A:128–133. doi: 10.1002/ajmg.a.20568. [DOI] [PubMed] [Google Scholar]
- Baptista J, Mercer C, Prigmore E, Gribble SM, Carter NP, Maloney V, Thomas NS, Jacobs PA, Crolla JA. Breakpoint mapping and array CGH in translocations: Comparison of a phenotypically normal and an abnormal cohort. Am J Hum Genet. 2008;82:927–936. doi: 10.1016/j.ajhg.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Sapir T, Shchelochkov OA, Zhang F, Withers MA, Hunter JV, Levy T, Shinder V, Peiffer DA, Gunderson KL, Nezarati MM, Shotts VA, Amato SS, Savage SK, Harris DJ, Day-Salvatore DL, Horner M, Lu XY, Sahoo T, Yanagawa Y, Beaudet AL, Cheung SW, Martinez S, Lupski JR, Reiner O. Increased LIS1 expression affects human and mouse brain development. Nat Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang SH, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK, Naqvi S, Garnica AD, Bernes SM, Fong CT, Summers A, Walters WD, Lupski JR, Stankiewicz P, Cheung SW, Patel A. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, Shaw C, Peacock S, Pursley A, Tavyev YJ, Ramocki MB, Nawara M, Obersztyn E, Vianna-Morgante AM, Stankiewicz P, Zoghbi HY, Cheung SW, Lupski JR. Complex rearrangements in patients with duplications of MECP2 can occur by Fork Stalling and template switching. Hum Mol Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, Li J, Ou Z, Patel A, Yatsenko SA, Cooper ML, Furman P, Stankiewicz P, Lupski JR, Chinault AC, Beaudet AL. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- De Gregori M, Ciccone R, Magini P, Pramparo T, Gimelli S, Messa J, Novara F, Vetro A, Rossi E, Maraschio P, Bonaglia MC, Anichini C, Ferrero GB, Silengo M, Fazzi E, Zatterale A, Fischetto R, Previdere C, Belli S, Turci A, Calabrese G, Bernardi F, Meneghelli E, Riegel M, Rocchi M, Guerneri S, Lalatta F, Zelante L, Romano C, Fichera M, Mattina T, Arrigo G, Zollino M, Giglio S, Lonardo F, Bonfante A, Ferlini A, Cifuentes F, Van Esch H, Backx L, Schinzel A, Vermeesch JR, Zuffardi O. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: A study of 59 patients. J Med Genet. 2007;44:750–762. doi: 10.1136/jmg.2007.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ravel T, Aerssens P, Vermeesch JR, Fryns JP. Trisomy of chromosome 16p13.3 due to an unbalanced insertional translocation into chromosome 22p13. Eur J Med Genet. 2005;48:355–359. doi: 10.1016/j.ejmg.2005.05.009. [DOI] [PubMed] [Google Scholar]
- del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, Berry SA, Pursley AA, Grebe TA, Freedenberg D, Martin RA, Hsich GE, Khera JR, Friedman NR, Zoghbi HY, Eng CM, Lupski JR, Beaudet AL, Cheung SW, Roa BB. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni M-C, de Mas P, Bieth E, Roge B, Heron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Fogu G, Bandiera P, Cambosu F, Carta AR, Pilo L, Serra G, Soro G, Tondi M, Tusacciu G, Montella A. Pure partial trisomy of 6p12.1–p22.1 secondary to a familial 12/6 insertion in two malformed babies. Eur J Med Genet. 2007;50:103–111. doi: 10.1016/j.ejmg.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Fryns JP, Kleczkowska A, Kenis H. De novo complex chromosomal rearrangement (CCR) in a severely mentally retarded boy. Ann Genet. 1984;27:62–64. [PubMed] [Google Scholar]
- Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, Mackay JP, Waddell LB, Cole AD, Hayward C, Keogh A, Macdonald P, Griffiths L, Fatkin D, Sholler GF, Zorn AM, Feneley MP, Winlaw DS, Harvey RP. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280–291. doi: 10.1086/519530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolomietz E, Ben-Omran T, Chitayat D, Mah M, Murphy J, Nie G, Teshima I. Array-based genomic delineation of a familial duplication 11q14.1–q22.1 associated with recurrent depression. Am J Med Genet Part B. 2006;141B:214–219. doi: 10.1002/ajmg.b.30297. [DOI] [PubMed] [Google Scholar]
- Lennon PA, Cooper ML, Peiffer DA, Gunderson KL, Patel A, Peters S, Cheung SW, Bacino CA. Deletion of 7q31.1 supports involvement of FOXP2 in language impairment: Clinical report and review. Am J Med Genet Part A. 2007;143A:791–798. doi: 10.1002/ajmg.a.31632. [DOI] [PubMed] [Google Scholar]
- Liang D, Wu L, Pan Q, Harada N, Long Z, Xia K, Yoshiura K, Dai H, Niikawa N, Cai F, Xia J. A father and son with mental retardation, a characteristic face, inv(12), and insertion trisomy 12p12.3–p11.2. Am J Med Genet Part A. 2006;140A:238–244. doi: 10.1002/ajmg.a.31077. [DOI] [PubMed] [Google Scholar]
- Lu X, Shaw CA, Patel A, Li J, Cooper ML, Wells WR, Sullivan CM, Sahoo T, Yatsenko SA, Bacino CA, Stankiewicz P, Ou Z, Chinault AC, Beaudet AL, Lupski JR, Cheung SW, Ward PA. Clinical implementation of chromosomal microarray analysis: Summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, Parkin G, Fichera M, Reitano S, Lo Giudice M, Li KE, Casuga I, Broomer A, Conrad B, Schwerzmann M, Raber L, Gallati S, Striano P, Coppola A, Tolmie JL, Tobias ES, Lilley C, Armengol L, Spysschaert Y, Verloo P, De Coene A, Goossens L, Mortier G, Speleman F, van Binsbergen E, Nelen MR, Hochstenbach R, Poot M, Gallagher L, Gill M, McClellan J, King MC, Regan R, Skinner C, Stevenson RE, Antonarakis SE, Chen C, Estivill X, Menten B, Gimelli G, Gribble S, Schwartz S, Sutcliffe JS, Walsh T, Knight SJ, Sebat J, Romano C, Schwartz CE, Veltman JA, de Vries BB, Vermeesch JR, Barber JC, Willatt L, Tassabehji M, Eichler EE. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Z, Kang SH, Shaw CA, Carmack CE, White LD, Patel A, Beaudet AL, Cheung SW, Chinault AC. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med. 2008a;10:278–289. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Z, Martin DM, Bedoyan JK, Cooper ML, Chinault AC, Stankiewicz P, Cheung SW. Branchiootorenal syndrome and oculoauriculovertebral spectrum features associated with duplication of SIX1, SIX6, and OTX2 resulting from a complex chromosomal rearrangement. Am J Med Genet Part A. 2008b;146A:2480–2489. doi: 10.1002/ajmg.a.32398. [DOI] [PubMed] [Google Scholar]
- Ramocki M, Peters S, Tavyev Y, Zhang F, Carvalho C, Schaaf C, Richman R, Fang P, Glaze D, Lupski J, Zoghbi H. Autism and other neuropsychiatric symptoms are prevalent in individuals with MECP2 duplication syndrome. Ann Neurol. 2009;66:771–782. doi: 10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg C, Knijnenburg J, de Chauffaille ML, Brunoni D, Catelani AL, Sloos W, Szuhai K, Tanke HJ. Array CGH detection of a cryptic deletion in a complex chromosome rearrangement. Hum Genet. 2005;116:390–394. doi: 10.1007/s00439-004-1248-x. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, Friedrich W, Seger RA, Hansen-Hagge TE, Desiderio S, Lieber MR, Bartram CR. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- Shinawi M, Erez A, Shardy DL, Lee B, Naeem R, Weissenberger G, Chinault AC, Cheung SW, Plon SE. Syndromic thrombocytopenia and predisposition to acute myelogenous leukemia caused by constitutional microdeletions on chromosome 21q. Blood. 2008;112:1042–1047. doi: 10.1182/blood-2008-01-135970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simovich MJ, Yatsenko SA, Kang SH, Cheung SW, Dudek ME, Pursley A, Ward PA, Patel A, Lupski JR. Prenatal diagnosis of a 9q34.3 microdeletion by array-CGH in a fetus with an apparently balanced translocation. Prenat Diagn. 2007;27:1112–1117. doi: 10.1002/pd.1841. [DOI] [PubMed] [Google Scholar]
- South ST, Rope AF, Lamb AN, Aston E, Glaus N, Whitby H, Maxwell T, Zhu XL, Brothman AR. Expansion in size of a terminal deletion: A paradigm shift for parental follow-up studies. J Med Genet. 2008;45:391–395. doi: 10.1136/jmg.2008.057315. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, Moraine C, Marynen P, Fryns JP, Froyen G. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hemel JO, Eussen HJ. Interchromosomal insertions. Identification of five cases and a review. Hum Genet. 2000;107:415–432. doi: 10.1007/s004390000398. [DOI] [PubMed] [Google Scholar]
- Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, Gatta LB, Ochs HD, Schwarz K, Notarangelo LD, Vezzoni P, Spanopoulou E. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93:885–896. doi: 10.1016/s0092-8674(00)81448-8. [DOI] [PubMed] [Google Scholar]