


Abstract
Experimental studies of complete mammalian genes and other genetic domains are impeded by the difficulty of introducing large DNA molecules into cells in culture. Previously we have shown that GST–Z2, a protein that contains three zinc fingers and a proline-rich multimerization domain from the polydactyl zinc finger protein RIP60 fused to glutathione S-transferase (GST), mediates DNA binding and looping in vitro. Atomic force microscopy showed that GST–Z2 is able to condense 130–150 kb bacterial artificial chromosomes (BACs) into protein–DNA complexes containing multiple DNA loops. Condensation of the DNA loops onto the Z2 protein–BAC DNA core complexes with cationic lipid resulted in particles that were readily transferred into multiple cell types in culture. Transfer of total genomic linear DNA containing amplified DHFR genes into DHFR– cells by GST–Z2 resulted in a 10-fold higher transformation rate than calcium phosphate co-precipitation. Chinese hamster ovarian cells transfected with a BAC containing the human TP53 gene locus expressed p53, showing native promoter elements are active after GST–Z2-mediated gene transfer. Because DNA condensation by GST–Z2 does not require the introduction of specific recognition sequences into the DNA substrate, condensation by the Z2 domain of RIP60 may be used in conjunction with a variety of other agents to provide a flexible and efficient non-viral platform for the delivery of large genes into mammalian cells.
INTRODUCTION
Functional elements of mammalian chromosomes, including genes, centromeres, replicons and telomeres, may encompass tens of thousands of base pairs. Studies on the control of gene expression have shown that boundary elements, promoters, enhancers, splicing signals, mRNA stability elements and other regulatory sequences are dispersed throughout genes and their flanking sequences. Cis-acting elements within gene loci may act in concert over very long distances (at least 50 kb), perhaps through DNA looping, DNA linking or sequestration in a common subnuclear compartment (reviewed in 1–4).
In the study of gene expression, transfection of plasmids bearing selected regulatory sequences into mammalian cells has proven remarkably valuable. However, plasmid-based transfection studies often fail to recapitulate the activity of sequences that reside in their native chromosomal context, perhaps because they do not reproduce the contributions of multiple cis-acting elements that are dispersed over long distances in genetic loci. For example, locus control regions may influence promoter activity at least 50 kb away, while promoters in turn may influence recruitment of factors to distal locus control regions (1–4). Additionally, some regulatory elements may not function when isolated as small sequence segments. For example, initiation sites for DNA synthesis have been mapped to specific chromosomal zones of 20–50 kb (5), yet plasmids bearing suspected replication origins generally do not replicate in the same manner as the chromosomal loci from which they were derived (6).
To dissect the role of specific cis-acting regulatory sequences in the control of a transcription unit, replicon or other genetic domain, it would be useful to employ an experimental system that allows precise modification of specific elements in the context of the complete locus. Homologous recombination in bacteria permits the introduction of precise modifications in bacterial and P1 artificial chromosomes (7–12), but the difficulty of introducing large DNA molecules bearing such alterations into cells has limited the utility of standard transfection assays for studying function.
We are interested in assessing the interaction of cis-acting elements at a distance in DNA synthesis and gene expression. Because of their stability, size and ability to be modified by homologous recombination, bacterial artificial chromosomes (BACs) represent an attractive experimental system for the study of entire genes and other chromosomal domains. Here we report the development of a simple gene transfer system that allows the introduction of BACs and other large DNA molecules into cultured cells with high efficiency. This system relies on the ability of a small protein fragment (Z2) of the polydactyl zinc finger protein RIP60 to bind at dispersed sites, multimerize on DNA and then induce DNA condensation by DNA looping. Collapse of DNA loops onto the GST–Z2–BAC DNA core complex with cationic lipids results in gene delivery complexes that are readily internalized and expressed by several cultured cell lines. Together with the ability to modify BACs by homologous recombination, DNA transfer systems that function with large DNA molecules should facilitate dissection of complex multipartite regulatory arrays within the context of entire gene loci.
MATERIALS AND METHODS
Preparation of BAC and plasmid DNA
BACs were isolated using a modified alkaline lysis method as described by Sinnett et al. (13). Plasmid DNA was isolated by alkaline lysis, purified by banding on CsCl gradients and tested for activity by transfection by calcium phosphate co-precipitation as before (14).
Purification of GST-tagged RIP60–Z2 fusion protein
GST–Z2 was purified from lysates of BL21 bacterial cultures by chromatography on glutathione–Sepharose beads (Pharmacia Biotech) as described previously (15).
Atomic force microscopy (AFM)
AFM studies were performed using a Nanoscope III AFM (Digital Instruments) equipped with a Plexiglass tapping-mode fluid cell as described previously (15).
Binding site selection assays
Two 60 base oligonucleotides (310 and 311) containing 16 randomized internal nucleotides were rendered double-stranded by primer extension with Taq polymerase using primers 312.5 and 312, as described previously (16). After primer extension, the products were amplified by a single cycle of PCR for 1 min at 94°C, 3 min at 62°C and 9 min at 72°C. Double-stranded products of 60 bp were purified on an 8% polyacrylamide gel, precipitated with glycogen and ethanol and resuspended in TE (10 mM Tris–HCl pH 7.0, 1 mM EDTA). Purified GST–Z2 was incubated with the oligonucleotide libraries as for RIP60 gel mobility shift reactions (15) for 45 min at room temperature, glutathione–Sepharose beads were added and the reaction was incubated at 4°C with shaking. The beads then were washed three times in 20 mM HEPES pH 7.9, 100 mM KCl, 0.2 mM EDTA, 0.2 mM EGTA and 20% glycerol. DNA was eluted by addition of 50 mM Tris–HCl pH 8.0, 100 mM sodium acetate, 5 mM EDTA and 0.5% SDS. After organic extraction, the DNA was recovered from the aqueous phase by precipitation with glycogen and ethanol, amplified by 20 cycles of PCR and double-stranded DNA products of 60 bp were isolated by gel electrophoresis as before. Binding of each pool of selected sequences was assessed by gel mobility shift assays. After four rounds of selection and amplification, no further increase in the specific activity of the libraries as assessed by gel mobility shift activity was detected (data not shown). The products arising from selection cycle 4 then were cloned into pBSK and sequenced.
BACs and BAC DNA preparation
BAC 269-21 containing Zipro1 tagged with an internal ribosome entry site–enhanced green fluorescent protein (IRES–EGFP) translation cassette in 3′-untranslated sequences and an SV40 promoter in the pBelo BAC vector was obtained from X.Yang and N.Heintz (Rockefeller University, New York, NY). BACs containing the human TP53 gene locus were identified in a library screen by Research Genetics using a PCR probe generated with the genomic primers spanning exon 2 described by Toguchida et al. (17). Candidate clones were purified to homogeneity and screened for exons 1–10 by single-stranded conformation polymorphism (SSCP) analysis and direct DNA sequencing (17). BAC DNA was prepared with Concert kits (Life Technologies) using conditions suggested by the supplier.
DNA transfer by GST–Z2
For studies with BAC 269-21, CHOC 400 cells (18) were plated in 35 mm dishes on coverslips in DMEM with 10% fetal bovine serum (FBS) such that they were ∼50% confluent the following day. On the day of transfection, 2.5 µg of BAC 269-21 DNA was mixed with 2.5 µg GST–Z2 in 20 mM Tris–HCl pH 7.5, 5 mM KCl, 5 mM MgCl2, 1 mM β-mercaptoethanol, 2 mM ZnCl in a total volume of 40 µl. After incubation for 20 min at room temperature, the binding reaction was diluted to 100 µl with serum-free DMEM containing penicillin/streptomycin (SFM). In parallel, 8 µl of Lipofectamine (Life Technologies) was mixed with 92 µl SFM. The diluted GST–Z2–DNA binding reaction was then mixed 1:1 with SFM containing Lipofectamine and incubated for 15 min. Fifteen minutes prior to transfection, medium was removed from the culture dishes and 0.8 ml SFM was added. The GST–Z2–Lipofectamine solution (200 µl) was then added to 0.8 ml of SFM on each plate and gently mixed. Three hours later, 1.5 ml of DMEM plus 10% FBS with penicillin/streptomycin was added. One to three days later, coverslips were removed, rinsed twice in phosphate-buffered saline (PBS) and the cells were fixed in PBS containing 3% para-formaldehyde. GFP expression was evaluated by confocal microscopy. Standard transfection with Lipofectamine of pK7-GFP, a pCMV-GFP plasmid expression vector, was used as a control.
For transfections with TP53 BAC 66H22, 1.5 × 105 CHOC 400 cells were plated in 35 mm dishes in DMEM containing 10% FBS. The following day, cells were incubated in serum- free medium with antibiotics for 30 min prior to the addition of BAC DNA–GST–Z2–Lipofectamine complexes prepared as above except that 3.0 µg of DNA was used per plate. Four days after transfection cells were harvested in SDS sample buffer and analyzed for p53 expression by immunoblotting with pAb240 as before (19).
Selection for DHFR activity in CHO DUKX cells
The CHO DUKX cell line, which lacks DHFR activity (20), was propagated in α-MEM supplemented with 10% FBS, 1 mM glutamine, 1 µg/ml adenosine and 1 µg/ml thymidine. Twenty-four hours after transfer of total genomic CHOC 400 DNA by either the GST–Z2–Lipofectamine method or calcium phosphate co-precipitation (15), the culture medium was changed to selection medium (α-MEM, 10% dialyzed FBS, 1 mM glutamine). In selection medium CHO DUKX cells lacking DHFR die within 10 days (21). After 14–21 days in selection medium, the cultures were washed with PBS, fixed in PBS with 2% para-formaldehyde and colonies were counted by phase contrast microscopy. Total genomic DNA was prepared from CHOC 400 cells as described previously (22).
RESULTS
Binding site specificity of GST–Z2
RIP60 contains 15 C2H2-Kruppel-like zinc finger (ZF) DNA binding motifs organized in three hands, which we have termed Z1 (ZFs 1–5), Z2 (ZFs 6–8) and Z3 (ZFs 9–15) (15). Located between hand Z2 and Z3 is a proline-rich region (18 of 58 residues are proline) that contains three segments of repeated (P-X-X)n motifs (n = 3 or 4) that are predicted to form three polyproline II helices. The smallest GST fusion of RIP60 with stable DNA binding and looping activities that we have identified to date is GST–Z2, which contains ZFs 6–8 and two of the predicted polyproline helices of RIP60. Upon binding to specific RIP60 sites, GST–Z2 forms multimers on DNA, protecting as much as 150 bp of flanking DNA from nuclease cleavage (15). AFM (see below) and ligation enhancement assays (data not shown) indicated that GST–Z2 bound avidly to substrates not known to contain consensus RIP60 sites.
To determine the sequence preference of GST–Z2, binding site selection experiments were performed (see Materials and Methods). While full-length RIP60 binds with strong preference to the dhfr oriβ sequence TTTTATTATTATTATTAG, selection experiments indicated GST–Z2 interacted preferentially with sequences containing one or more ATT motifs, or stretches of four or more thymidines, or both (data not shown). As suggested by other studies (15), the binding site selection assays indicate the binding specificity of GST–Z2 is relaxed as compared to full-length RIP60.
Binding of GST-Z2 to plasmids and BACs
AFM was used to examine the binding of GST–Z2 to a variety of supercoiled, linear or relaxed circular substrates. Binding of GST–Z2 to pCH127 (1), a plasmid that contains both the upstream (URS) and downstream (DRS) RIP60 binding sites from dhfr oriβ, results in a protein–DNA complex with three prominent lobes (Fig. 1A). The plasmid DNA within these complexes appeared to be either supercoiled (Fig. 1A, filled arrows) or relaxed (Fig. 1A, open arrows). For complexes containing supercoiled plasmids, the DNA appeared to be wound around or between the protein lobes (Fig. 1B). GST–Z2 also bound pOICAT plasmid DNA that lacks RIP60 binding sites, but binding did not result in the formation of trilobular complexes (Fig. 1C). Binding to linear DNA fragments resulted in complexes with 2–6 DNA molecules (Fig. 1D). Linking of linear molecules did not require the presence of specific RIP60 binding sites (Fig. 1D and data not shown).
Figure 1.

AFM of Z2–DNA complexes. GST–Z2 was incubated with plasmid DNA substrates and the resulting complexes were examined by AFM. (A) GST–Z2–pCH127 complexes. pCH127, a derivative of pOICAT, contains a 1.1 kb fragment from dhfr oriβ that includes both the DRS and URS RIP60 binding sites (15). Filled arrows, binding to supercoiled plasmid DNA; open arrows, complexes with relaxed circular DNA. (B) Higher magnification of GST–Z2 bound to supercoiled pCH127 DNA: note the trilobular structure of the protein complexes. (C) Binding of GST–Z2 to pOICAT. pOICAT is not known to contain consensus RIP60 binding sites, and trilobular protein complexes were not observed on this substrate. (D) Linking of linear DNA by GST–Z2. GST–Z2 was bound to linear pOICAT DNA using the same conditions as for pCH127. Complexes were observed that contained as many as six linear plasmid molecules, suggesting GST–Z2 is able to link DNA molecules.
Condensation and gene transfer of BAC DNA by GST–Z2
Transfection of DNA into cells involves a number of steps, most of which are poorly understood. Calcium phosphate, poly(ethylenimine), cationic lipids and other transfection reagents condense DNA into particles that are adsorbed to the cell surface and internalized, either by endocytosis, receptor-mediated transport or direct penetration of the membrane (reviewed in 23,24). The ability of GST–Z2 to mediate DNA looping and linking in molecules lacking known RIP60 binding sites suggested that GST–Z2 might act as a condensation reagent suitable for the transfer of large DNA molecules into mammalian cells.
When examined by AFM, preparations of BAC DNA contained a mixture of supercoiled, relaxed circular and linear molecules (Fig. 2A). When incubated with purified BAC 269-21 DNA (∼130 kb), GST–Z2 induced the formation of complexes containing multiple DNA loops (Fig. 2B). Estimation of the size of individual DNA loops from photomicrographs ranged between 10 and 25 kb (Fig. 2B). The ratio of protein to DNA and the concentration of DNA in the binding reaction had an effect on the formation of looped complexes: at high protein to DNA ratios intermolecular linking was favored, while at lower protein to DNA concentrations intramolecular condensation was observed (data not shown).
Figure 2.

Looping of BAC DNA by GST–Z2. (A) BAC 269-21 DNA was isolated by modified alkaline lysis and visualized by AFM. A single linear BAC molecule is shown. (B) BAC 269-21 DNA was incubated with GST–Z2 and examined by AFM. Individual BAC–GST–Z2 complexes displayed prominent protein cores with multiple DNA loops, which ranged in size from 5 to 25 kb.
BAC 269-21 contains an IRES–EGFP translation cassette in the 3′-untranslated region of Zipro1, a gene encoding a ZF transcription factor expressed in the brain and epidermis (25). BAC 269-21 also contains an SV40 promoter in the BAC vector such that Zipro1 is expressed in other cell types. BAC 269-21 DNA was incubated first with GST–Z2 for 15 min, and the protein–DNA complexes were then diluted in SFM containing Lipofectamine (Life Technologies) as described in the Materials and Methods. AFM indicated addition of cationic lipid condensed the DNA loops onto the GST–Z2–BAC DNA core (data not shown).
When incubated with BAC 269-21–GST–Z2–Lipofectamine complexes GFP-positive cells were observed as early as 24 h after addition of medium containing DNA. By 4 days post-transfection, transfer of BAC 269-21 into CHOC 400 cells resulted in a high proportion of GFP positive cells (Fig. 3D) as compared to control preparations prepared without GST–Z2 (Fig. 3C). In contrast to the diffuse GFP signal observed by standard transfection with the pCMV-GFP plasmid expression vector pK7-GFP (Fig. 3B), the GFP signal from BAC 269-21 was punctate in nature. Examination of cells expressing GFP from BAC 269-21 at high magnification showed that the GFP-positive cells were dead or dying from apoptosis: the cells were rounded up, extruded from the culture plate and displayed cell remnants typical of apoptotic cells (Fig. 3E and F). These results indicate that elevated expression of Zipro1 induces apoptosis, and that both EGFP and Zipro1 were expressed upon transfer into CHOC 400 cells.
Figure 3.

Gene transfer by GST–Z2 and Lipofectamine. GST–Z2 was incubated with BAC 269-21 DNA and then diluted in SFM containing Lipofectamine. The gene transfer complexes were added to subconfluent CHOC 400 cells and expression of EGFP from bicistronic Zipro1–IRES–EGFP mRNA was evaluated by confocal microscopy 4 days later. (A) Untransfected cells. (B) Cells transfected with a CMV-GFP expression plasmid (pK7-GFP) with Lipofectamine as a control for GFP expression. (C) Cells transfected with BAC 269-21 DNA using Lipofectamine and Plus reagent. (D) Cells transfected with BAC 269-21 using GST–Z2 and Lipofectamine. (E) Phase contrast image of cells in (D) at higher magnification. (F) GFP expression in the field of cells shown in (E). Note condensation and extrusion of GFP-positive cells from the culture dish, a morphology consistent with apoptosis.
Other experiments show BAC 269-21 was readily transferred by complexes containing GST–Z2 into several cell types, including CHO, COS and U2OS cells (data not shown). When the number of GFP-positive cells was estimated as a percentage of cells present at the time of transfection, efficiency of gene transfer in four experiments ranged from 45 to 70%. In typical experiments, GFP-positive cells were observed after 2 days at a level near 70% of the initial cell population (data not shown).
Transfer of total genomic DNA by GST–Z2
Mammalian DNA is known to bear covalent modifications (i.e. methylated cytosines) that have important consequences for gene expression. Upon cloning in bacteria post-replication modifications are lost, and procaryotic modifications of sequences may be introduced, perforce resulting in loss of specific patterns of methylation or other modifications. To test direct transfer of genomic DNA from cell to cell, total DNA from CHOC 400 cells was transferred by either GST–Z2 or calcium phosphate coprecipitation into DHFR– CHO DUKX cells (20). The average size of the genomic DNA by pulse field electrophoresis was ∼100 kb (Fig. 4A). After 2–3 weeks of selection for DHFR activity, colonies were counted. With 10 µg of total genomic DNA per 100 mM culture dish, gene transfer with GST–Z2 yielded ∼10-fold more primary DHFR+ colonies (∼500 colonies per 100 mM dish) than did calcium phosphate co-precipitation (Fig. 4B). Remarkably, the DHFR+ colonies generated by GST–Z2–Lipofectamine method appeared 7–10 days earlier during selection than did those that arose from calcium phosphate co-precipitation (data not shown).
Figure 4.
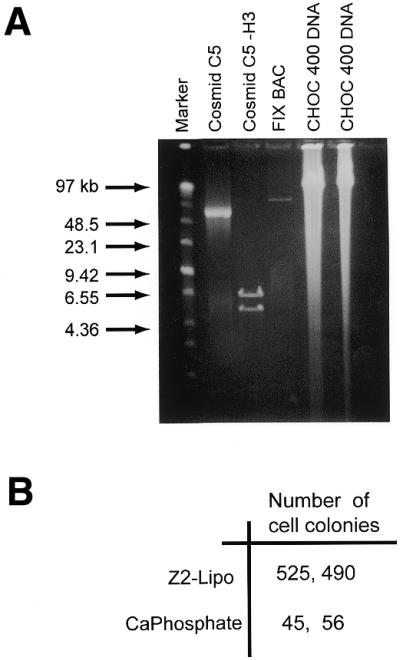
Transfer of genomic DNA by GST–Z2–Lipofectamine or calcium phosphate co-precipitation. (A) Total genomic DNA was prepared from CHOC 400 cells and analyzed by pulse field gel electrophoresis. Cosmid C5 (55 kb) and a BAC that includes the mouse Factor IX gene (90 kb) were used as markers. (B) Replicate plates of CHO DUKX cells were transfected with 10 µg of total CHOC 400 DNA using either the GST–Z2–Lipofectamine or calcium phosphate co-precipitation method. Two to three weeks after selection for DHFR activity, cell colonies were counted. Well-defined colonies were visible after 14 days in the cultures transfected by GST–Z2–Lipofectamine method, whereas discrete colonies were not visible until 21 days after transfection by the calcium phosphate co-precipitation method.
Expression of human p53 from TP53 BAC 66H22
Elevated expression of tumor suppressor gene products, transcription factors, cyclins or other cell cycle regulators from plasmid vectors with viral promoters often leads to supraphysiological levels of protein expression, and consequently profound alterations in cell growth control. For example, expression of the checkpoint protein p53 from plasmid constructs can lead to transcriptional squelching, cell cycle arrest and apoptosis, even in the absence of cellular insult (26–29). In an effort to restore p53 expression to wild-type levels in cells lacking p53, we screened a human BAC DNA library for the TP53 gene locus. From a series of candidate clones, analysis by SSCP and direct sequencing showed BAC clone 66H22 contained wild-type sequences for the entire coding region of the human TP53 gene (Fig. 5).
Figure 5.

Analysis of TP53 BACs by SSCP and direct DNA sequencing. Purified DNA from the indicated TP53 BACs was analyzed by SSCP and direct DNA sequencing. (A) Shown is the SSCP analysis for exons 2–5. Exon 10 appears wild-type in all BACs by SSCP but was not sequenced. Arrows indicate altered migration of PCR products for exons with nucleotide polymorphisms (exons 2–4) or mutations (exon 5). (B) Summary of analysis of TP53 BACs by SSCP and direct DNA sequencing. SFT indicates band shifts that were correlated with sequence polymorphisms or mutations as detected by direct DNA sequencing. BAC 66H22 appears to contain the full-length, wild-type human TP53 gene.
While CHOC 400 cells express a mutant form of Chinese hamster p53 (30), the hamster protein is not recognized by several antibodies to human p53 (data not shown). As a test for p53 expression from its own promoter region, we transfected CHOC 400 cells with BAC 66H22 and examined cell lysates for p53 by immunoblotting. When transfected with 2.0 µg of pCMV-p53, CHOC 400 cells expressed very large amounts of p53, much of which was constitutively nuclear, polyubiquitinated or degraded (Fig. 6, lanes 1–4, and data not shown). In addition, cells transfected with pCMV-p53 died from apoptosis due to elevated expression of p53, and consequently could not be propagated (data not shown). In contrast, CHOC 400 cells transfected with BAC 66H22 using the GST–Z2–Lipofectamine co-condensation method expressed full-length p53 at levels that did not lead to significant ubiquitination or degradation of the protein (Fig. 6, lanes 7–8). Moreover, in contrast to cells transfected with pCMV-p53, cells transfected with BAC 66H22 showed no signs of overt cytotoxicity. While treatment of cells transfected with BAC 66H22 with cis-platinum for 4 h appeared to induce a modest increase in p53 levels, further studies will be required to determine if human p53 expressed from 66H22 is phosphorylated and stabilized in response to DNA damage, and consequently mediates a DNA damage checkpoint in hamster or human cells. As expected, shorter exposures of the blot shown in Figure 6 showed cis-platinum had no effect on the expression profile of p53 from pCMV-p53 (data not shown).
Figure 6.

Expression of p53 from TP53 BAC 66H22. CHOC 400 cells were transfected with either pCMV-p53 or BAC 66H22 using Plus Reagent and Lipofectamine (Plus/Lipo) or GST–Z2 and Lipofectamine (Z2/Lipo). Plus Reagent and Lipofectamine were from Life Technologies, Inc. (see Materials and Methods). After 4 days, total cell lysates were examined for expression of p53 by immunoblotting with pAb240. Replicate cultures were treated with cis-platinum for 4 h as indicated. Arrow, full-length p53; bracket, ubiquitinated p53.
From a series of BAC transfection experiments similar to those shown here we estimate that condensation by GST–Z2 improves the efficiency of transfer of large molecules into cells in culture ∼50–100-fold (Fig. 6, compare lanes 5 and 6 with lanes 7 and 8). We have recently generated modified alleles of BAC 66H22 that contain a CMV–RFP expression cassette in the pBelo BAC vector (S.Phelps and N.H.Heintz, unpublished data). Introduction of these and other modified BAC alleles into cells will permit us to directly measure transfection efficiency, expression of p53 or other proteins and the fate of transfected BAC DNA over time.
DISCUSSION
An efficient method for the transfer of BACs, yeast artificial chromosomes (YACs) and other large DNA molecules into mammalian cells would facilitate genetic analysis of specific cis-acting regulatory elements that act over large distances in the context of native flanking sequences. Moreover, the ability to transfer BACs containing targeted modifications into cells in culture would permit evaluation of modified alleles prior to generating BAC transgenic mice. The ability to transfer imprinted genes or other modified sequences directly from one cell to another should also prove useful. Here we have described a small recombinant fusion protein that facilitates transfer of large linear or circular DNA molecules into mammalian cells.
A working model for gene transfer by GST–Z2 is presented in Figure 7. Based on the binding site selection experiments, the protein appears to bind to ATT or T-rich sequences, which are likely to occur at random with considerable frequency in DNA from mammalian cells. Upon initial binding to ATT or T-rich sequences, GST–Z2 nucleates the formation of multimers on neighboring DNA, leading to protection of 150–200 bp of sequence. AFM suggests GST–Z2 does not saturate DNA, even at high protein concentrations, but rather forms discrete multimeric complexes, even on substrates that lack known RIP60 sites (Fig. 1C). On BAC 269 molecules the GST–Z2 protein complexes were located at 5–25 kb intervals along the entire 130 kb BAC DNA substrate. Based on the ability of RIP60 to mediate DNA looping (15), we suggest protein–protein interactions between multimeric GST–Z2 complexes then foster the formation of DNA loops, resulting in a complex with a single protein core and multiple free DNA loops. Addition of cationic lipid (or other agents) may then be used to collapse the DNA loops onto the protein core, resulting in a stable, compact particle. Upon binding the cell surface, the complex is internalized, ferried to the cell nucleus and the DNA is transcribed. Because GFP–Z2 locates to the cell nucleus (15), it is also possible that GST–Z2 promotes nuclear import. Preliminary experiments indicate that phosphorylation of the proline-rich region of GST–Z2 in vitro by casein kinase II prevents multimerization and DNA binding (J.Gilbert, T.Messit, C.Houchens and N.H.Heintz, unpublished data), suggesting phosphorylation of the multimerization domain may release GST–Z2 from DNA in vivo. If so, this feature of the transfer system would provide a mechanism for the efficient dissolution of gene delivery complexes upon cell entry.
Figure 7.
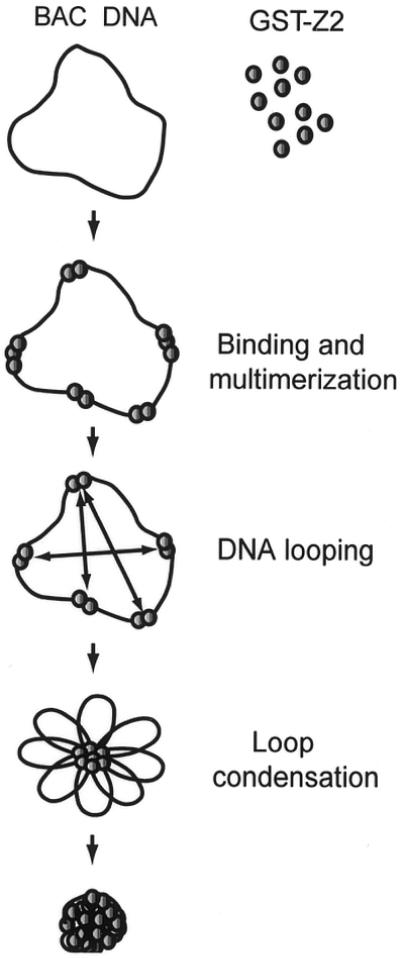
Model for DNA looping by GST–Z2 in gene transfer. Shown is a working model for DNA looping and condensation of BAC DNA by GST–Z2 and Lipofectamine. See text for details.
Transfer of multiple BACs and total genomic DNA into cells shows that the DNA substrates need not contain RIP60 binding sites for condensation by GST–Z2, thereby obviating introduction of specific binding sites into the substrate. In addition, linear, relaxed circular or supercoiled substrates are all bound by GST–Z2. As a recombinant protein, GST–Z2 may be modified by protein engineering to contain specific motifs, such as nuclear localization sequences, protein degradation domains or cell targeting sequences. Moreover, the DNA loops emanating from GST–Z2–DNA complexes may be decorated with peptides, polymers or conjugates to encourage uptake by specific cell types. Hence, combined with other methods, GST–Z2 may represent one element of a flexible, non-viral platform for gene delivery in vitro and in vivo.
Acknowledgments
ACKNOWLEDGEMENTS
We thank W.Yang and N.Heintz (Rockefeller University, New York, NY) for the BAC 269-21 bacterial strain, Jon Horowitz for the library of oligonucleotides containing randomized sequences and T.Quinn and D.Taatjes of the UVM Cell Imaging Facility for assistance with AFM. This work was supported by the Vermont Cancer Center and the Lake Champlain Cancer Research Organization (LCCRO). Y.-C.C. was supported in part by a J. Walter Juckett Fellowship from the LCCRO, J.G. by a mini-grant from the UVM HELiX program supported by the Howard Hughes Medical Institute and E.C. by an internship from the UVM Department of Microbiology and Molecular Genetics.
References
- 1.Bulger M. and Groudine,M. (1999) Looping versus linking: toward a model for long-distance gene activation. Genes Dev., 13, 2465–2477. [DOI] [PubMed] [Google Scholar]
- 2.Cockell M. and Gasser,S.M. (1999) Nuclear compartments and gene regulation. Curr. Opin. Genet. Dev., 9, 199–205. [DOI] [PubMed] [Google Scholar]
- 3.Gerasimova T.I. and Corces,V.G. (1996) Boundary and insulator elements in chromosomes. Curr. Opin. Genet. Dev., 6, 185–192. [DOI] [PubMed] [Google Scholar]
- 4.Blackwood E.M. and Kadonaga,J.T. (1998) Going the distance: a current view of enhancer action. Science, 281, 61–63. [DOI] [PubMed] [Google Scholar]
- 5.DePamphilis M.L. (1999) Replication origins in metazoan chromosomes: fact or fiction? Bioessays, 21, 5–16. [DOI] [PubMed] [Google Scholar]
- 6.Heintz N.H. (1996) Mammalian DNA replication. In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, New York, NY, pp. 983–1004.
- 7.Yang X.W., Model,P. and Heintz,N. (1997) Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol., 15, 859–865. [DOI] [PubMed] [Google Scholar]
- 8.Narayanan K., Williamson,R., Zhang,Y., Stewart,A.F. and Ioannou,P.A. (1999) Efficient and precise engineering of a 200 kb β-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther., 6, 442–447. [DOI] [PubMed] [Google Scholar]
- 9.Muyrers J.P., Zhang,Y., Testa,G. and Stewart,A.F. (1999) Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res., 27, 1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jessen J.R., Meng,A., McFarlane,R.J., Paw,B.H., Zon,L.I., Smith,G.R. and Lin,S. (1998) Modification of bacterial artificial chromosomes through χ-stimulated homologous recombination and its application in zebrafish trans-genesis. Proc. Natl Acad. Sci. USA, 95, 5121–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Bucholz,F., Muyrers,J. and Stewart,A. (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet., 20, 123–128. [DOI] [PubMed] [Google Scholar]
- 12.Orford M., Nefedov,M., Vadolas,J., Zaibak,F., Williamson,R. and Ioannou,P. (2000) Engineering EGFP reporter constructs into a 200 kb human β-globin BAC clone using GET recombination. Nucleic Acids Res., 28, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinnett D., Richer,C. and Baccichet,A. (1998) Isolation of stable bacterial artificial chromosome DNA using a modified alkaline lysis method. Biotechniques, 24, 752–754. [DOI] [PubMed] [Google Scholar]
- 14.Magae J., Wu,C.-L., Illenye,S., Harlow,E. and Heintz,N.H. (1996) Nuclear localization of DP and E2F transcription factors by heterodimeric partners and retinoblastoma protein family members. J. Cell Sci., 109, 1717–1726. [DOI] [PubMed] [Google Scholar]
- 15.Houchens C.R., Montigny,W., Zeltser,L., Dailey,L., Gilbert,J. and Heintz,N.H. (2000) The dhfr oriβ-binding protein RIP60 contains 15 zinc fingers: DNA binding and looping by the central three fingers and an associated proline-rich region. Nucleic Acids Res., 28, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tao Y., Kassatly,R.F., Cress,W.D. and Horowitz,J.M. (1997) Subunit composition determines E2F DNA-binding site specificity. Mol. Cell. Biol., 17, 6994–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toguchida J., Yamaguchi,T., Ritchie,B., Beauchamp,R., Dayton,S., Herrera,G., Yamamuro,K., Sasaki,M., Little,J., Weichselbaum,R., Ishizaki,K. and Yandell,D. (1992) Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res., 52, 6194–6199. [PubMed] [Google Scholar]
- 18.Milbrandt J.D., Heintz,N.H., White,W.C., Rothman,S.M. and Hamlin,J.L. (1981) Methotrexate-resistant Chinese hamster ovary cells have amplified a 135-kilobase-pair region that includes the gene for dihydrofolate reductase. Proc. Natl Acad. Sci. USA, 78, 6042–6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang Y.-C., Lee,Y.-S., Tejima,T., Tanaka,K., Omura,S., Heintz,N.H., Mitsui,Y. and Magae,J. (1998) Mdm2 and bax, downstream mediators of the p53 response, are degraded by the ubiquitin-proteosome pathway. Cell Growth Differ., 9, 79–84. [PubMed] [Google Scholar]
- 20.Urlaub G. and Chasin,L.A. (1980) Isolation of Chinese hamster cell mutants deficient in dihydrofolate reductase activity. Proc. Natl Acad. Sci. USA, 77, 4216–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brinton B.T. and Heintz,N.H. (1995) Plasmid amplification-promoting sequences from the origin region of Chinese hamster dihydrofolate reductase gene do not promote position-independent chromosomal gene amplification. Chromosoma, 104, 143–151. [DOI] [PubMed] [Google Scholar]
- 22.Heintz N.H. and Hamlin,J.L. (1982) An amplified chromosomal sequence that includes the gene for dihydrofolate reductase initiates replication within specific restriction fragments. Proc. Natl Acad. Sci. USA, 79, 4083–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilaro A.M. and Serabian,M.A. (1999) Preclinical development strategies for novel gene therapeutic products. Toxicol. Pathol., 27, 4–7. [DOI] [PubMed] [Google Scholar]
- 24.Beutler E. (1999) Gene therapy. Biol. Blood Marrow Transplant., 5, 273–276. [DOI] [PubMed] [Google Scholar]
- 25.Yang X.W., Wynder,C., Doughty,M.L. and Heintz,N. (1999) BAC-mediated gene-dosage analysis reveals a role for Zipro1 (Ru49/Zfp38) in progenitor cell proliferation in cerebellum and skin. Nat. Genet., 22, 327–335. [DOI] [PubMed] [Google Scholar]
- 26.Magae J., Illenye,S., Tejima,T., Chang,Y.-C., Mitsui,Y., Tanaka,K., Omura,S. and Heintz,N.H. (1997) Transcriptional squelching by ectopic expressio of E2F-1 and p53 is alleviated by proteosome inhibitors MG-132 and lactacystin. (1997) Oncogene, 15, 759–769. [DOI] [PubMed] [Google Scholar]
- 27.Chowdary D., Dermody,J., Jha,K. and Ozer,H. (1994) Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol. Cell. Biol., 14, 1997–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maki C., Huibregtse,J. and Howley,P. (1996) In vivo ubiquitination and proteosome-mediated degradation of p53. Cancer Res., 56, 2649–2654. [PubMed] [Google Scholar]
- 29.Yu J., Zhang,L., Hwang,P.M., Rago,C., Kinzler,K. and Vogelstein,B. (1999) Proc. Natl Acad. Sci. USA, 96, 14517–14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H., Larner,J.M. and Hamlin,J.L. (1997) Cloning and characterization of Chinese hamster p53 cDNA. Gene, 184, 177–183. [DOI] [PubMed] [Google Scholar]