

This study demonstrated a reproducible protocol for reprogramming blood cells into transgene-free induced pluripotent stem cells (iPSCs) using Sendai viral vectors. Creation of iPSCs, without integration of exogenous DNA, helps preserve genomic integrity and fosters the development of therapeutic-grade stem cells for regenerative medicine.
Keywords: CD34+, Reprogramming, Hematopoietic progenitors, Differentiation
Abstract
The discovery of induced pluripotent stem cells (iPSCs) holds great promise for regenerative medicine since it is possible to produce patient-specific pluripotent stem cells from affected individuals for potential autologous treatment. Using nonintegrating cytoplasmic Sendai viral vectors, we generated iPSCs efficiently from adult mobilized CD34+ and peripheral blood mononuclear cells. After 5–8 passages, the Sendai viral genome could not be detected by real-time quantitative reverse transcription-polymerase chain reaction. Using the spin embryoid body method, we showed that these blood cell-derived iPSCs could efficiently be differentiated into hematopoietic stem and progenitor cells without the need of coculture with either mouse or human stromal cells. We obtained up to 40% CD34+ of which ∼25% were CD34+/CD43+ hematopoietic precursors that could readily be differentiated into mature blood cells. Our study demonstrated a reproducible protocol for reprogramming blood cells into transgene-free iPSCs by the Sendai viral vector method. Maintenance of the genomic integrity of iPSCs without integration of exogenous DNA should allow the development of therapeutic-grade stem cells for regenerative medicine.
Introduction
Terminally differentiated somatic cells can be reprogrammed by defined transcription factors into induced pluripotent stem cells (iPSCs) that are morphologically and functionally similar to embryonic stem cells (ESCs) [1, 2]. The development of iPSC technology opens up a new avenue for the field of personalized regenerative medicine since it is possible to generate patient-specific pluripotent stem cells for modeling disease [3–5] and for developing cell therapies [6–8]. iPSCs that maintain original genome integrity and do not carry integrated viral vector sequences or transcription factor DNA are highly desirable if they are to be considered for clinical application. Many reprogramming methods are being investigated to achieve this goal; examples include the use of adenoviral vectors [9], episomal vectors [10], plasmids [11, 12], minicircle vectors [13], synthetic mRNA [14], miRNAs [15], protein transduction [16, 17], and chemical compounds. All of these methods have some restrictions, such as having a low reprogramming efficiency, requiring serial transgene deliveries, or being successful only with some specific types of somatic cells, such as the commonly used fibroblasts.
Although fibroblasts are a popular source for deriving iPSCs, they require skin biopsy and cell culture for several passages to provide enough cells for reprogramming. Early passages of primary cells are required as late passage cells are difficult to reprogram. In contrast to these requirements, peripheral blood-derived mononuclear cells (MNCs) can be obtained by routine and safe procedures. As several studies have suggested that donor cell type can influence the epigenome and differentiation potential of iPSCs [18, 19], blood cell-derived iPSCs appear to differentiate back to the hematopoietic lineages more efficiently than fibroblast-derived iPSCs [20]. With these advantages, blood cells may be the ideal source for generation of iPSCs from blood disorders. Previously, peripheral blood CD34+ and mononuclear cells from adults have been reprogrammed into iPSCs using integrating and nonintegrating viral [21–23] or DNA vectors [24]. In this study we explored the use of a nonintegrating RNA virus to reprogram easily accessible adult blood cells.
Sendai virus is a negative sense single-stranded RNA virus that replicates only in the cytoplasm and does not integrate into the host genome [25]. Sendai viral vectors (SeVs) encoding OCT4, SOX2, KLF4, and c-MYC have been reported to reprogram a small number of blood cells with high efficiency [23]. Temperature-sensitive SeVs generated by specific mutations of the viral genome allow rapid removal of residual viral genomic RNA from the reprogrammed cells [26]. Thus, the transcription factor- and virus-free iPSCs have been generated from terminally differentiated T lymphocytes [23] and from CD34+ cells from cord blood [27].
Our laboratory has been interested in studying sickle cell anemia and thalassemia for some years and is now exploring the feasibility of using the iPSC technology to treat these diseases [28]. The proposed approach is to generate iPSCs from these patients, correct the β-globin-gene mutations, and differentiate the iPSCs to hematopoietic cells for autotransplantation. In this study, we explored the conditions for reprogramming either mobilized CD34+ cells or peripheral blood MNCs from adults. We found that MNCs can be efficiently reprogrammed into iPSCs as readily as CD34+ cells. Furthermore, these MNCs derived iPSCs can be terminally differentiated into mature blood cells.
Materials and Methods
Cell Preparation and Culture
Frozen human peripheral blood mobilized CD34+ cells were purchased from AllCells (Emeryville, CA, http://www.allcells.com). After thawing, CD34+ cells were cultured with serum-free medium (SFM) as described by Ye and coworkers [24, 29], containing 50% Iscove's modified Dulbecco's medium, 50% Ham's F12, 2 mM glutamine, 1× CD lipid concentrate (all from Invitrogen, Carlsbad, CA, http://www.invitrogen.com), 1× insulin-transferrin-selenium supplement, and 50 μg/ml ascorbic acid (both from Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com), supplemented with the recombinant human cytokines: 100 ng/ml stem cell factor (SCF), 100 ng/ml Flt3 ligand (FL), 20 ng/ml thrombopoietin (TPO), and 10 ng/ml interleukin 3 (IL-3) (all from R&D Systems Inc., Minneapolis, MN, http://www.rndsystems.com). For isolation of MNCs, 2 ml of peripheral blood was collected by venipuncture from one male and one female healthy donor. Two milliliters of heparinized blood was diluted with an equal volume of Dulbecco's phosphate-buffered saline (D-PBS) and layered over 4 ml of Ficoll-Hypaque Premium (GE Healthcare, Little Chalfont, U.K., http://www.gehealthcare.com) gradient, according to the manufacturer's instructions, and centrifuged at 1,200g for 40 minutes, yielding 5 × 106 MNCs. Of the MNCs obtained, 1–2 × 106 were cultured with SFM supplemented with the following cytokines: 50 ng/ml SCF, 10 ng/ml IL-3, 2 U/ml erythropoietin (EPO), 40 ng/ml insulin-like growth factor 1 (IGF-1) (all from R&D Systems), and 1 μg/ml dexamethasone (Sigma-Aldrich). The medium was changed on day 3 and day 6. The day 5 and day 8 cultured cells were used for reprogramming. The human iPSCs were maintained in human embryonic stem (ES) medium containing 20% knockout serum replacement, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, 50 U/ml penicillin, 50 μg/ml streptomycin, and 8 ng/ml basic fibroblast growth factor (bFGF) in Dulbecco's Modified Eagle's Medium: Nutrient Mixture F-12 (DMEM/F12) (all from Invitrogen).
Generation of iPSCs With Sendai Viral Vectors
The thawed mobilized peripheral blood CD34+ cells cultured for 2 days in CD34+ culture medium described in the previous section were then infected with SeV. The freshly prepared MNCs were cultured in MNC medium for 5–8 days. Then, 1–2 × 104 CD34+ cells or MNCs were placed in 1 well of a 96-well plate and infected with the CytoTune-iPS reprogramming kit (kindly provided by the DNAVEC Corporation, Tsukuba, Japan, http://www.dnavec.co.jp/en/) containing five F-deficient Sendai virus vectors (Sev/ΔF) encoding OCT4, SOX2, KLF4, cMYC, and green fluorescent protein (GFP) in SFM at a multiplicity of infection (MOI) of 5 or 10 for each factor. One day after infection, the cells were harvested and plated onto two wells layered with mouse embryonic fibroblast (MEF) feeders in a six-well plate and cultured in the same medium for an additional day. On day 2, the medium was changed to human ES medium supplemented with 8 ng/ml bFGF and replenished every day with fresh medium. Colonies with morphology similar to that of ES colonies started to appear on day 13 after infection; they were picked on day 21 or 28, expanded, and examined for pluripotency markers. The frequency of expandable clones was measured by counting the colonies that could be expanded in the first two passages among the total number of TRA-1-60-positive clones that were picked up from each reprogramming experiment.
TRA-1-60 Live Staining and Immunofluorescence Staining
TRA-1-60 antibody (Millipore, Billerica, MA, http://www.millipore.com) and Alexa 555-conjugated anti-mouse IgM secondary antibody (Invitrogen) were mixed in the human ES medium and added to the reprogramming plate. The cells were incubated at 37°C for 1 hour, washed once with fresh medium, and examined for positive TRA-1-60 stain under an inverted fluorescence microscope. Additionally, immunofluorescence staining of iPSC colonies was performed using the following primary antibodies: NANOG (R&D Systems), stage-specific embryonic antigen 4 (SSEA4) (Abcam, Cambridge, MA, http://www.abcam.com), SSEA3, TRA-1-60, and TRA-1-81 (Millipore). For detection of three germ layer differention of iPSCs, the following antibodies were used: IIIβ-tubulin (Tuj) (Covance, San Diego, CA, http://www.covance.com), α-fetoprotein and Sox17 (R&D Systems), and actin α-smooth muscle (Sigma-Aldrich). The staining protocol was used as previously described [28].
Global Gene Expression Analysis
The GeneChip microarray processing was performed by the Genomics Core Laboratory and statistical analysis was performed by the Bioinformatics Core facility at the J. David Gladstone Institutes. The GeneChips we used were GeneChip Human Gene 1.0 ST arrays from Affymetrix (Santa Clara, CA, http://www.affymetrix.com). The detailed procedure is shown at http://labs.gladstone.ucsf.edu/genomics/.
Reverse Transcription-Polymerase Chain Reaction and Real-Time Polymerase Chain Reaction
Total RNA was extracted from iPSCs and treated with DNase using the RNeasy mini kit (Qiagen, Hilden, Germany, http://www.qiagen.com). Random-primed RNA was reverse transcribed to cDNA using the Superscript III (Invitrogen). AccuPower PCR-Premix (Bioneer, Alameda, CA, http://us.bioneer.com) was used for polymerase chain reaction (PCR) amplification using the primer sets provided by the manufacturer to detect Sendai viral vector and each of the reprogramming genes. The other primer sets for detecting ES marker gene expression were used as previously described [30]. For real-time PCR to detect Sendai viral genome, cDNA was diluted 1:5, and 2 μl was used in a total of 20 μl of PCR using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, http://www.bio-rad.com).
Immunoglobulin Heavy Chain and T-Cell Gene Rearrangements
Genomic DNA was extracted from MNC-derived iPSC lines, and PCR amplification was performed using immunoglobulin heavy chain (IGH)/T cell receptor (TCR) clonality assay kits that use the European BIOMED-2 primer sets from InVivoScribe Technologies (San Diego, CA, http://www.invivoscribe.com). For identification of clonal T-cell receptor β chain, δ chain, and γ chain gene rearrangements, the TCRB, TCRD, and TCRG gene clonality assay kits were used. For identification of clonal immunoglobulin heavy chain rearrangements, the IGH B-cell clonality assay kit was used. Six percent polyacrylamide gel electrophoresis was used to resolve the different-sized amplicon products, and ethidium bromide was used to stain and detect the products.
In Vivo Teratoma Formation and In Vitro Differentiation
For teratoma assays, iPSCs from one 10-cm dish were collected from each line by collagenase IV treatment (1 mg/ml), scraped, and collected in DMEM/F12. Cells were washed twice with DMEM/F12, aggregated by spinning at 200g for 5 minutes, and resuspended in 200 μl of DMEM/F12, and 100 μl of Matrigel was added. The total 300 μl of suspension was injected intramuscularly into the hind legs of NOD/SCID mice. Tumors were formed in 8–10 weeks, removed before they exceeded 1.5 cm in diameter, and fixed in 10% formalin for hematoxylin and eosin staining.
The protocol for spontaneous in vitro differentiation has been described previously [28]. Briefly, iPSC colonies were treated with 1 mg/ml collagenase IV (Invitrogen) for 10 minutes, washed with D-PBS, and dissociated into clumps by scraping and pipetting a few times. iPSC clumps were plated into Lipidure coat plate (NOF Corporation, Irvine, CA, http://nofamerica.net) in ES medium without bFGF. The medium was changed every other day. After 8 days of suspension culture, embryoid bodies (EBs) were transferred into a gelatin-coated plate and cultured in the same medium for an additional 8–10 days.
Hematopoietic Differentiation
The hematopoietic differentiation of CD34+- and MNC-derived iPSCs was conducted using the recombinant protein-based, animal product-free medium StemDiff APEL (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com) in combination with the spin EB method [31, 32]. One 10-cm dish of iPSCs was treated with Accutase (Millipore) for 3 minutes to dissociate into single cells. The cells were collected and washed once with StemDiff APEL and then suspended at a cell density of 5–10 × 104 cells per milliliter in StemDiff APEL medium supplemented with the following cytokines: 40 ng/ml SCF, 20 ng/ml bone morphogenetic protein 4 (BMP4), and 20 ng/ml vascular endothelial growth factor (VEGF) (all from R&D Systems). One hundred microliters of the cell suspension was aliquoted into each well of a 96-well plate with a V-shaped bottom (no. 3894; Costa; Sigma-Aldrich) using only the central 60 wells; the suspension was then spun at 480g for 2 minutes at room temperature to initiate the formation of spin EBs. One 10-cm dish was enough to make three plates. After the spinning, the plates were incubated at 37°C with 5% CO2 in air 4 days. On day 5, EBs were harvested and plated into Lipidure-coated 35-mm dishes (NOF Corporation), and the culture was continued in the same conditions. Four more cytokines (20 ng/ml FL, 20 ng/ml IL-6, 20 ng/ml TPO [all from Prospec, Ness-Ziona, Israel, http://www.prospecbio.com], and 20 ng/ml IGF-II [R&D Systems]) were added to this medium, and the medium was changed every other day.
On day 14, EBs were harvested, washed once with D-PBS, and treated with TrypLE Select (Invitrogen) for 30 minutes in a 37°C water bath. They were then passed through a 23-gauge, 1-inch needle attached to a 3-ml syringe three times to make single-cell suspensions. For the colony forming assay, 5–10 × 104 single cells were plated into MethoCult H4434 (StemCell Technologies) and cultured for 10 more days. For flow cytometric analysis and cell staining, terminally differentiated cells were developed by transferring 14-day EBs to a gelatin-coated plate and cultured in StemDiff APEL medium supplemented with 50 ng/ml SCF, 30 ng/ml VEGF, 30 ng/ml IL-3 (Prospec), 30 ng/ml IL-6, 30 ng/ml TPO, and 5 U/ml EPO (R&D Systems) for 2–3 more weeks. Single cells were collected from each well, cytospun on glass slides, and stained with Accustain Wright-Giemsa stain (Sigma-Aldrich).
Flow Cytometry Analysis
Single cells prepared from EBs were stained with fluorochrome-conjugated monoclonal antibodies (mAbs): phycoerythrin-cyanine (Cy7)-labeled anti-human CD34 (clone 581; BioLegend, San Diego, CA, http://www.biolegend.com), fluorescein isothiocyanate-labeled anti-human CD43 (clone MEM-59; BioLegend), and allophycocyanin-labeled anti-human CD45 (clone HI30; Invitrogen). The mAbs and their corresponding nonspecific isotype-matched controls were used at 1–2 μg/ml. Cells were held on ice and stained with mAbs for at least 30 minutes in D-PBS supplemented with 0.01% NaN3 (Sigma-Aldrich) and 5% normal mouse serum. Cells were then washed twice and suspended in D-PBS supplemented with 0.3% bovine serum albumin (Roche Diagnostics, Basel, Switzerland, http://www.roche-applied-science.com) and 0.01% NaN3. This buffer also contained 2 μg/ml propidium iodide (Invitrogen) to stain dead cells. Samples were analyzed using an LSR II flow cytometer (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com). Data were analyzed using FlowJo version 9 (Tree Star, Ashland, OR, http://www.treestar.com). Dead cells were eliminated from the analysis by gating on cells not stained with propidium iodide, and light-scatter emissions were used to draw gates that avoided inclusion of cell clumps and debris.
Bisulfite Sequencing
Bisulfite conversion was performed using the EpiTect Bisulfite kit (Qiagen) following the manufacturer's instructions. PCR of human OCT4 and NANOG promoter regions was performed using the specific primer sets [1]. The PCR products were cloned into pCR2.1Topo vector (Invitrogen). Seven to 10 randomly chosen clones from each induced pluripotent stem (iPS) line and 3–5 clones from parental MNC were sequenced for each gene. There were not enough CD34+ cells for genomic DNA extraction to perform this analysis; instead, the parental MNCs were used for this analysis.
Karyotyping
Two to three individual iPSC lines derived from each donor were randomly chosen for karyotyping. Standard G-banded karyotypes were conducted and interpreted by the University of California, San Francisco, cytogenetics laboratory.
Results
Reprograming Adult Peripheral Blood CD34+ and Mononuclear Cells Into iPSCs by Sendai Viral Vectors
To generate blood-derived iPSCs, we used two sources of blood cells from adults: frozen CD34+ hematopoietic stem/progenitor cells isolated from mobilized peripheral blood and MNCs isolated from 2 ml of peripheral blood by Ficoll-Hypaque density gradient centrifugation. Two CD34+ cell samples, one male and one female, were thawed and cultured in SFM containing a combination of SCF, FL, TPO, and IL-3 for 2 days to stimulate cell proliferation before they were challenged with SeV containing the reprogramming factors. To reprogram MNCs, as it has been reported that reprogramming with SeV could efficiently generate iPSCs from T lymphocytes [23], we minimized reprogramming cells of the lymphocyte lineages by culturing the MNCs in SFM containing a combination of SCF, IL-3, EPO, IGF-1, and dexamethasone for 5–8 days before SeV infection. This culture condition has been reported to support myeloerythroid survival and proliferation [24]. To test the infection efficiency of SeV for the MNCs, the 2-day and 8-day precultured MNCs were infected with 10 MOI SeV GFP vector. Twenty-four hours later, the samples were analyzed by flow cytometry. We could detect only 1.47% GFP-positive cells in the 2-day precultured MNCs, whereas the 8-day precultured cells became 91% GFP-positive (data not shown). This result indicates that preculture of blood cells for this period increased the efficiency of SeV infection.
SeVs carrying human OCT4, SOX2, KLF4, c-MYC, and GFP were used to infect 1–2 × 104 2-day precultured CD34+ cells or 5- or 8-day precultured MNCs at 5 or 10 MOI according to the protocol illustrated in Figure 1A. Twenty-four hours after infection, the cells were harvested and seeded onto MEF feeder cells and cultured with the same medium as that used before infection. On the second day after plating, the culture medium was replaced by human ES medium with bFGF. Colonies were first observed approximately 13 days after transduction and identified through positive live staining with TRA-1-60 antibody. Some colonies showed almost complete extinction of GFP fluorescence at day 25 (supplemental online Fig. 1). The reprogramming efficiencies for CD34+ and MNCs were 0.05%–0.06% and 0.02%–0.27%, respectively, as scored by TRA-1-60-positive and ES-like morphology on day 20 (Table 1). The TRA-1-60-positive colonies were picked from day 21 or day 28 after infection. The colonies picked on days 21 and 28 were found to be different in their expandable rate (16% [4 of 25] vs. 50% [8 of 16]). When the cells were replated on day 15 and picked on day 28, the expandable rate could reach 90% (18 of 20). This observation indicates that there were still many colonies that were not fully reprogrammed on day 21, and replating cells on day 15 may help to eliminate those partially reprogrammed cells.
Figure 1.

Reprogramming blood cells into transgene-free iPS cells (iPSCs) by Sendai vectors. (A): Schematic diagram of the reprogramming protocols used to generate iPSCs from CD34+ and MNCs. (B): Expression of the embryonic stem cell-related pluripotency surface markers was confirmed by the immunostaining using specific antibodies. Magnification, ×10. (C): Endogenous pluripotency markers expression was further confirmed by reverse transcription-polymerase chain reaction in several MNC- and CD34+-derived iPSC clones and two CD34+ cells after days 28 and 32 after infection. (D): Pluripotency of CD34+- and MNC-derived iPSCs was verified by spontaneous differentiation in vitro into three germ layers identified by antibodies against βIII-Tuj (ectoderm marker), aFP and Sox17 (endoderm markers), and α-SMA (mesoderm marker). Magnification, ×20 (all). (E): Pluripotency of CD34+- and MNC-derived iPSCs was confirmed by differentiation in vivo into three germ layers. Shown are neuron pigmented epithelium (ectoderm), cartilage and bone (mesoderm), and respiratory epithelium (endoderm). Magnification, ×10. Abbreviations: aFP, α-fetoprotein; a-SMA, α-smooth muscle actin; βIII-Tuj, βIII-tubulin; D, days after infection; DDW, double-distilled water; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescence protein; hES, human embryonic stem; HSC, hematopoietic stem cell; iPS, induced pluripotent stem; MEF, mouse embryonic fibroblast; MNC, mononuclear cell; OSKM, OCT3, SOX2, KLF4, and cMYC; PL, pooled CD34-induced pluripotent stem clones; SSEA, stage-specific embryonic antigen.
Table 1.
Efficiency of blood-derived induced pluripotent stem cell generation by Sendai viral vector
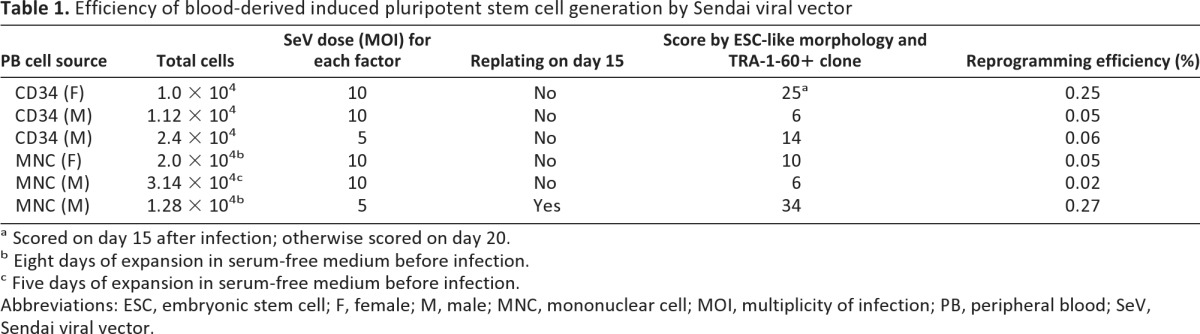
a Scored on day 15 after infection; otherwise scored on day 20.
b Eight days of expansion in serum-free medium before infection.
c Five days of expansion in serum-free medium before infection.
Abbreviations: ESC, embryonic stem cell; F, female; M, male; MNC, mononuclear cell; MOI, multiplicity of infection; PB, peripheral blood; SeV, Sendai viral vector.
After expansion, the TRA-1-60-positive clones were positive for pluripotency markers NANOG, SSEA3, SSEA4, and TRA-1-81 staining (Fig. 1B). Reverse transcription-polymerase chain reaction (RT-PCR) analysis revealed that iPSCs generated from both sources had expression of endogenous pluripotent markers NANOG, OCT4, REX1, DNMT-3B, ABCG2, and hTERT, whereas the original CD34+ cells had expression of only OCT4 (Fig. 1C). The pluripotency of iPSCs generated from them was further confirmed by EB formation and their successful differentiation into the three germ layers in vitro as well as the formation of teratomas containing tissues of the three germ layers in vivo (Fig. 1D, 1E). The promoters of NANOG and OCT4 were demethylated in both iPSCs as compared with original MNCs (supplemental online Fig. 2). We randomly chose two to three clones from each donor's reprogrammed iPSC lines for karyotyping. Karyotypes showed that all the clones from the MNCs from the male and female donors were normal (supplemental online Fig. 3). From the CD34+ cells, the karyotype from the male donor was normal, whereas that from the female donor showed trisomy 12 (supplemental online Fig. 3). We examined two other independent iPSC clones reprogrammed from this female CD34+ cells. All three clones showed the trisomy 12.
Global gene expression analysis using RNA microarrays revealed that the patterns of both CD34+- and MNC-derived iPSCs were similar overall to those of human H9 ESCs as well as to each other, and they were all highly divergent from the primary CD34+ cell and MNC (Fig. 2A, 2B). Consistent with this result, the dendrogram of unsupervised one-way hierarchical clustering analysis showed that iPSCs derived from both CD34+ and MNCs formed a branch with H9 ESCs, distinct from the branch of the original CD34+ and MNCs (Fig. 2C). Notably, global gene expression profiles failed to distinguish between CD34+-derived iPSCs and MNC-derived iPSCs. These results indicated SeV containing reprogramming factors could successfully reprogram CD34+ and MNCs from adult blood cells into iPSCs with very similar efficiency and qualities.
Figure 2.

Global gene expression analysis of blood-derived iPS cells (iPSCs). (A): Heat map comparison of 8,577 genes between primary CD34+ cells, MNCs, three lines each of iPSCs reprogrammed from CD34+ cells and MNCs, and H9 human ES cells. The horizontal top panel shows the color key and the histogram of the distribution of heat map values with a dashed line at the median value (4.47). (B): Global gene expression patterns of CD34+- and MNC-derived iPSC clones were compared with H9 ES cells, with each other (left three panels), and with parental primary cells (right two panels). R2 represents the Pearson correlation rate of two samples in log2 transformed value. (C): Dendrogram showing the unsupervised hierarchical clustering of the blood cell-derived iPSC lines, H9 human ES cells, the parental primary CD34+ cells, and MNCs. Abbreviations: ES, embryonic stem cell; hES, human embryonic stem cell; iPS, induced pluripotent stem cell; MNC, mononuclear cell.
Infrequent Reprogramming of Lymphoid Cells From the MNCs
It has been reported that peripheral blood lymphoid cells from MNCs have been reprogrammed into iPSCs [23, 33]. However, if iPSCs are to be used for future regenerative medicine, those derived from B or T lymphocytes exhibiting immunoglobulin or T-cell receptor gene rearrangements will not be desirable. To determine the effectiveness of limiting reprogramming lymphocytes from MNCs after using the preculture condition described, PCR analysis to detect immunoglobulin gene rearrangements was performed using IGH framework 1–3 primer sets and T-cell receptor gene rearrangement using TCRB, TCRD, and TCRG primer sets designed by the European BIOMED-2 consortium (supplemental online Fig. 4A, 4B). Of 17 independent iPSC clones analyzed, only one had a positive signal in the immunoglobulin frame 3 test and none had TCRD and TCRG rearrangements. For the TCRB rearrangement Vβ-Jβ1/2 test (supplemental online Fig. 4B), all tested samples showed multiple bands in PCR products within the valid size range. Since the patterns were similar to those of an iPSC line derived from skin fibroblast that served as the negative control sample, these PCR bands could have arisen from nonspecific amplification. Using Southern blot analysis, we confirmed that none of the 17 lines had TCRB receptor rearrangement (data not shown). These results show that preculture of blood MNCs under the conditions used greatly limited the reprogramming of lymphoid cells.
Absence of SeV and Reprogramming Transcription Factor Sequences in the iPSCs Derived From Blood Cells
SeV is a negative-sense single-stranded RNA virus that replicates only in the cytoplasm and does not integrate into the host genome. The recombinant SeV we used had deletion of the F-protein and mutations that facilitate the removal of its genome from the infected cells. These properties should generate iPSCs free of residual viral vectors carrying reprogramming factors. To determine when the SeVs become undetectable during the process of reprogramming, RT-PCRs for vector as well as vector-delivered transcription factor sequences were performed in the CD34+ cells soon after infection and followed in the 15 CD34+- and MNC-generated iPSC lines several passages later. SeV genome and reprogramming factor sequences could be detected at 32 days after infection when the iPSC colonies were not yet mature (Fig. 3A). The time of the loss of viral sequences was variable, depending on the construct of the virus. Since we did not see the SeV-Sox2 genome on day 28, we analyzed the day 10 infected CD34+ cells and found that they had been infected with this vector. However, from passage 5 onward, no SeV or reprogramming factor sequences could be detected (Fig. 3B). Similarly, SeV sequences were undetectable in iPSCs reprogrammed from MNCs after passage 8 (supplemental online Fig. 5). These results indicate that iPSCs reprogrammed by SeV rapidly became transcription factor DNA- and viral genome-free.
Figure 3.
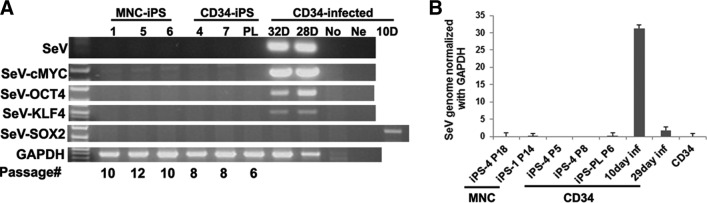
Complete loss of reprogramming factors and Sendai virus sequences in the blood cell-derived iPS cells (iPSCs). (A): Reverse transcription-polymerase chain reaction (RT-PCR) analysis of SeV and reprogramming factor sequences in blood-derived iPSCs starting from day 10 after infection to passage 12. A 10-day infected CD34 cell group was added only for detecting transgene SeV-Sox2 expression. The primers used for RT-PCR were specific for detecting the SeV reprogramming factors. (B): Real-time RT-PCR analysis of Sendai vector sequences in blood cell-derived iPSC lines from day 10 after infection to passage 18 and uninfected CD34 cells. Abbreviations: D, days after infection; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Inf, after infection; iPS, induced pluripotent stem cell; MNC, mononuclear cell; No, no infection; Ne, water control; PL, pooled induced pluripotent stem clones; SeV, Sendai viral vector.
Differentiation of the Reprogrammed Blood iPSCs Into Hematopoietic Stem Cells
Several studies have suggested that donor cell type can influence the epigenetics and differentiation potential of iPSCs. To test whether transcription factor-free blood-derived iPSCs can be differentiated to hematopoietic cell lineages, we used a recombinant protein-based, animal product-free medium StemDiff APEL (StemCell Technologies) in combination with the spin EB method to differentiate blood-derived iPSCs.
We modified the protocol described by Ng et al. [31, 32] by first growing the spin EBs that were generated by centrifugation in 96 wells for 4 days with the combination of SCF, BMP4, and VEGF to favor hematopoietic mesoderm differentiation, followed by the addition of four more cytokines (FL, TPO, IL-6, and IGF-II) to promote hematopoietic differentiation for 10 more days (Fig. 4A). From day 8 forward, single small round cells were observed and thought to be released from the EB. On day 14, EBs (Fig. 4B) were harvested and dissociated to generate single cells for flow cytometric analysis. With this protocol, we obtained two lines derived from MNCs (MNC-iPS-2 and MNC-iPS-12) that had 38%–45% CD34+ cells, of which 22%–25% coexpressed CD45 and 17%–23% coexpressed CD43 (Fig. 4C; supplemental online Fig. 6).
Figure 4.

Hematopoietic differentiation of mononuclear cell (MNC)-derived induced pluripotent stem cell (iPSC) line. (A): Schematic diagram of the protocol for hematopoietic differentiation of iPSCs. (B): Top left image shows the morphology of day 9 spin cultured EBs in StemDiff APEL medium supplemented with cytokines (magnification, ×4). Bottom left: day 14 EBs cultured in a gelatin-coated plate for additional 7 days (magnification, ×10). Images on right: Wright-Giemsa staining of the terminally differentiated blood cells. Black arrows, monocytes; red arrows, mast cells; brown arrows, neutrophils; blue arrows, eosinophils; gray arrows, erythrocytes. Scale bars = 15 μm. (C): Flow cytometry analysis of single cells dissociated from day 14 MNC-iPS-2 EBs demonstrating the differentiation into hematopoietic stem and progenitor cells that are CD34+CD43+ and CD34+CD45+. (D): Colony-forming unit assay from single cells differentiated from day 14 EBs from MNC-iPS-2. Magnification, ×4. Abbreviations: BMP, bone morphogenetic protein; CFU-E, colony-forming unit-erythroid; CFU-GM, colony-forming unit-granulocyte and monocyte; CFU-M, colony-forming unit-macrophage; EB, embryoid body; EPO, erythropoietin; FACS, fluorescence-activated cell sorting; FL, Flt3 ligand; IGF, insulin-like growth factor; IL, interleukin; SCF, stem cell factor; TPO, thrombopoietin; VEGF, vascular endothelial growth factor.
We further demonstrated that the single cells isolated from EBs could form colony-forming unit (CFU)-erythroid and CFU-granulocyte and monocyte in methylcellulose culture, thus confirming their hematopoietic potential (Fig. 4D). The EBs were seeded on a gelatin-coated plate for terminal differentiation (Fig. 4B, bottom left) with a cytokine combination of SCF, VEGF, FL, IL-3, IL-6, TPO, and EPO. Many single and round cells were observed starting from day 4 after seeding, The single cells were collected after 1–3 weeks, displayed on slides by cytocentrifugation, and Wright-Giemsa stained. We could see a range of blood cells belonging to the myeloid lineage, including neutrophils, monocytes (macrophages), mast cells, and nucleated red blood cells (Fig. 4B, right four panels). Since the differentiation conditions that we used are not known to support lymphocyte differentiation, we did not check for lymphoid lineage markers such as CD3, CD19, or CD56. Nevertheless, we conclude that mature blood cells of the myeloerythroid series could be readily differentiated from blood cell-derived iPSCs.
Discussion
In this study, we tested the efficiency of Sendai viral vectors in generating iPSCs from peripheral blood CD34+ and mononuclear cells and found that both cell types could be reprogrammed efficiently. For practical reasons, using peripheral blood MNCs is advantageous over CD34+ cells as collection of enough CD34+ cells from the adult would usually require a large volume of blood. Often, administration of granulocyte colony-stimulating factor (G-CSF) is required to mobilize CD34+ cells, a procedure that is not without untoward side effects [34]. In contrast, more than enough MNCs for reprogramming can be obtained from only 2 ml of peripheral blood. A simple venipuncture to obtain MNCs for reprogramming into iPSCs should be much more acceptable to patients or donors, particularly pediatric patients.
Several studies have reported that derivation and prolonged culture of iPSCs may cause recurrent aneuploidies, including trisomy 12, trisomy 8, and 20q [35–37]. Trisomy 12 is a predominant abnormality found in iPSC lines and occurs at a frequency of 31.9% [36]. The explanation for gaining this abnormality may be that chromosome 12 contains the NANOG gene, which, as one of the key pluripotent genes, may provide a selective advantage for iPSCs in long-term culture [38]. In our study we observed that all three independent iPSC lines derived from the same donor showed the same trisomy 12 abnormality. If this had happened during reprogramming, the abnormality should be found in some independent clones and not in others. The same three out of three abnormalities observed in our study could also be due to the presence of trisomy already in the starting CD34+ cells, as it has been reported that chromosome aneuploidy can occur in cells mobilized with G-CSF [39]. Thus, it would be preferable to use CD34+ cells without mobilization. A recent study shows that 2 × 104 CD34+ cells could be obtained from 8 ml of blood without putting the patient through G-CSF mobilization [40]. This quantity of cells would be adequate for generating iPSCs using the protocol we describe in this paper.
Whereas other transgene-free reprogramming methods, such as the EBNA1/OriP plasmid method, require a large amount of blood to prepare enough MNCs (1–2 × 106) for reprogramming [24], Sendai viral vectors need many fewer blood cells (2–3 × 104). In this study, MNCs prepared from 2 ml of blood are more than enough to generate functional iPSCs.
For iPSCs to be used for cell biology studies in vitro or for regenerative medicine in the future, those derived from B and T cells that possess intrinsic DNA rearrangements should be avoided. Using preculture conditions that favor proliferation of myeloid progenitor cells [24] before SeV infection, we were able to obtain few iPSCs with immunoglobulin or T-cell receptor gene rearrangements. iPSCs without integration of foreign DNA in the genome should be ideal for future clinical applications. Episomal vector-mediated reprogramming could generate blood iPSCs without integration. However, as these DNA vectors are introduced into the nucleus, occasional integration into the host genome cannot be ruled out. In contrast, SeV-mediated reprogramming is efficient and, as an RNA virus that only replicates in cytoplasm, it does not enter the nucleus and therefore will not integrate into the host genome.
A protocol similar to our own was published online under the NIH Center for Regenerative Medicine (CRM) Protocols (http://crm.nih.gov/stemcell_types/ESC_iPSC/UPenn_ESC_iPSC.asp) while we were conducting our study. The protocol we described here used only 2 ml of blood, whereas the published protocol relied on a larger volume. The published protocol also used a longer preculture period before exposure to reprogramming factors, and it used fetal bovine serum during the reprogramming process. For possible future clinic application, culture with bovine serum is to be avoided because it contains animal products. In short, our protocol relies on fewer cells, is faster, and is more clinically applicable. However, the NIH CRM protocol reports a higher frequency of reprogramming (1% vs. 0.27% in this study) that may be influenced by the longer incubation periods and the use of animal serum.
Several comparison studies have suggested that iPSCs retain the epigenetic memory of the differentiated state of their tissue origin because of the inherent limitation of the technique for reprogramming [20]. This property, however, may favor their differentiation back to the lineages of the donor cells. With a modified protocol, we showed that the MNC-derived iPSCs were readily differentiated back to hematopoietic cells. We could obtain 38%–45% of CD34+ cells, about half of which coexpressed CD43+ or CD45+. These cells are hematopoietic stem and progenitor cells that are potentially capable of differentiating into mature blood cells, including lymphoid, myeloid, and erythroid cells. Furthermore, we demonstrated that the iPSCs reprogrammed from MNCs can be terminally differentiated into mature blood cells.
Conclusion
Our study demonstrated a reproducible protocol for reprogramming blood cells into transgene-free iPSCs by using the Sendai viral vector and the subsequent differentiation of the iPSCs into hematopoietic cells. Maintaining genomic integrity of iPSCs should allow the development of therapeutic-grade stem cells for regenerative medicine.
See www.StemCellsTM.com for supporting information available online.
Supplementary Material
Acknowledgments
We thank Yanxia Hao, Linda Ta (Genomic Core), and Alexander Williams (Bioinformatic Core) from the Gladstone Institute for excellent work for conducting microarray analysis experiments. This work was supported by NIH Grant P01-DK088760.
Author Contributions
L.Y.: design and direction of experiment, data acquisition and analysis, manuscript discussion and writing; M.O.M. and A.I.B.: flow cytometry; N.F: provision of SeV; J.W.: gene expression analysis; Z.Q and J.Y: karyotyping; Y.W.K.: design of experiments, discussion, manuscript writing.
Disclosure of Potential Conflicts of Interest
N.F. has compensated employment from DNAVEC Corporation and compensated research funding from JST PRESTO.
References
- 1.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 3.Carvajal-Vergara X, Sevilla A, D'Souza SL, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hargus G, Cooper O, Deleidi M, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc Natl Acad Sci USA. 2010;107:15921–15926. doi: 10.1073/pnas.1010209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanna J, Wernig M, Markoulaki S, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 7.Lee G, Papapetrou EP, Kim H, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchetto MC, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stadtfeld M, Nagaya M, Utikal J, et al. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okita K, Nakagawa M, Hyenjong H, et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- 12.Kaji K, Norrby K, Paca A, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia F, Wilson KD, Sun N, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7:197–199. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anokye-Danso F, Trivedi CM, Juhr D, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim D, Kim CH, Moon JI, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou H, Wu S, Joo JY, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bar-Nur O, Russ HA, Efrat S, et al. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K, Zhao R, Doi A, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staerk J, Dawlaty MM, Gao Q, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010;7:20–24. doi: 10.1016/j.stem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loh YH, Agarwal S, Park IH, et al. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476–5479. doi: 10.1182/blood-2009-02-204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seki T, Yuasa S, Oda M, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–14. doi: 10.1016/j.stem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Chou BK, Mali P, Huang X, et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21:518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li HO, Zhu YF, Asakawa M, et al. A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J Virol. 2000;74:6564–6569. doi: 10.1128/jvi.74.14.6564-6569.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fusaki N, Ban H, Nishiyama A, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ban H, Nishishita N, Fusaki N, et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci USA. 2011;108:14234–14239. doi: 10.1073/pnas.1103509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye L, Chang JC, Lin C, et al. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc Natl Acad Sci USA. 2009;106:9826–9830. doi: 10.1073/pnas.0904689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye Z, Zhan H, Mali P, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye L, Chang JC, Lin C, et al. Generation of induced pluripotent stem cells using site-specific integration with phage integrase. Proc Natl Acad Sci USA. 2010;107:19467–19472. doi: 10.1073/pnas.1012677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ng ES, Davis RP, Azzola L, et al. Forced aggregation of defined numbers of human embryonic stem cells into embryoid bodies fosters robust, reproducible hematopoietic differentiation. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- 32.Ng ES, Davis R, Stanley EG, et al. A protocol describing the use of a recombinant protein-based, animal product-free medium (APEL) for human embryonic stem cell differentiation as spin embryoid bodies. Nat Protoc. 2008;3:768–776. doi: 10.1038/nprot.2008.42. [DOI] [PubMed] [Google Scholar]
- 33.Loh YH, Hartung O, Li H, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7:15–19. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cashen AF, Lazarus HM, Devine SM. Mobilizing stem cells from normal donors: Is it possible to improve upon G-CSF? Bone Marrow Transplant. 2007;39:577–588. doi: 10.1038/sj.bmt.1705616. [DOI] [PubMed] [Google Scholar]
- 35.Mayshar Y, Ben-David U, Lavon N, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 36.Taapken SM, Nisler BS, Newton MA, et al. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat Biotechnol. 2011;29:313–314. doi: 10.1038/nbt.1835. [DOI] [PubMed] [Google Scholar]
- 37.Martins-Taylor K, Nisler BS, Taapken SM, et al. Recurrent copy number variations in human induced pluripotent stem cells. Nat Biotechnol. 2011;29:488–491. doi: 10.1038/nbt.1890. [DOI] [PubMed] [Google Scholar]
- 38.Martins-Taylor K, Xu RH. Concise review: Genomic stability of human induced pluripotent stem cells. Stem Cells. 2012;30:22–27. doi: 10.1002/stem.705. [DOI] [PubMed] [Google Scholar]
- 39.Nagler A, Korenstein-Ilan A, Amiel A, et al. Granulocyte colony-stimulating factor generates epigenetic and genetic alterations in lymphocytes of normal volunteer donors of stem cells. Exp Hematol. 2004;32:122–130. doi: 10.1016/j.exphem.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Mack AA, Kroboth S, Rajesh D, et al. Generation of induced pluripotent stem cells from CD34+ cells across blood drawn from multiple donors with non-integrating episomal vectors. PLoS One. 2011;6:e27956. doi: 10.1371/journal.pone.0027956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.