

This study found that stable allograft survival could be achieved following third-party multipotent adult progenitor cell (MAPC) infusion in a rat model of fully allogeneic, heterotopic heart transplantation. MAPC-based immunomodulation represents a promising pathway for clinical immunotherapy and has prompted the initiation of a phase I clinical trial for testing safety and feasibility of third-party MAPC therapy after liver transplantation.
Keywords: Immunosuppression, Mesenchymal stem cells, Transplantation, T cells
Abstract
Multipotent adult progenitor cells (MAPCs) are an adherent stem cell population that belongs to the mesenchymal-type progenitor cell family. Although MAPCs are emerging as candidate agents for immunomodulation after solid organ transplantation, their value requires further validation in a clinically relevant cell therapy model using an organ donor- and organ recipient-independent, third-party cell product. We report that stable allograft survival can be achieved following third-party MAPC infusion in a rat model of fully allogeneic, heterotopic heart transplantation. Furthermore, long-term accepted heart grafts recovered from MAPC-treated animals can be successfully retransplanted to naïve animals without additional immunosuppression. This prolongation of MAPC-mediated allograft acceptance depends upon a myeloid cell population since depletion of macrophages by clodronate abrogates the tolerogenic MAPC effect. We also show that MAPC-mediated allograft acceptance differs mechanistically from drug-induced tolerance regarding marker gene expression, T regulatory cell induction, retransplantability, and macrophage dependence. MAPC-based immunomodulation represents a promising pathway for clinical immunotherapy that has led us to initiate a phase I clinical trial for testing safety and feasibility of third-party MAPC therapy after liver transplantation.
Introduction
Multipotent adult progenitor cells (MAPCs) are a distinct population of bone marrow-derived, nonhematopoietic, adherent stem cells that has been extensively characterized in the last decade [1–4]. MAPCs display a defined range of regenerative abilities and share functional features with mesenchymal stromal cells (MSCs). The immunological attributes of MSCs and MAPCs make both populations promising candidates for providing immunosuppressive support post-organ transplantation [5]. In contrast to MSCs, MAPCs can be cultured more long-term, potentially making them the better choice for routine clinical use [6]—especially in a setting in which the cell donor is unrelated to the organ donor and recipient (the so-called third-party scenario).
Evidence of cell-derived immunomodulation and its beneficial effects in preclinical solid organ transplant models is compelling and has a history that dates back to donor-type blood transfusions [7]. However, most previous preclinical studies have always used either donor-type MSCs [8–13] or hematopoietic cell preparations [14–16]. Both of these technologies share inherent weaknesses affecting their broader adoption into routine clinical use, principally that sufficient cell expansion or selection is impractical given the time constraints of routine post-mortem organ allocation and transport. Furthermore, the preparation of personally tailored cell therapies is costly and challenging to standardize, and the application of donor cells harbors the risk of recipient antidonor sensitization.
To address these concerns and to use a clinically relevant preclinical model, we show here that MAPCs mediate long-term acceptance of allogeneic, vascularized heart grafts when administered concurrently with low-dose pharmacological immunosuppression in rats. In contrast to grafts treated with immunosuppressive drug alone, MAPC-exposed allografts can be retransplanted into naïve recipients without additional immunosuppression, indicating that MAPC tolerance is a localized event within the heart graft. We further detail the tolerogenic features of MAPCs on a gene expression level and show that MAPCs induce T regulatory cells (Tregs) via a macrophage intermediate and that depletion of macrophages functionally abrogates the tolerogenic effect of MAPCs.
Taken together, we present a model for MAPC-mediated graft acceptance and demonstrate that the achieved immune privilege is transferable to naïve recipients via the graft. Based on these findings and others, we have initiated a phase I clinical trial to assess the safety and feasibility of applying third-party MAPCs to liver allograft recipients [17].
Materials and Methods
Animals
Lewis (LEW) (major histocompatibility complex [MHC] haplotype: RT1l; Charles River, Sulzfeld, Germany, http://www.criver.com) and ACI (MHC haplotype: RT1a) rats weighing approximately 200 g were maintained in the animal center at Regensburg University Medical Center. All animal procedures were approved by regional authorities and conducted under appropriate general anesthesia whenever necessary.
Heterotopic Heart Transplantation Model
A well-established, fully allogeneic Lewis-into-ACI model of heterotopic heart transplantation was used as described before [18–20]. LEW rats were used as heart graft donors and ACI rats served as recipients. This strain combination has been previously used by us [13, 21] and others [19, 20] as a reliable model system. Injections of 5 × 106 MAPCs into the tail vein were carried out 4 days prior to heart transplantation, and in some groups immediately after heart transplantation with an additional injection on day 0 as indicated. In some experimental groups the cells were injected directly into the spleen instead. Mycophenolate mofetil (Roche, Basel, Switzerland, http://www.roche-applied-science.com) diluted in a 5% glucose solution was injected into the peritoneal cavity at a dose of 20 mg/kg body weight (BW) per day from day 0 to day 7, and ciclosporin A (CsA; Novartis Pharma, Basel, Switzerland, http://www.novartis.com) was injected into the intraperitoneal cavity at a dose of 5 mg/kg BW from day 0 to day 15 in designated experiments. Heart grafts were checked daily for loss of function by abdominal palpation in the first weeks, and then twice a week. Grafts were considered rejected when no cardiac contractions were palpable, with verification of rejection by direct inspection of the allograft via laparotomy in selected animals. Retransplantation of accepted hearts was conducted in the same way after carefully harvesting the graft from the previous recipient without any additional immunosuppressive drugs or cell injections.
To deplete macrophages in designated groups, clodronate-filled liposomes were administered intravenously 5 days prior to transplantation (1 ml/100 g of BW) as previously described by one of the investigators (N.v.R.) [22, 23]. When injected i.v., clodronate-filled liposomes deplete ED1+ and ED2+ cells in the circulation, in bone marrow, liver and spleen. ED1 (CD68) and ED2 (CD163) are expressed on mature macrophages in the rat. CD68 staining is predominantly found on tissue macrophages and the lysosomal membrane of myeloid cells, where there is also weak cell surface expression. CD163 is primarily present on resident rat macrophages (e.g., Kupffer cells in the liver) [22, 23]. Control animals received the same amount of empty liposomes. All transplantation groups are summarized in Table 1.
Table 1.
Group design of heart transplantation experiments
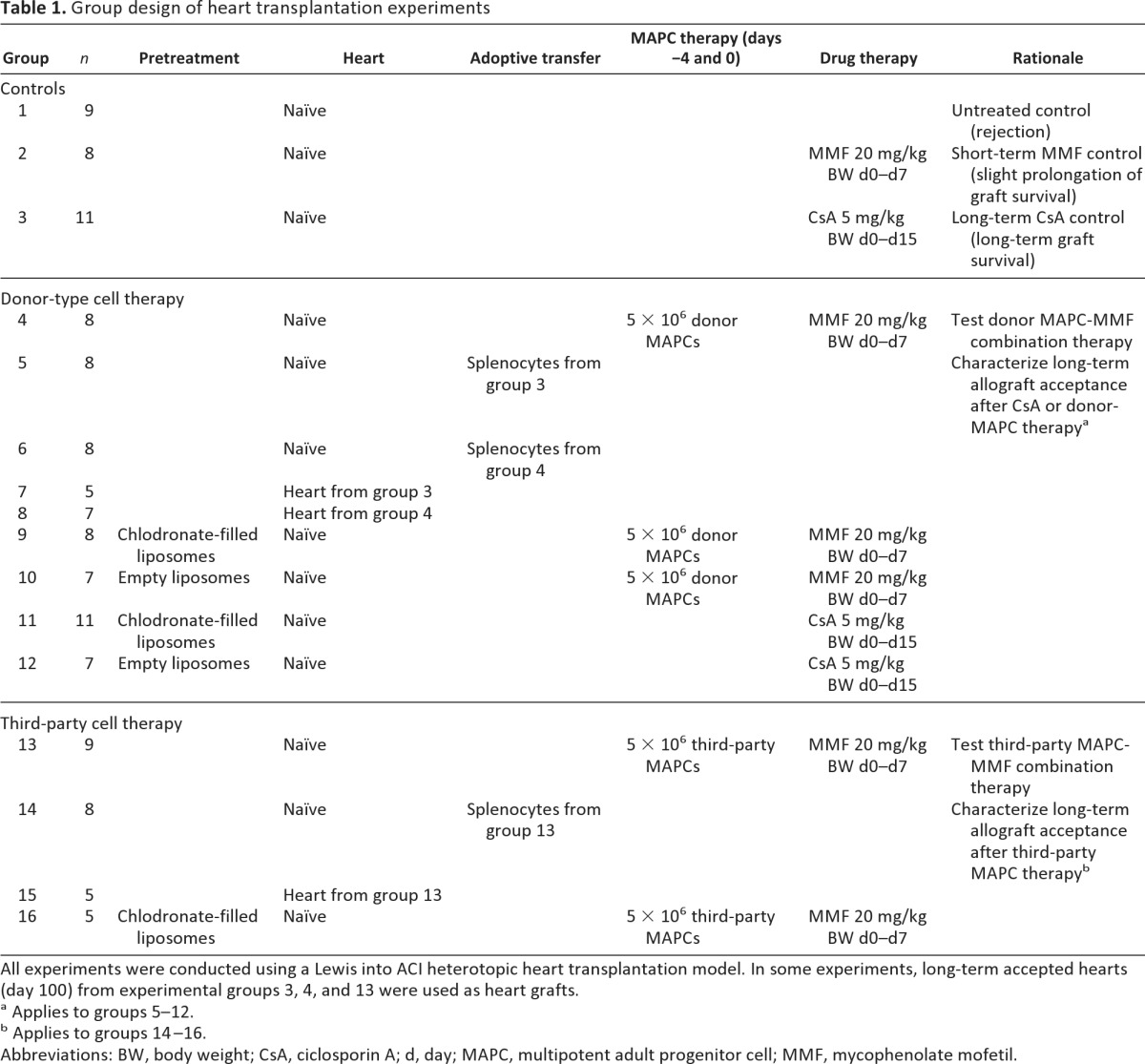
All experiments were conducted using a Lewis into ACI heterotopic heart transplantation model. In some experiments, long-term accepted hearts (day 100) from experimental groups 3, 4, and 13 were used as heart grafts.
a Applies to groups 5–12.
b Applies to groups 14–16.
Abbreviations: BW, body weight; CsA, ciclosporin A; d, day; MAPC, multipotent adult progenitor cell; MMF, mycophenolate mofetil.
Skin Transplantation
Skin grafting was performed on day 100 onto long-term heart graft accepting animals. A graft bed (2.5 cm in diameter) was prepared on the back of a recipient rat. Skin grafts (2.5 cm in diameter) were derived from the ventral body surface of the donor rat and fixed with eight interrupted sutures of 5-0 silk. The graft was covered with a protective tape, and the first inspection was performed 7 days after the procedure. Graft diameter was measured daily from day 7, and the graft was considered rejected when its size was less than 1.25 cm in diameter (<50%).
Mesenchymal Stromal Cells
MSCs were isolated according to protocols described previously [13]. After bone marrow cells were collected by flushing the long bones of donor animals with Hanks' balanced saline solution medium (Biochrom AG, Berlin, Germany, http://www.biochrom.de), these cells were plated in 175-cm2 flasks and cultured in expansion medium (high-glucose Dulbecco's modified Eagle's medium [DMEM] with GlutaMAX, without pyruvate; Biochrom), supplemented with 10% fetal bovine serum (FBS; Biochrom), 1% penicillin/streptomycin, and 1% glutamine. After 24, 48, and 72 hours, nonadherent cells were removed by changing the culture medium. Adherent cells were trypsinized (0.5% trypsin-EDTA), harvested, and replated into new flasks 2 weeks after starting the culture, or each time when cell confluence exceeded 80%. Flow cytometrical analysis of MSCs and differentiation into adipocytes, chondrocytes, and osteoblasts was performed as previously described [24]. MSCs from passages 6–8 were used for experimentation.
Multipotent Adult Progenitor Cells
We used rat MAPCs expanded to large scale. Lewis-derived (donor-type) and Sprague-Dawley-derived MAPCs (genetically unrelated to donor and recipient in the current system, thus third party) were expanded to more than 500 million cells per expansion and analyzed for viability, sterility, phenotype, karyotypic stability, in vitro multilineage differentiation potential, and maintenance of telomere length [25] (good manufacturing practice [GMP]-grade rat MAPCs analogous to human Multistem; Athersys Inc., Cleveland, OH, http://www.athersys.com). Expanded MAPCs displayed stable karyotypes, stable expression of Oct-4 and telomerase genes, and no evidence of telomere length shortening. Multilineage differentiation potential was confirmed by subjecting cells to various in vitro differentiation protocols (routine quality control data not shown). When cultured under the appropriate conditions, cells used in this study displayed significant expression of the endothelial markers PECAM-1 (platelet endothelial cell adhesion molecule-1), TEK (tyrosine kinase, endothelial), and von Willebrand factor; the hepatic markers albumin, cytokeratin-18, Cyp2B6, and HNF-1α (hepatocyte nuclear factor-1α); and the neuronal markers GFAP (glial fibrillary acidic protein), NF-200 (neurofilament-200), Tau, and nestin. Overall, the description of these cells as having extensive replicative capacity, stable karyotypes and telomeres, expression of the stemness markers Oct4 and telomerase, and the capacity for differentiation into cells from lineages representative of the three primitive germ layers supports the description of these cells as MAPCs, as originally reported by Jiang et al. [1]. On each infusion day, the required number of Lewis rat MAPCs were thawed from frozen stocks and washed in phosphate-buffered saline (PBS) to remove dimethyl sulfoxide. Viable cells were counted by trypan blue staining and were resuspended and applied in a volume of 900 μl.
Cell Size Determination
Two million MSCs and MAPCs were plated separately in 175-cm2 flasks and cultured in expansion medium for 2 days. Adherent cells were trypsinized (0.5% trypsin-EDTA), harvested, and analyzed under a light microscope. The diameter of 50 cells of each cell type were measured using a Zeiss AxioObserver microscope with Zeiss AxioVision software (Carl Zeiss, Jena, Germany, http://www.zeiss.com).
Proliferation Assays
The proliferation of splenocytes in response to concanavalin A (ConA) or alloantigen was determined by carboxyfluorescein succinimidyl ester (CFSE) dilution, as described previously [21]. Briefly, 2 × 105 CD3-negative LEW stimulator spleen cells were seeded into 96-well round-bottomed plates in triplicates. CFSE-stained responder cells (1 × 105 cells) from spleens of experimental animals were added in a total volume of 200 μl of RPMI medium (supplemented with 50 mM 2-mercaptoethanol, minimal essential medium [MEM]-nonessential amino-acids, 1 M HEPES (pH 7.36–7.39), 100 mM Na-pyruvate, antibiotic, MEM vitamin, 200 mM l-glutamine, FBS). ACI splenocytes without stimulation served as controls. In further experiments cells from ACI spleens were stimulated nonspecifically with ConA (2 ng/ml). MAPCs were used as modulator cells in various dilutions as indicated. In some experiments, CD11b+ cells sorted by magnetic-activated cell sorting from spleens or heart grafts from MAPC- or CsA-treated animals were used as modulators in a responder:modulator ratio of 5:1. Plates were cultured for 3 days (ConA) or 5 days (allogeneic stimulus) (37°C/5% CO2), and proliferation was monitored by measuring CFSE dilution to responder daughter cells by flow cytometry. Histograms were characterized and the division index was calculated using FlowJo V7.1.3 software (Tree Star, Ashland, OR, http://www.treestar.com). The division index is the average number of cell divisions that a cell in the original population has statistically undergone.
Migration Assays
Matrigel Invasion Chambers (Becton, Dickinson and Company, Franklin Lakes, NJ, http://www.bd.com) were hydrated for at least 2 hours in a tissue culture incubator with 500 μl of medium in the bottom of the well and 500 μl in the top of the chamber. After hydration of Matrigel, the medium in the bottom of the well was replaced with DMEM containing ConA-stimulated splenocytes from ACI rats. MAPCs (5 × 104) were then plated in 500 μl of medium in the top of the chamber. The invasion assay was carried out for 24 hours in the incubator. Cells were fixed and stained by the DiffQuick staining kit (Dade Behring Inc., Newark, NJ, http://www.dadebehring.com). After the chambers were washed five times by being dipped into a large beaker filled with distilled H2O, cells at the top of the Matrigel membrane were removed with several cotton swabs. Membranes were placed on microscopy slides and covered with Entellan (Merck & Co., Whitehouse Station, NY, http://www.merck.com) and coverslips. The remaining cells on the bottom of the membrane were counted using an inverted microscope equipped with a ×20 objective and plotted as the percentage of cells from the control.
Flow Cytometry
Heart grafts and spleens were washed in PBS to remove blood cells. Single-cell suspensions were isolated by GentleMACS (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com) according to the manufacturer's protocol. Briefly, tissues were homogenized using a GentleMACS C-tube (Miltenyi Biotec). Cardiac allograft tissues were additionally digested at 37°C in buffer containing collagenase II (600 U/ml) and DNase I (60 U/ml) for 30 minutes. Lymphocytes were isolated by gradient centrifugation followed by hypotonic red blood cell lysis. Before fixation and permeabilization, cells were stained with anti-CD4-fluorescein isothiocyanate (FITC), anti-CD25-phycoerythrin (PE) (eBioscience, San Diego, CA, http://www.ebioscience.com) and propidium iodide at 4°C for 20 minutes. FoxP3-allophycocyanin (APC) staining was achieved using a FoxP3 staining Kit (eBioscience) according to the manufacturer's protocol. Flow cytometric analysis was performed with a FACSCalibur (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com) and FlowJo V7.1.3 software (Tree Star).
Histochemistry and Immunohistochemistry
Heart grafts from groups 1, 2, 3, 4, and 13 (n = 5) were removed on day 100 post-transplantation or (where applicable) at the day of rejection. Sections were stained with hematoxylin and eosin as described before [21]. Graft rejection was graded on the basis of the extent of infiltration and the anatomical localization of inflammatory cells, according to the International Society of Heart and Lung Transplantation (ISHLT) standard, described by Billingham et al. [26].
For identification of myeloid-derived suppressor cells (MDSCs), graft samples were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA, http://www.sakura.com), snap-frozen in liquid nitrogen, cut into 5-μm sections, and fixed in acetone. Sections were blocked with 10% normal goat serum for 10 minutes, washed, and stained with the rabbit anti-inducible nitric oxide synthase (iNOS) (primary antibody by Abcam, Cambridge, MA, http://www.abcam.com) for 3 hours at room temperature. After washing, sections were incubated with a monoclonal Alexa 488-conjugated goat anti-rabbit antibody (Ab) (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) diluted in normal rat serum for 30 minutes. After being washed, sections were incubated with purified CD11b/c (OX42) monoclonal Ab (BD Biosciences) for 40 minutes. After being washed, sections were then incubated for 30 minutes with Alexa 594-conjugated anti-mouse (secondary antibody by Invitrogen) and DAPI (1:20,000), mounted with Dako medium (Dako, Glostrup, Denmark, http://www.dako.com), and analyzed using a immunofluorescence technique (Zeiss AxioObserver microscope). Control sections were performed by replacing the primary Abs with dilution buffer.
Microarray and Quantitative Real-Time Polymerase Chain Reaction
Microarray of rat graft tissue was conducted as contracted research by AROS Applied Biotechnology (Aarhus, Denmark, http://www.arosab.com) using their established technique. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in a LightCycler 480 Real-Time PCR system (Roche) using SYBR Green reagents. Primers for the following genes were used: Clqtnf1, DPPIV, CD79b, Pnoc, FoxP3, iNOS, Arg-1, and tlr6. All primers were designed and purchased from Qiagen (QuantiTect Primer Assays; Qiagen, Hilden, Germany, http://www.qiagen.com). Glyceraldehyde-3-phosphate dehydrogenase was used as an endogenous control gene to normalize varying starting amounts of RNA. Relative expression between a given sample and a control sample, used for all experiments, was calculated with the 2−ΔΔCT method. All samples were analyzed in triplicate. Expression of genes of interest was compared between CsA- and MAPC-treated animals.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism, version 5 (GraphPad Software, Inc., San Diego, CA, http://www.graphpad.com). Survival data were compared using the log-rank test (GraphPad Prism 5). Proliferation and cell size determination data were analyzed by comparing group means using Student's test. Statistical comparison of Treg frequencies among three groups was analyzed using analysis of variance, followed by a post test using Bonferroni comparison of all pairs. Differences with a p value <.05 were considered significant.
Results
MAPCs Are Significantly Smaller Than and Differ Phenotypically From MSCs
The MAPCs used in this work are positive for CD29, CD90, CD44, and MHC class I and lack expression of MHC class II, CD45, CD106, and the costimulatory molecules CD80 and CD86, indicating that these cells are clearly not derived from the hematopoietic lineage (Fig. 1A, flow cytometry). For the current transplant model, we have further outlined that rat MAPCs are smaller than rat MSCs (Fig. 1B; 23 μm vs. 13 μm). In a mixed lymphocyte reaction between LEW (RT1l) and ACI (RT1a) splenocytes, stimulator-type MAPCs dose-dependently inhibit T-cell proliferation upon allogeneic stimulation up to a 1:10 dilution (Fig. 1C). MAPCs suppress T-cell proliferation by downregulation of activation marker CD25. This mechanism is not MHC-restricted, since inhibition with third-party MAPCs has the same effect (data not shown). This finding has been confirmed in the literature [27, 28].
Figure 1.

Phenotypic analysis of MAPCs. (A): Representative surface marker analysis of MAPCs from all strains used (Lewis, Sprague-Dawley). MAPCs stained positive for CD29, CD90, and MHC class I and negative for MHC class II, CD45, CD106, CD80, and CD86 using single-channel flow cytometry with appropriate isotype controls. (B): Microscopic analysis of MAPC size. In culture, MAPCs were significantly smaller than MSCs with an average of 13 μm versus 23 μm (n = 30). (C): Mixed lymphocyte suppression assay with MAPCs. Proliferation of rat splenocytes stimulated with irradiated allogeneic splenocytes could be effectively suppressed by increasing doses of MAPCs. The suppression was strictly dose-dependent (mean of three independent experiments). (D): Migratory capacity of MAPCs versus MSCs. MSCs actively migrated toward activated splenocytes in a Boyden chamber assay; MAPCs, on the contrary, did not (mean of three independent experiments). Proliferation and cell size determination data were analyzed by comparing group means using Student's test. **, p < .01; ***, p < .001. Abbreviations: MAPC, multipotent adult progenitor cell; MHC, major histocompatibility complex; MSC, mesenchymal stromal cell; n.s., not significant; w/o, without.
Since it has recently been reported that the migratory pattern of MAPCs differed from that of MSCs and that MAPCs were able to suppress graft-versus-host disease only when localized to sites of allopriming [6], we compared the migratory potential of the current population of rat MAPCs toward activated lymphocytes with that of MSCs. We can show that MAPCs were substantially less effective at migrating toward activated lymphocytes than MSCs (Fig. 1D).
MAPCs or Ciclosporin A Induces Long-Term Systemic Allograft Acceptance; However, the Effects on the Graft Are Fundamentally Different
To test the immunosuppressive capacity of MAPCs in vivo we chose an established rat heart transplantation model [12, 13]. In this particular rat model, we have shown previously that mycophenolate (MPA)-based immunosuppression prolongs donor-MSC-induced allograft survival [12], whereas calcineurin inhibitor (CNI)-based immunosuppression reduces the positive immunomodulatory effect of MSCs [13]. A rat model appeared beneficial since rat MAPCs can easily be propagated under GMP-like conditions in culture. Murine MSCs as well as MAPCs, in contrast, are notoriously difficult to culture and harbor species-specific genetic instability that limits the applicability of results to a clinical situation [25, 29]. Thus, combining MAPCs with low-dose MPA in a rat model was a reasonable approach, especially considering that clinical practice aims to avoid CNI-based immunosuppression because of the unfavorable side effects of long-term CNI treatment, irrespective of cellular therapy [30]. The strain combination used in this study (LEW to ACI) models acute cellular rejection, and, in the absence of immunosuppressive treatment, all heterotopic heart grafts were rejected by a median of 9 days. Low-dose MPA treatment alone is capable of prolonging median graft survival to day 15 (Fig. 2A). Intravenous injection of a single dose of donor-type MAPCs on day −4 increased graft survival in some animals, but not in a clearly dose-dependent manner (supplemental online Fig. 1). Injection of two doses of donor-type MAPCs, on the other hand, markedly improved allograft survival (data not shown). When we subsequently administered the first dose of donor-type MAPCs into the portal circulation by intrasplenic injection, 80% of allografts survived to 100 days. In comparison, high-dose treatment with CsA induced long-term allograft acceptance in all animals (Fig. 2A). At day 100 after the heart transplantation, the grafts were harvested and histologically analyzed. The ISHLT scores of the hearts harvested from CsA-treated as well as MAPC-treated animals did not show any signs of acute and chronic rejection, whereas grafts from the control group showed high levels of lymphocyte infiltration and severe tissue damage (data not shown).
Figure 2.

Donor-type MAPCs induced long-term allograft survival. (A): Graft survival after allogeneic heart transplantation in a LEW to ACI rat transplant model with intrasplenic injection of donor-type MAPCs. Untreated control animals rejected the grafts between day 7 and day 12 (n = 9). Short-term treatment with MPA slightly prolonged allograft survival to a median of day 15 (n = 8). When 5 × 106 donor MAPCs were injected into the spleen on day −4 and on day 0, followed by short-term MPA treatment, 80% of heart grafts survived (n = 8). In high-dose CsA-treated animals, 100% survived long-term (n = 11). (B): Microarray analysis of tolerance-associated gene markers. When analyzing differential gene expression by array technique, heart grafts that had been tolerized by MAPC versus CsA monotherapy showed a trend toward the expression of genes associated with operational tolerance (heat map outlining fold gene of target gene as indicated). Real-time polymerase chain reaction of Pnoc and Tlr6 confirmed upregulation in MAPC-treated animals (column plot outlining fold upregulation). (C): Secondary donor skin grafts (LEW) were rejected by ACI recipients that had previously accepted an allogeneic LEW heart. Syngeneic ACI skin was accepted, and the survival of the primary LEW heart grafts was not impaired (color photographs of representative animals on days 0, 15, and 20; n = 4). (D): Survival of heart grafts after splenocyte adoptive transfer. Splenocytes from LEW recipients that had previously accepted an allogeneic ACI heart graft through either donor MAPC therapy or CsA monotherapy were adoptively transferred into secondary, naïve ACI recipients that then received a secondary LEW graft with no further immunosuppression (n = 8 animals each group). Adoptive splenocyte transfer resulted in partial acceptance of secondary heart grafts. There was no significant difference between splenocytes from MAPC- or CsA-treated primary recipients. When LEW heart grafts were retransplanted to secondary, naïve ACI recipients, all grafts survived without further immunosuppression (n = 5), whereas more than 50% of CsA-treated grafts were rejected (n = 7). Survival data were compared using the log-rank test; &, p < .05 versus untreated control group; §, p < .05 versus MPA-treated group. Abbreviations: AT, adoptive transfer; CsA, ciclosporin A; d, day; LEW, Lewis; MAPC, multipotent adult progenitor cell; MPA, mycophenolate; ns, not significant; ReTx, retransplantation; SC, splenocytes.
To detail the immunological mechanism behind MAPC-induced allograft acceptance, we performed a set of further experiments. To get an initial indication of the potential molecular factors involved, we assessed differential gene expression within accepted allografts on day 100 and compared MAPC- and CsA-treated animals. Genes recently postulated in the literature for their association with operational tolerance were a primary screening goal [31, 32]. And indeed, we observed that there was a trend toward the expression of markers related to tolerance, such as HS3ST1, CD79b, MS4A1, Pnoc, SLC81, TLR4, and TLR6 in MAPC-treated animals. For Pnoc and TLR6, this array data could be confirmed by individual qRT-PCR (Fig. 2B).
Considering the assumption that the T-cell response can be silenced in a localized and time-restricted mode by an array of stem cell-derived factors [10, 13, 33, 34], MAPC-derived allograft acceptance could be a peripheral phenomenon. And indeed, rejection of additional donor skin (LEW) grafted on long-term accepting recipients (ACI) was observed in all MAPC-treated rats. Skin grafts were surrounded by inflammatory tissue starting on day 15, and all grafts were rejected by day 20 ± 2 days (raised skin flaps with underlying necrosis). Control syngeneic ACI skin grafts, in contrast, were accepted (Fig. 2C). Interestingly, the initial heart graft (LEW) also survived in this setting.
We further analyzed whether MAPC-induced graft acceptance can be adoptively transferred to a naïve secondary recipient (ACI), via the graft or via the primary recipient's splenocyte pool (Fig. 2D). Adoptive transfer of splenocytes isolated from a tolerant primary recipient (ACI) mediated graft acceptance of naïve grafts (LEW) in 50% of cases. As a control, splenocytes were also isolated from animals of the same recipient strain (ACI) that had been tolerized by CsA monotherapy; in this case 35% of the allografts were accepted, which is significantly different from totally untreated control animals. However, there was no significant difference in graft survival either between these two groups of secondary recipients receiving splenocytes infusions or between each of these groups and the MPA control. When entire heart grafts of tolerant animals (LEW in ACI) were transplanted into naïve secondary recipients (ACI), fewer than half of CsA-treated grafts were accepted, whereas significantly more (100%) MAPC-treated grafts survived (Fig. 2D).
MAPCs Maintain Allograft Acceptance Through Myeloid-Derived Suppressor Cell-Induced Tregs
It was recently established that MSCs can modulate macrophages in immunological diseases such as abdominal sepsis [35]. To investigate whether such generation of a tolerogenic microenvironment also plays a role in our model, we depleted ED1+ and ED2+ macrophages in the circulation, bone marrow, liver, and spleen by intravenous administration of clodronate-filled liposomes in organ graft recipients before MAPC infusion. MAPC-mediated long-term acceptance of allografts was subsequently significantly diminished (p = .004; Fig. 3A). Ninety percent of these recipients rejected their heart grafts before day 22 after transplantation. In contrast, CsA-mediated long-term allograft acceptance was not abolished by macrophage depletion (Fig. 3A), suggesting that two distinct immunological mechanisms of long-term allograft acceptance are involved for MAPC- versus CsA-treated animals.
Figure 3.

Mechanism of donor MAPC-induced allograft acceptance. (A): Survival of heart grafts after macrophage depletion. When recipient animals that were tolerized through donor-type MAPCs plus short-term MPA treatment received monocyte-depleting CLOD liposomes, 88% of the animals rejected the grafts (n = 8). Controls that received empty liposomes (n = 7) or ciclosporin A (CsA)-based immunosuppression survived (n = 7 with empty liposomes or clodronate liposomes). (B): Immunohistochemistry on splenic tissue from MAPC-treated animals on day 2 showed single events of CD11b/c+ INOS+ cells (representative microscopic images from four independent animals). (C): CD11b/c+ cells isolated from spleens and grafts on day 2 from MAPC-treated animals suppressed allogeneic lymphocyte proliferation. CD11b/c+ cells from CsA-treated and control animals were less suppressive (CFSE dilution assay, histograms, representative plots from three independent experiments). (D): Intragraft lymphocytes isolated from primary and secondary heart grafts that had been subjected to MAPC therapy in the primary recipient contained a significantly larger population of CD4+/CD25+/foxp3-positive cells than control grafts; the grafts subjected to CsA monotherapy did not contain more CD4+/CD25+/foxp3-positive cells than control grafts (representative contour plot and mean percentage of total lymphocytes, at least n = 4 for each group). Statistical comparison of T regulatory cell frequencies between three groups was analyzed using analysis of variance, followed by a post test using Bonferroni comparison of all pairs. *, p < .05; **, p < .01. Survival data were compared using the log-rank test. Abbreviations: APC, allophycocyanin; CFSE, carboxyfluorescein succinimidyl ester; CLOD, clodronate-filled; ctr, control; DAPI, 4′,6-diamidino-2-phenylindole; iNOS, inducible nitric oxide synthase; is, intrasplenic; LEW, Lewis; MAPC, multipotent adult progenitor cell; Mio, million; MPA, mycophenolate; n.s., not significant; ReTx, retransplantation; w/o, without.
MDSCs have been known to be immunosuppressive in other rat transplantation models [36], and they are affected by the macrophage depletion with clodronate [37]. Therefore, these cells are a likely candidate to mediate a peripheral MAPC-induced immunoprivilege as was observed in the current model. MDSCs are further positive for CD11b/c+ and produce iNOS. We were not able to identify these cells by immunohistochemistry in grafts of long-term accepting animals on day 100 (data not shown). But when analyzed early after transplantation (day 2), CD11b/c+/iNOS+ lymphocytes were detected in spleens of tolerant animals (Fig. 3B). To characterize the function of these MDSCs, we isolated splenic and graft infiltrating CD11b/c+ cells from recipients treated with either CsA, MAPCs, or PBS (control) on day 2 and tested their suppressive ability in a mixed lymphocyte reaction. And indeed, CD11b/c+ cells from MAPC-treated animals expressed an increased suppressive potential over CD11b/c+ cells from CsA-treated recipients (Fig. 3C). CD11b/c+ cells from control animals had almost no suppressive effect. Since MDSCs are also described to induce Tregs, we characterized graft infiltrating lymphocytes of tolerant animals on day 100. Significantly more CD4+CD25+foxP3+ Tregs were found in MAPC-treated primary grafts than in controls, whereas CsA-treated grafts did not contain significantly more CD4+CD25+foxP3+ Tregs as compared with control (Fig. 3D). This was also true (and even more pronounced) when retransplanted grafts were analyzed (Fig. 3D), stressing the notion that MAPCs induce a local microenvironment of immunoprivilege that can be carried to a secondary recipient by the graft itself.
MAPC-Mediated Allograft Acceptance Is Not MHC-Restricted
Donor-derived MAPCs have shown promising immunological features, but their potential for clinical use is limited to living-related donation because of time constraints of the post-mortem donation process. Furthermore, GMP-grade autologous or organ donor-specific cell preparation is costly, as a new cell product has to be generated for each donor-recipient pair. We therefore introduced third-party-derived MAPCs instead of donor-type cells into our model. When MAPCs of an unrelated strain (Sprague-Dawley) were administered to recipients under the same conditions as above, two cell doses significantly prolonged survival of almost 60% of allogeneic grafts (Fig. 4A). Allograft prolongation induced by third-party MAPCs was completely abrogated by depleting macrophages by administration of clodronate-filled liposomes before transplantation (Fig. 4A). In keeping with the observation that MAPCs act locally and can have an MHC-independent impact, adoptive transfer of splenocytes from accepting, MAPC-treated animals tolerized 70% of the recipients of secondary naïve heart grafts; splenocytes from CsA-treated primary recipients induced long-term allograft acceptance in only one-third of the cases (not significantly different) (Fig. 4B). Regrafting of third-party MAPC-treated grafts (LEW) into secondary recipients (ACI) was successful in all cases with no further pharmacological immunosuppression. Grafts from animals treated with CsA monotherapy and transplanted into secondary recipients were rejected in 60% of cases, as indicated above (Fig. 4B).
Figure 4.

Allogeneic heart transplantation in the LEW to ACI rat model using third-party multipotent adult progenitor cells (MAPCs). (A): Survival of heart grafts after third-party MAPC infusion. When recipient rats (ACI) received intrasplenic injections of 5 × 106 sdMAPCs on days −4 and 0 plus short-term treatment with MPA followed by allogeneic heart transplantation (LEW heart), allograft survival was prolonged in 60% of recipients (n = 9). When recipient animals that were tolerized through sdMAPCs received monocyte-depleting, clodronate-filled liposomes, all animals rejected the grafts before day 16 (n = 5). 100% of allogeneic grafts from CsA-treated control animals survived (n = 11). (B): When splenocytes from graft-accepting animals were transferred into secondary, naïve recipients, 38% of the secondary grafts survived (n = 8). Splenocytes transferred from MAPC-treated recipients were more effective in this setting, (63%, n = 8). When heart grafts were retransplanted all third-party MAPC-treated grafts survived with no further immunosuppression in secondary recipients (n = 5), whereas the majority of CsA-pretreated grafts were rejected (n = 7). (C): Intragraft lymphocytes isolated from secondary heart grafts that had been subjected to MAPC therapy in the primary recipient contained a significantly larger population of CD4+/CD25+/foxp3-positive cells than control grafts; the grafts subjected to CsA monotherapy did not contain more CD4+/CD25+/foxp3-positive cells than control grafts (representative contour plot and mean percentage of total lymphocytes, at least n = 4 for each group). Statistical comparison of Treg frequencies between three groups was analyzed using analysis of variance, followed by a post test using Bonferroni comparison of all pairs. *, p < .05; **, p < .01. Survival data were compared using the log-rank test: &, p < .05 versus untreated control group; §, p < .05 versus MPA-treated group. Abbreviations: AT, adoptive transfer; CLOD, clodronate-filled; CsA, ciclosporin A; ctr, control; is, intrasplenic; Mio, million; MPA, mycophenolate; ns, not significant; ReTx, retransplantation; SC, splenocytes; sdMAPC, Sprague-Dawley multipotent adult progenitor cell; Tregs, T regulatory cells.
To verify that third-party MAPCs share their immunological potency with donor-derived MAPCs in this model, we repeated the functional analysis as above. Analysis of graft infiltrating lymphocytes from secondary heart grafts on day 100 confirmed that significantly more CD4+CD25+foxP3+ Tregs were found in third-party MAPC-treated grafts than in controls or in CsA-treated secondary grafts (Fig. 4C). Together, these results demonstrate that MAPCs induce long-term allograft acceptance of allogeneic heart grafts by a myelomonocytic intermediate.
Discussion
Standard-of-care immunosuppressive pharmacotherapy allows the transplantation of solid organ grafts with reasonable patient and graft survival rates. However, long-term continuous exposure to immunosuppressive drugs, such as calcineurin inhibitors (CNIs), mTOR inhibitors, and steroids, also harbors significant clinical side effects. Indeed, the overall success of organ transplantation as a definitive therapy can depend on the management of deleterious side effects. We therefore believe that cellular therapy with potent immunomodulative cell populations represents a very promising adjunct to drug-based regimens—especially in the early post-transplant phase. Through this approach we hope to (a) shift the balance of immunity versus tolerance toward a tolerogenic state through an early immunomodulative intervention, (b) exploit additional regenerative capacities that adult off-the-shelf stem cells may provide, and (c) reduce unwanted intrinsic side effects of drug therapy such as nephrotoxicity.
Adherent adult stem cells from the mesenchymal lineage are one especially promising cell population that can be used for immunomodulation after solid organ transplantation. MSCs are the prototypic cell population from this group. As they can be produced from bone marrow and other tissue using scalable GMP-techniques, they are especially suited to clinical applications. In this current study, we used MAPCs, functionally a close relative of MSCs. To assess the immunosuppressive capacity of MAPCs in vivo, we chose an established rat heart transplantation model [12, 13, 18]. The rationale behind selecting a rat rather than an equivalent mouse model comes from the fact that rat MAPCs can be routinely maintained in culture and retain cytogenetic integrity. In contrast, murine MSCs are notoriously difficult to propagate and harbor species-specific genetic instability [25, 29]. Our earlier studies in this rat model demonstrated that MPA-based immunosuppression prolonged donor-MSC-induced allograft survival [13], whereas CNI-based immunosuppression reduced the positive immunomodulatory effect of MSCs [12].
In consideration of these previous findings, and together with the current drive to reduce the clinical use of CNI-based immunosuppression independently of cellular therapy [30], we designed this current study to investigate the effects of combining MAPCs with low-dose MPA suppression. As the strain combination used in this study models acute cellular rejection [12], in the absence of immunosuppressive treatment all heterotopic heart grafts were rejected. However, rats treated with donor-derived MAPCs tolerated grafts and exhibited prolonged allograft survival. Considering that in a clinical setting the potential for use of donor-derived cells is limited to living-related donation and as the routine production of a GMP-manufactured, organ recipient-tailored cell treatment is likely to be cost-prohibitive, we explored the potential of using a third-party-derived MAPCs to act as a universal MAPC donor. For the first time in a fully allogeneic model system, we established that these off-the-shelf MAPC preparations were nearly as effective as donor MAPCs at prolonging allograft survival. We are currently in the process of applying these preclinical findings to a clinical study of human allogeneic liver transplantation and will be eager to report the first clinical findings.
We then set out to further characterize the acceptance of allogeneic grafts mediated by MAPCs. Using a differential gene expression approach between MAPC- and CsA-treated accepted allografts on day 100, we observed a trend toward the expression of markers previously published as related with operational tolerance, such as HS3ST1, CD79b, MS4A1, Pnoc, SLC81, TLR4, and TLR6, in MAPC-treated animals [31, 32]. For Pnoc and TLR6, these array data could be confirmed by individual qRT-PCR; however, it is not yet clear which cells contribute to the high gene expression levels or whether this has functional relevance.
Furthermore, some evidence has been presented that adherent stem cells (or their degradation products) home to filter organs, such as the liver or lung [33], or to sites of immunological engagement after intravenous injection [38, 39]. This ability may be less pronounced in MAPCs, which can require injection directly into the respective microenvironment for optimal effect [6]. Our findings support this observation as donor-type MAPCs applied into the portal circulation [40, 41] were most effective. Most likely, MAPCs are then activated by inflammatory cytokines, such as interferon-γ and tumor necrosis factor-α, at the site of engagement [42, 43]. As shown by us and others recently, mesenchymal stromal cells are short-lived after in vivo administration, even when used in a syngeneic setting [44], suggesting that MSCs (and MAPCs) can prime host immune cells through an array of yet undefined cytokines, which, in turn, adopt a regulatory phenotype. Recently, a review article was published describing the so far known effects of MSCs on macrophages [45]. In our present work, we have shown that depletion of macrophages by clodronate was sufficient to abrogate the tolerogenic effect of MAPCs, which supports the hypothesis of a macrophage intermediate. Work from various disease systems shows that the T-cell response can be suppressed in a spatiotemporal manner by an array of adherent stem cell- or macrophage-derived factors, such as IDO [13], PGE2 [34], iNOS [10], or TSG-6 [33], as the last step of the immunomodulative cascade. Supporting this idea, we can show an elevated number of Tregs in the graft as a result.
Conclusion
When these data are taken together, our current approach advances the concept of cell-based immunomodulation in solid organ transplantation by demonstrating that third-party, adherent, adult stem cells from the bone marrow are capable of acting as a universal cell product that mediates long-term survival of fully allogeneic organ grafts. Furthermore, the resulting immunological privileged state is maintained locally to the graft, allowing secondary transplantation of the graft into a naïve, fully immunocompetent host. Previous work in this field had shown that (a) passenger leukocytes are important for the survival of allogeneic grafts and depletion of these cells negatively impacts graft survival [46], (b) donor embryonic stem cells can prolong allograft survival [14], and (c) appropriately stimulated peripheral T regulatory cells prolong allograft survival when adoptively transferred [15]. We add a realistic clinical component to these findings. To study the third-party MAPCs used in this preclinical work in a clinical setting, we have recently obtained regulatory approval for a phase I clinical trial in liver transplant recipients to assess the safety profile of MAPC infusion in the early post-transplant phase [17]. This study is now recruiting.
See www.StemCellsTM.com for supporting information available online.
Supplementary Material
Acknowledgments
Expert technical assistance was provided by Irina Kučuk and Teresa Mark. We thank the statistician Florian Zeman for support and Dr. Christian Johnson for proofreading the manuscript. The work was supported by a grant of the Deutsche Forschungsgemeinschaft to M.H.D. (DA572/12-1). Athersys Inc. (Cleveland, OH) provided the cell product (MAPCs, Multistem) and limited additional funds.
Author Contributions
E.E. and F.C.P.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; M.M.: conception and design, data analysis and interpretation, manuscript writing; P.S.: collection and assembly of data; W.v.H.: data analysis and interpretation; P.R.: conception and design, collection and assembly of data, data analysis and interpretation; M.J.H.: data analysis and interpretation, final approval of manuscript; J.P. and N.v.R.: provision of study material; E.K.G. and H.J.S.: financial support, manuscript writing, final approval of manuscript; R.D.: financial support, provision of study material, final approval of manuscript; M.H.D.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
W.v.H. and R.D. have compensated employment and stock options from Athersys, Inc. W.v.H. also has uncompensated intellectual property rights. J.P. is a compensated consultant of Athersys. M.H.D. has compensated research funding.
References
- 1.Jiang Y, Vaessen B, Lenvik T, et al. Multipotent progenitor cells can be isolated from postnatal murine bone marrow, muscle, and brain. Exp Hematol. 2002;30:896–904. doi: 10.1016/s0301-472x(02)00869-x. [DOI] [PubMed] [Google Scholar]
- 2.Keene CD, Ortiz-Gonzalez XR, Jiang Y, et al. Neural differentiation and incorporation of bone marrow-derived multipotent adult progenitor cells after single cell transplantation into blastocyst stage mouse embryos. Cell Transplant. 2003;12:201–213. doi: 10.3727/000000003108746768. [DOI] [PubMed] [Google Scholar]
- 3.Pelacho B, Nakamura Y, Zhang J, et al. Multipotent adult progenitor cell transplantation increases vascularity and improves left ventricular function after myocardial infarction. J Tissue Eng Regen Med. 2007;1:51–59. doi: 10.1002/term.7. [DOI] [PubMed] [Google Scholar]
- 4.Ross JJ, Hong Z, Willenbring B, et al. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest. 2006;116:3139–3149. doi: 10.1172/JCI28184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahlke MH, Hoogduijn M, Eggenhofer E, et al. Toward MSC in solid organ transplantation: 2008 position paper of the MISOT study group. Transplantation. 2009;88:614–619. doi: 10.1097/TP.0b013e3181b4425a. [DOI] [PubMed] [Google Scholar]
- 6.Highfill SL, Kelly RM, O'Shaughnessy MJ, et al. Multipotent adult progenitor cells can suppress graft-versus-host disease via prostaglandin E2 synthesis and only if localized to sites of allopriming. Blood. 2009;114:693–701. doi: 10.1182/blood-2009-03-213850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flye MW, Burton K, Mohanakumar T, et al. Donor-specific transfusions have long-term beneficial effects for human renal allografts. Transplantation. 1995;60:1395–1401. doi: 10.1097/00007890-199560120-00004. [DOI] [PubMed] [Google Scholar]
- 8.Bartholomew A, Sturgeon C, Siatskas M, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42–48. doi: 10.1016/s0301-472x(01)00769-x. [DOI] [PubMed] [Google Scholar]
- 9.Casiraghi F, Azzollini N, Cassis P, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. 2008;181:3933–3946. doi: 10.4049/jimmunol.181.6.3933. [DOI] [PubMed] [Google Scholar]
- 10.Chabannes D, Hill M, Merieau E, et al. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. Blood. 2007;110:3691–3694. doi: 10.1182/blood-2007-02-075481. [DOI] [PubMed] [Google Scholar]
- 11.Ge W, Jiang J, Baroja ML, et al. Infusion of mesenchymal stem cells and rapamycin synergize to attenuate alloimmune responses and promote cardiac allograft tolerance. Am J Transplant. 2009;9:1760–1772. doi: 10.1111/j.1600-6143.2009.02721.x. [DOI] [PubMed] [Google Scholar]
- 12.Inoue S, Popp FC, Koehl GE, et al. Immunomodulatory effects of mesenchymal stem cells in a rat organ transplant model. Transplantation. 2006;81:1589–1595. doi: 10.1097/01.tp.0000209919.90630.7b. [DOI] [PubMed] [Google Scholar]
- 13.Popp FC, Eggenhofer E, Renner P, et al. Mesenchymal stem cells can induce long-term acceptance of solid organ allografts in synergy with low-dose mycophenolate. Transpl Immunol. 2008;20:55–60. doi: 10.1016/j.trim.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Fändrich F, Lin X, Chai GX, et al. Preimplantation-stage stem cells induce long-term allogeneic graft acceptance without supplementary host conditioning. Nat Med. 2002;8:171–178. doi: 10.1038/nm0202-171. [DOI] [PubMed] [Google Scholar]
- 15.Joffre O, Santolaria T, Calise D, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med. 2008;14:88–92. doi: 10.1038/nm1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsui TY, Jager MD, Deiwick A, et al. Induction of peripheral tolerance by posttransplant infusion of donor splenocytes. Transplant Proc. 2001;33:187–188. doi: 10.1016/s0041-1345(00)01969-2. [DOI] [PubMed] [Google Scholar]
- 17.Popp FC, Fillenberg B, Eggenhofer E, et al. Safety and feasibility of third-party multipotent adult progenitor cells for immunomodulation therapy after liver transplantation: A phase I study (MISOT-I) J Transl Med. 2011;9:124. doi: 10.1186/1479-5876-9-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ono K, Lindsey ES. Improved technique of heart transplantation in rats. J Thorac Cardiovasc Surg. 1969;57:225–229. [PubMed] [Google Scholar]
- 19.Stegall MD, Tezuka K, Oluwole SF, et al. Interstitial class II-positive cell depletion by donor pretreatment with gamma irradiation: Evidence of differential immunogenicity between vascularized cardiac allografts and islets. Transplantation. 1990;49:246–251. doi: 10.1097/00007890-199002000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Oluwole SF, Chowdhury NC, Fawwaz RA. Induction of donor-specific unresponsiveness to rat cardiac allografts by pretreatment with intrathymic donor MHC class I antigens. Transplantation. 1993;55:1396–1402. doi: 10.1097/00007890-199306000-00035. [DOI] [PubMed] [Google Scholar]
- 21.Eggenhofer E, Steinmann JF, Renner P, et al. Mesenchymal stem cells together with mycophenolate mofetil inhibit antigen presenting cell and T cell infiltration into allogeneic heart grafts. Transpl Immunol. 2011;24:157–163. doi: 10.1016/j.trim.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Van Rooijen N, Kors N, vd Ende M, et al. Depletion and repopulation of macrophages in spleen and liver of rat after intravenous treatment with liposome-encapsulated dichloromethylene diphosphonate. Cell Tissue Res. 1990;260:215–222. doi: 10.1007/BF00318625. [DOI] [PubMed] [Google Scholar]
- 23.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 24.Popp FC, Slowik P, Eggenhofer E, et al. No contribution of multipotent mesenchymal stromal cells to liver regeneration in a rat model of prolonged hepatic injury. Stem Cells. 2007;25:639–645. doi: 10.1634/stemcells.2006-0515. [DOI] [PubMed] [Google Scholar]
- 25.Luyckx A, De Somer L, Rutgeerts O, et al. Mouse MAPC-mediated immunomodulation: Cell-line dependent variation. Exp Hematol. 2010;38:1–2. doi: 10.1016/j.exphem.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 26.Billingham ME, Cary NR, Hammond ME, et al. A working formulation for the standardization of nomenclature in the diagnosis of heart and lung rejection: Heart Rejection Study Group. The International Society For Heart Transplantation. J Heart Transplant. 1990;9:587–593. [PubMed] [Google Scholar]
- 27.Duffy MM, Ritter T, Ceredig R, et al. Mesenchymal stem cell effects on T-cell effector pathways. Stem Cell Res Ther. 2011;2:34. doi: 10.1186/scrt75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groh ME, Maitra B, Szekely E, et al. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp Hematol. 2005;33:928–934. doi: 10.1016/j.exphem.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Ren G, Su J, Zhang L, et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells. 2009;27:1954–1962. doi: 10.1002/stem.118. [DOI] [PubMed] [Google Scholar]
- 30.Grinyó JM, Cruzado JM. Mycophenolate mofetil and calcineurin-inhibitor reduction: recent progress. Am J Transplant. 2009;9:2447–2452. doi: 10.1111/j.1600-6143.2009.02812.x. [DOI] [PubMed] [Google Scholar]
- 31.Newell KA, Asare A, Kirk AD, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest. 2010;120:1836–1847. doi: 10.1172/JCI39933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagoo P, Perucha E, Sawitzki B, et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest. 2010;120:1848–1861. doi: 10.1172/JCI39922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee RH, Pulin AA, Seo MJ, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu G, Zhang L, Ren G, et al. Immunosuppressive properties of cloned bone marrow mesenchymal stem cells. Cell Res. 2007;17:240–248. doi: 10.1038/cr.2007.4. [DOI] [PubMed] [Google Scholar]
- 35.Németh K, Leelahavanichkul A, Yuen PS, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dugast AS, Haudebourg T, Coulon F, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008;180:7898–7906. doi: 10.4049/jimmunol.180.12.7898. [DOI] [PubMed] [Google Scholar]
- 37.Fischer MA, Davies ML, Reider IE, et al. CD11b(+), Ly6G(+) cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog. 2011;7:e1002374. doi: 10.1371/journal.ppat.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.François S, Bensidhoum M, Mouiseddine M, et al. Local irradiation not only induces homing of human mesenchymal stem cells at exposed sites but promotes their widespread engraftment to multiple organs: A study of their quantitative distribution after irradiation damage. Stem Cells. 2006;24:1020–1029. doi: 10.1634/stemcells.2005-0260. [DOI] [PubMed] [Google Scholar]
- 39.Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhanireddy KK, Bruno DA, Weaver TA, et al. Portal venous donor-specific transfusion in conjunction with sirolimus prolongs renal allograft survival in nonhuman primates. Am J Transplant. 2009;9:124–131. doi: 10.1111/j.1600-6143.2008.02448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breous E, Somanathan S, Vandenberghe LH, et al. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50:612–621. doi: 10.1002/hep.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren G, Zhang L, Zhao X, et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–150. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 43.Ren G, Zhao X, Zhang L, et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol. 2010;184:2321–2328. doi: 10.4049/jimmunol.0902023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eggenhofer E, Benseler V, Kroemer A, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front Immunol. 2012;3:297. doi: 10.3389/fimmu.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eggenhofer E, Hoogduijn MJ. Mesenchymal stem cell-educated macrophages. Transplant Res. 2012;1:12. doi: 10.1186/2047-1440-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ko S, Deiwick A, Jager MD, et al. The functional relevance of passenger leukocytes and microchimerism for heart allograft acceptance in the rat. Nat Med. 1999;5:1292–1297. doi: 10.1038/15248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.