

Abstract
Dysfunctional bone morphogenetic protein receptor-2 (BMPR2) signaling is implicated in the pathogenesis of pulmonary arterial hypertension (PAH). We used a transcriptional high-throughput luciferase reporter assay to screen 3,756 FDA-approved drugs and bioactive compounds for induction of BMPR2 signaling. The best response was achieved with FK506 (tacrolimus), via a dual mechanism of action as a calcineurin inhibitor that also binds FK-binding protein-12 (FKBP12), a repressor of BMP signaling. FK506 released FKBP12 from type I receptors activin receptor-like kinase 1 (ALK1), ALK2, and ALK3 and activated downstream SMAD1/5 and MAPK signaling and ID1 gene regulation in a manner superior to the calcineurin inhibitor cyclosporine and the FKBP12 ligand rapamycin. In pulmonary artery endothelial cells (ECs) from patients with idiopathic PAH, low-dose FK506 reversed dysfunctional BMPR2 signaling. In mice with conditional Bmpr2 deletion in ECs, low-dose FK506 prevented exaggerated chronic hypoxic PAH associated with induction of EC targets of BMP signaling, such as apelin. Low-dose FK506 also reversed severe PAH in rats with medial hypertrophy following monocrotaline and in rats with neointima formation following VEGF receptor blockade and chronic hypoxia. Our studies indicate that low-dose FK506 could be useful in the treatment of PAH.
Introduction
Idiopathic pulmonary arterial hypertension (IPAH) is a rare disorder thought to develop following a genetic and/or environmental insult that triggers endothelial cell (EC) apoptosis, loss of distal vessels, and occlusive vascular remodeling (1). These pathological changes increase resistance to pulmonary flow and cause progressive right heart failure. Current therapies mainly include drugs with vasodilatory properties that improve cardiopulmonary function (2). However, the obliterative vascular pathology usually continues to progress (3), leaving heart-lung transplantation as the only option for many patients. Therefore, new approaches are needed that focus on activating cellular mechanisms to reverse vascular remodeling. One strategy could be to improve function of the bone morphogenetic protein receptor-2 (BMPR2) signaling pathway. Germline mutations causing loss of BMPR2 function are found in >80% of familial and approximately 20% of sporadic cases of IPAH (4, 5). Acquired somatic chromosomal abnormalities in the BMPR2 signaling pathway have also been described (6). The low penetrance of pulmonary arterial hypertension (PAH) found in nonaffected family members with a BMPR2 mutation has been attributed to a higher level of BMPR2 expression from the normal allele (7). In addition, patients with IPAH without a BMPR2 mutation or with PAH associated with other conditions have reduced expression of BMPR2 in pulmonary arteries (8). Furthermore, estrogen can reduce BMPR2 expression, perhaps explaining the propensity of females to develop PAH (9). IL-6, a cytokine increased in the blood of patients with IPAH, can reduce BMPR2 expression via a STAT3-miR17/92–mediated mechanism (10). Furthermore, patients with a BMPR2 mutation have worse pulmonary vascular remodeling (3). The importance of BMPR2 dysfunction in PAH is supported by studies in transgenic mice. Mice with deletion of BMPR2 in ECs (11) develop PAH, as do mice expressing a dominant-negative Bmpr2 gene after birth in vascular SMCs (12). Mice heterozygous for BMPR2 develop exaggerated PAH in response to hypoxia and serotonin (13). Reduced BMPR2 expression also occurs in monocrotaline (14) and chronic hypoxic (15) rat models of PAH, and delivery of BMPR2 by intravenous gene therapy attenuates the disease in both models (16, 17). Moreover reconstitution of athymic rats with regulatory T cells also prevents PAH resulting from blockade of the VEGF receptor, coincident with an increase in BMPR2 expression in ECs (18).
Since clinical application of BMPR2 gene therapy poses challenges, we sought to identify an FDA-approved drug with a known pharmacokinetic and toxicity profile that increases signaling through the BMPR2 pathway. We applied a high-throughput assay to screen available libraries, and agents that appeared to activate the receptor were further tested in human pulmonary arterial ECs (PAECs), including those from patients with dysfunctional BMPR2 signaling. The mechanism of BMPR2 activation by the most effective compound was investigated. Then the compound was tested in experimental PAH animal models for its efficacy in rescuing dysfunctional EC signaling and gene regulation and in preventing and reversing PAH.
Results
High-throughput screen yields activators of BMPR2 signaling.
Like other members of the TGF-β family, BMPs elicit their effects through activation of receptor complexes, composed of a type II receptor (BMPR2, ActivinR2A, or ActivinR2B) and a type I receptor (ALK3 [also known as BMPR1A], ALK6 [also known as BMPR1B], ALK2, or ALK1). This results in activation of multiple signaling pathways, such as MAPK, PI3K (pAKT), and pSMAD, and downstream transcription factors, such as PPARγ (19) and inhibitor of differentiation 1 (ID1) (20). The latter is a basic helix-loop-helix (HLH) transcription factor that binds other HLH transcription factors and acts as a dominant-negative antagonist (21). As substrates of the ubiquitin/26S proteasome system that regulate protein stability, ID proteins are short-lived, with a half-life of 30 minutes (22). As an early response gene (23), ID1 is transcribed in a pSMAD1/5-dependent manner (24) and thus is used commonly as a read out for BMPR2 signaling in vascular cells (20, 25, 26). Furthermore, ectopic ID1 expression induces EC migration and tube formation (26).
In developing the high-throughput assay to assess activation of BMPR2 signaling, we used the C2C12 mouse myoblastoma cell line stably transfected with a construct in which the BMP/SMAD elements of the ID1 promoter were linked to luciferase (BRE-luc). We optimized conditions to improve the luciferase signal related to varying the cell number, adding test compounds and the luciferase substrate. Plating 1,500 cells in 384-well plates without exogenous ligand permitted the assessment of activators of BMPR2 signaling in the presence of only endogenous ligand produced by the cells. Adding 250 pM BMP4, the concentration resulting in 20% of maximal response (EC-20), allowed distinction of activators requiring the presence of exogenous ligand (Figure 1A). Although BRE-luc C2C12 cells respond to all BMP ligands (BMP2, BMP4, BMP6, BMP7, and BMP9), we chose BMP4 for this assay because of excellent assay sensitivity. The following libraries available at the Stanford High-Throughput Bioscience Center were screened: NIH-CC, LOPAC, Biomol ICCB Known Bioactives, Microsource spectrum, and Biomol FDA-approved drug library.
Figure 1. High-dose FK506, the main activator of BMP signaling found in the high-throughput screen, recapitulates BMP4 signaling and function.

(A) BRE-luciferase activity in different numbers of C2C12 cells at different concentrations of BMP4. BMP4 at the EC-20 concentration of 250 pM for 1,500 cells per 60 μl per well permitted identification of coactivators. (B) Percentage BRE-luc activation, compared with 250 pM BMP4, in 6-point serial dilution for FK506 and rapamycin. (C) BRE-luciferase activity in C2C12 cells comparing FK506 (FK; 2 μg/ml), rapamycin (Rapa; 4 μg/ml), cyclosporine (Cyclo; 2 μg/ml), and Shield-1 (1 μg/ml) (n = 8; ***P < 0.001 vs. CON, ###P < 0.001 vs. FK506, §§§P < 0.001 vs. cyclosporine, 1-way ANOVA, Bonferroni multiple-comparison test). (D) In human PAECs, ID1 expression relative to B2M mRNA 24 hours after stimulation with vehicle (CON), FK506 (15 ng/ml), or rapamycin (10 ng/ml) (n = 3; **P < 0.01 vs. vehicle, 1-way ANOVA, Dunnett’s post test). (E and F) Representative Western immunoblot and relative densitometric analysis of PAECs showing (E) pSMAD1/5/8 and (F) ID1 relative to β-actin following stimulation with BMP4 (10 ng/ml) or FK506 (15 ng/ml) (n = 3; *P < 0.05; **P < 0.01 vs. CON, 2-way ANOVA). (G) Assessment of apoptosis with measurement of caspase-3/7 luminescence 24 hours after serum starvation and treatment with BMP4 (10 ng/ml) or FK506 (15 ng/ml). (n = 5; *P < 0.05; **P < 0.01 vs. CON, 1-way ANOVA, Dunnett’s post test). (H) Representative images from a Matrigel tube formation assay. Tube number and length were assessed after 8 hours in unstimulated cells (CON) or following stimulation with VEGF (20 ng/ml), BMP4 (10 ng/ml), or FK506 (15 ng/ml). (n = 6; *P < 0.05; **P < 0.01 vs. CON, 1-way ANOVA, Dunnett’s post test). Scale bar: 100 μm. Mean ± SEM.
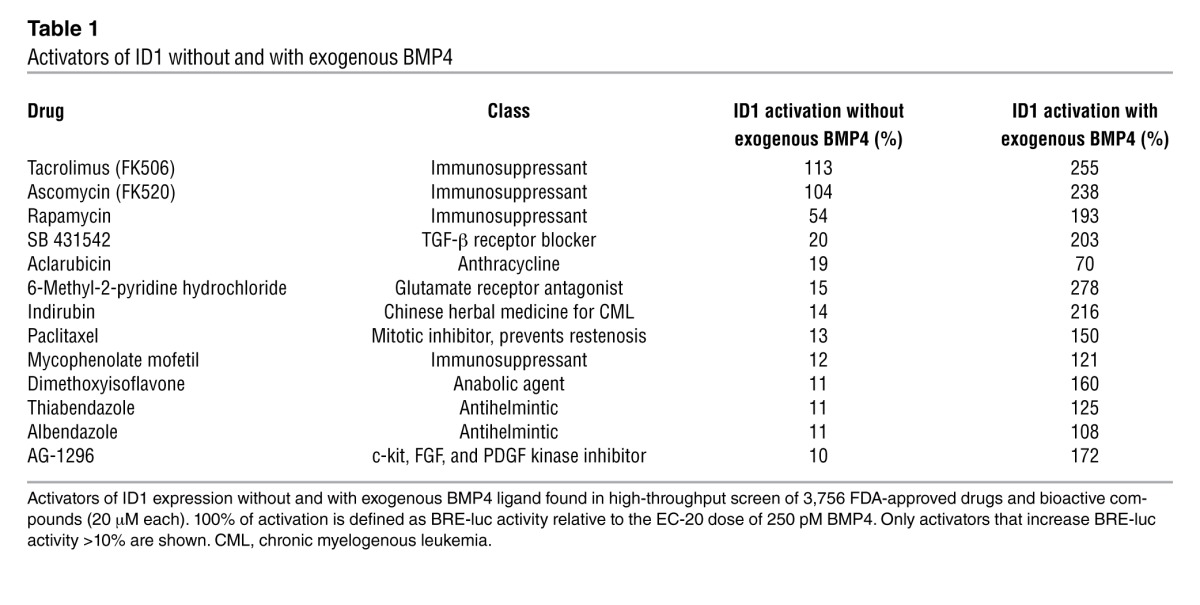
The major activators of BMPR2 signaling identified, with and without additional ligand, are listed in Table 1. Values of percentage of activation were defined as luminescence relative to the EC-20 dose of BMP4, and agents that increased BRE-luc reporter activity greater than 10% are shown. Tacrolimus (FK506) was the main activator of BRE-luc (113%). A similar response was seen with a less commonly used immunosuppressive agent, ascomycin (FK520). This compound, like FK506, interacts with FK-binding protein-12 (FKBP12) to inhibit calcineurin and NF-ATc–mediated IL-2 expression in T cells. Rapamycin, a third immunosuppressant that also interacts with FKBP12 and inhibits the mTOR pathway, but does not inhibit calcineurin, was a much weaker activator of signaling. Other agents included the TGF-β receptor blocker SB431542. The promising “hits” were tested in a 6-point serial dilution assay (Figure 1B), and the superiority of FK506 over rapamycin was shown with respect to the magnitude of BRE-luc activity reflecting BMPR2-mediated ID1 expression. FK506 induced ID1 at low doses, and a high plateau of activation was rapidly achieved. We then compared the effect of FK506 and rapamycin to another calcineurin inhibitor, cyclosporine, that does not interact with FKBP12 but binds the immunophilin, cyclophilin A, as well as shield-1 (27). Shield-1 binds FKBP12 but does not have any immunosuppressive properties (Figure 1C). We showed that the effect of FK506 on increasing BMP signaling could only partially be replicated by an agent that binds FKBP12 or that inhibits calcineurin. Furthermore, we showed that FK506 is selective in increasing BMP signaling in a dose-dependent fashion (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI65592DS1), without inducing TGF-β signaling (Supplemental Figure 1B).
Table 1.
Activators of ID1 without and with exogenous BMP4
FK506, like BMP4, induces ID1 and pSMAD1/5 in PAECs.
We also compared FK506 with rapamycin-mediated BMPR2 signaling in human PAECs. These cells have higher BMPR2 expression than other pulmonary artery vascular cells (8), and PAEC dysfunction has been implicated in the pathogenesis of PAH (11, 19, 28). Rapamycin attenuates chronic hypoxic PAH (29), but its effect on monocrotaline-induced PAH in rats is not unequivocal (30, 31). FK506 had not been previously tested in experimental PAH. We stimulated PAECs with 15 ng/ml FK506 or 10 ng/ml rapamycin, concentrations used to induce immunosuppression in the clinical setting. Consistent with the BMP/SMAD transcriptional luciferase assay, we confirmed a significant increase in ID1 mRNA 24 hours after stimulation with FK506, but only a trend was evident with rapamycin (Figure 1D). We then compared BMPR2 signaling induced by FK506 with that induced by the natural ligand BMP4. Both agents resulted in similar induction of pSMAD1/5 at 15 minutes and ID1 at 1 hour (Figure 1E), consistent with the fact that ID1 is an early target gene of SMAD1/5-dependent and -independent BMP signaling (23, 32). ID1 levels returned to control values 6 hours after BMP4 stimulation but remained elevated following FK506 stimulation (Figure 1F). We then showed that FK506 was similar to BMP4 in promoting PAEC survival following serum withdrawal, as judged by the Caspase-Glo 3/7 apoptosis assay (Figure 1G). In addition, BMP4 and FK506 were comparable to VEGF (20 ng/ml) in inducing formation of PAEC tubular networks in Matrigel (Figure 1H).
FK506 binds to and removes FKBP12 from BMP type I receptors.
Previous data suggested that a low subimmunosuppressive dose of FK506 can prevent neointima formation in a carotid artery injury model in Apoe–/– mice (33, 34). We confirmed in microvessel PAECs (mvPAECs) that FK506, in concentrations as low as 0.2 ng/ml, increased Id1 mRNA and protein (Figure 2, A and B, respectively). The mechanism of action of FK506 was then investigated. Previous studies have shown that a BMP ligand (BMP2, BMP4, BMP6, BMP7, and BMP9) can initiate the interaction of a type II receptor with serine/threonine kinase activity (BMPR2, ActivinR2A, or ActivinR2B) with a type I receptor (ALK1, ALK2, ALK3, and ALK6), resulting in its phosphorylation (35). We therefore determined whether FK506 requires the kinase activity to induce BMP signaling. When we pretreated C2C12 BRE-luc cells with a BMP type I receptor kinase inhibitor, LDN-193189, FK506 failed to induce BMP signaling, as judged by BRE-luciferase (ID1 expression) (Figure 2C) as well as pSMAD1/5 (Supplemental Figure 2A). These features were replicated by LDN-193189 inhibition of BMP6 activation of BMP signaling (BRE-luciferase) as a positive control. When BMPR2 was reduced by siRNA (documented in Supplemental Figure 2B), there was only a partial decrease in ID1 expression (Figure 2D), suggesting that another type II receptor such as Activin R2A could substitute. In fact, we were able to show that reducing both ActivinR2A and BMPR2 by siRNA caused an additive decrease in ID1 expression (Figure 2D), consistent with previous reports (36).
Figure 2. Low-dose FK506 induces pSMAD1/5/8 and ID1 in human mvPAECs related to FKBP12 interaction.

(A) Id1 mRNA, normalized to 18S mRNA, in human unstimulated mvPAECs (CON) or 1 hour after the addition of BMP4 (10 ng/ml) or FK506 (0.2 ng/ml and 2.0 ng/ml). (B) Representative immunoblot and densitometric analysis of ID1 protein relative to β-actin at time points and doses indicated in A. (A and B) (n = 3; *P < 0.05; **P < 0.01 vs. CON, 1-way ANOVA, Dunnett’s post test). (C) BRE-luciferase activity in C2C12 cells after stimulation with the BMPR2 ligand BMP6 (50 ng/ml) and FK506 (1 μg/ml) for 24 hours, with or without preincubation for 30 minutes with kinase inhibitor LDN-193189 (120 nM) (n = 6; ***P < 0.001 vs. CON; §§§P < 0.001 vs. BMP6, 1-way ANOVA, Bonferroni’s multiple-comparison test). (D) BRE-luciferase activity in C2C12 cells treated with nontargeting siRNA (NTsi), BMPR2 siRNA (BMPR2si), BMPR2si plus Activin2A siRNA (ActA2si), and BMPR2si plus Activin2B siRNA (ActBsi) and stimulated with FK506 (1 μg/ml) (n = 6; §§P < 0.01 vs. BMPR2si and FK treatment; ##P < 0.01 vs. BMPR2si-Act2Asi, 1-way ANOVA, Bonferroni’s multiple-comparison test). (E) Immunoprecipitation with an antibody to FLAG and immunoblot for HA and FLAG in 293T cells transfected with plasmids HA-ALK1, HA-ALK2, and HA-ALK3 as well as FKBP12-FLAG and stimulated with FK506 (100 ng/ml for 30 minutes). (F–H). BRE-luciferase activity in C2C12 cells treated with NTsi, ALK1 siRNA (ALK1si), ALK2si, and ALK3si and stimulated with (F) BMP4 (250 pM), (G) FK506 (2 μg/ml), and (H) rapamycin (n = 8; ##P < 0.01 vs. NTsi plus BMP4, NTsi plus FK506, NTsi plus rapamycin, 1-way ANOVA, Bonferroni’s multiple-comparison test). Mean ± SEM.
FKBP12 interacts with TGF-β type I family receptors and blocks signaling by occupying the phosphorylation site at its glycine-serine–rich region (37). We hypothesized that FK506-mediated BMP signaling was related to its interaction with and removal of FKBP12 (38) from type I receptors involved in BMP signaling, in addition to its known function as an inhibitor of the phosphatase calcineurin (39). By coimmunoprecipitation of 293T cell lysates transfected with plasmids containing FLAG-tagged FKBP12 as well as HA-tagged ALK1, ALK2, or ALK3, we showed that FKBP12 binds to type I receptors involved in BMP signaling and that FK506 releases FKBP12 from all 3 tested (Figure 2E). We then assessed which of the type I receptors was predominantly involved in FK506-mediated BMP signaling. We knocked down ALK1, ALK2, and ALK3 by siRNA in BRE-luc cells and assessed whether the loss of any one of the above receptors would compromise BMP4- or FK506-induced BRE luciferase activity. While BMP4 predominantly signaled via ALK3 and, to a lesser extent, ALK1 (Figure 2F), FK506 was able to interact with all 3 receptors (Figure 2G). Rapamycin exclusively recruited ALK2 (Figure 2H), a receptor that is not involved in BMP4-induced BMP signaling and therefore does not appear to preferentially interact with BMPR2. We confirmed that cyclosporine did not interact with type I receptors (Supplemental Figure 2C).
FK506 binds and removes FKBP12 from multiple type I receptors that interact with BMPR2 or ActivinR2A, whereas rapamycin only engages FKBP12 bound to ALK2. This explains, perhaps, why BRE-luc activity is greater with FK506 than with rapamycin combined with the calcineurin inhibitor cyclosporine (Supplemental Figure 2D). We furthermore tested whether removing FKBP12 by siRNA would mimic FK506 function, allowing for an increase in ID1 expression. However, reducing levels of FKBP12 to 30% of control values (Supplemental Figure 2B) did not enhance baseline BRE-luciferase (Supplemental Figure 2E). This suggests that incomplete depletion of FKBP12 is either not sufficient to activate type I BMP receptors (BRE-luc) or that engaging FKBP12 bound to the receptor and then removing it by FK506 may be required to activate the signaling machinery. We also assessed whether other FKBPs could inhibit ligand-independent type I receptor activation. However, reducing levels of 6 other FKBPs (FKBP12.6, FKBP13, FKBP25, FKBP52, FKBP54, and FKBP38) by siRNA, one at a time, did not induce BRE-luciferase activity (Supplemental Figure 2, F and G). These data are consistent with a redundancy in FKBPs as binding partners of type I receptors. We confirmed FK506 activation of pSMAD1/5 as one read out for BMP signaling as well as others, such as pERK, pAKT, and p-c-JUN (Supplemental Figure 2, H–J).
FK506 rescues endothelial dysfunction in PAECs from patients with IPAH.
To determine whether FK506 could rescue BMPR2 dysfunction in PAECs, we reduced BMPR2 protein levels to 50% of control values using RNAi (Figure 3A). As expected, these cells were unresponsive to BMP4 (10 ng/ml), but FK506 (15 ng/ml) increased ID1 in keeping with its ability to engage another type II receptor, specifically ActivinR2A (40). We then screened mvPAECs cultured from lungs harvested at transplant from 6 patients with IPAH that were obtained through the Pulmonary Hypertension Breakthrough Initiative Network. We found that mvPAECS from 3 out of the 6 patients did not increase pSMAD1/5 or ID1 in response to normally activating doses of BMP4 (10 ng/ml). One of these patients had a documented mutation in BMPR2 (c.961C>CT; p.321R>R/X). In contrast, all mvPAEC cultures that did not respond to BMP4 showed an increase in pSMAD1/5 and ID1 1 hour after stimulation with FK506 (15 ng/ml) (Figure 3B). Interestingly, 4 hours after low-dose FK506 (0.2 ng/ml), ID1 mRNA was elevated, and at 8 hours, apelin mRNA was increased (Figure 3C). Apelin is regulated by BMPR2-mediated signaling (19) and is important in endothelial homeostasis. We further documented that FK506 (0.2 ng/ml) rescued dysfunction of IPAH PAECs by reversing their previously described impaired ability to form tubular networks in Matrigel (41). FK506 induced tube formation 4 hours after stimulation, in a manner comparable to that of VEGF (20 ng/ml), whereas BMP4 failed to do so (Figure 3D).
Figure 3. Low-dose FK506 rescues IPAH mvPAEC dysfunction.

(A) Representative immunoblot and densitometry showing BMPR2 and ID1/β-actin in human PAECs stimulated with BMP4 (10 ng/ml) or FK506 (15 ng/ml) following knockdown of BMPR2 with BMPR2si compared with NTsi as control (n = 3; *P < 0.05 vs. CON, 2-way ANOVA). (B) Representative immunoblot and densitometry of mvPAECs from 3 patients with IPAH showing ID1/β-actin at baseline (CON) or at various time points after stimulation with BMP4 (10 ng/ml) or FK506 (15 ng/ml) (n = 3; *P < 0.05 vs. CON, 2-way ANOVA). (C) ID1/18S mRNA 4 hours after and apelin/18S mRNA 8 hours after stimulation with BMP4 (10 ng/ml) and low-dose FK506 (0.2 ng/ml) in the same IPAH mvPAECs as in B (n = 3; *P < 0.05; **P < 0.01, vs. CON, 1-way ANOVA, Dunnett’s post test). (D) Representative images and quantitative analysis of tube number and length of tubes formed in Matrigel with mvPAECs from a patient with IPAH 4 hours after unstimulated CON, VEGF (10 ng/ml), BMP4 (10 ng/ml), and FK506 (0.2 ng/ml) (n = 6; ***P < 0.001; **P < 0.01 vs. CON, 1-way ANOVA, Dunnett’s post test; scale bar: 100 μm). (E) Proposed model of activation of BMPR2 in the presence of a subactivating dose of BMPs and an activating dose of BMPs and mutated or dysfunctional BMPR2 receptor with an activating dose of BMPs or with a subactivating dose of BMPs and FK506. Mean ± SEM.
Taken together, we propose the following mechanism by which FK506 activates BMP signaling (see schema in Figure 3E). In the presence of BMPs at a subactivating dose, FKBP12 acts as inhibitor of BMP signaling. An activating dose of BMP ligand initiates removal of FKBP12 from the type I receptor and allows for normal SMAD- and non-SMAD–dependent signaling. When BMPR2 is mutated or dysfunctional, FK506 restores normal signaling by the dual action of removing FKBP12 from the type I receptor and inhibiting calcineurin.
Low-dose FK506 prevents hypoxic PAH in mice with deletion of BMPR2 in ECs.
Next, we asked whether FK506 would prevent PAH in a mouse with BMPR2 deleted in ECs to show — as proof of concept — that FK506 could rescue downstream BMP signaling, even in the absence of a functional BMPR2 receptor. We crossed SCL-CreERTM, R26 LacZfl/fl, and Bmpr2fl/fl mice and created an SCL-CreERTM+/R26R/Bmpr2–/– (EC-Bmpr2–/–) mouse by administering 2 mg tamoxifen per day intraperitoneally for 10 days when the mice were 8–12 weeks of age. We documented BMPR2 deletion by positive LacZ staining in lung ECs (Figure 4, A and B) and in ECs of other tissues (data not shown). Whole lung lysates of EC-Bmpr2–/– mice showed reduced BMPR2 protein expression compared with those of Cre–/– mice and Cre+/+ mice without tamoxifen treatment (Figure 4C). ECs harvested from the lungs of EC-Bmpr2–/– mice using CD31 antibody-coated beads showed no BMPR2 expression in contrast to PAECs from littermate controls (Figure 4D).
Figure 4. Low-dose FK506 prevents the development of PAH in mice with a deletion of BMPR2 in ECs.

(A and B) LacZ staining documenting BMPR2 deletion in (A) distal PAECs and (B) capillary ECs in EC-Bmpr2–/– mice. Scale bar: 100 μm. (C) Reduced BMPR2 in whole lung lysates by Western immunoblot comparing wild-type mice with tamoxifen (TAM) with floxed Bmpr2 Cre-ERT mice with or without tamoxifen. (D) Absence of Bmpr2 mRNA in ECs harvested from EC-Bmpr2–/– (KO) vs. wild-type mice (n = 3; ***P < 0.001 WT vs. EC-Bmpr2–/–, 1-way ANOVA, Dunnett’s). (E) RVSP in wild-type and EC-Bmpr2–/– mice treated with vehicle or FK506 (0.05 mg/kg/d) in normoxia or hypoxia (10% O2) for 3 weeks (n = 8 per group; **P < 0.01 compared with vehicle normoxia; §§P < 0.01 vehicle hypoxia wild-type vs. EC-Bmpr2–/–, 2-way ANOVA, Dunnett’s). (F) RVH (Fulton index, weight of RV/LV and septum) in same groups as in E. (G) Representative histology of barium-injected lungs of wild-type and EC-Bmpr2–/– mice after normoxia/hypoxia with or without vehicle or FK506. The decreased number of vessels in hypoxia is ameliorated by FK506 (arrows). Scale bar: 100 μm. (H) Percentage muscularized alveolar wall and alveolar duct arteries per total arteries in wild-type and EC-Bmpr2–/– mice after normoxia/hypoxia with or without vehicle or FK506 (n = 8 mice;mean of 5 ×20 fields per mouse; **P < 0.01; ***P < 0.001 vs. normoxia wild-type and EC-Bmpr2–/–, 2-way ANOVA, Dunnett’s). (I) Arteries per 100 alveoli in same animals as in H. (J) Apelin/18S mRNA and (K) eNOS/18S mRNA in mvPAECs from EC-Bmpr2–/– mice, preconditioned in 1% hypoxia and harvested 8 hours after BMP4 (10 ng/ml) or FK506 (0.2 ng/ml) vs. vehicle (CON) (n = 3; *P < 0.05 vs. CON, 1-way ANOVA, Dunnett’s). Mean ± SEM.
Male animals generally have a greater chronic hypoxia-induced pulmonary hypertensive response (42), so we used them exclusively in our study. EC-Bmpr2–/– and littermate control mice were housed in room air or in 10% oxygen (chronic hypoxia) for 3 weeks, during which time half the mice of each genotype were given a continuous s.c. infusion of low-dose FK506 (0.05 mg/kg/day) by mini-osmotic pump and the other half were given vehicle. EC-Bmpr2–/– mice and littermate controls maintained in room air had similar values for right ventricular systolic pressure (RVSP), and FK506 had no effect on this measurement. The elevation in RVSP in chronic hypoxia-exposed mice, as a measure of PAH, was more severe in the EC-Bmpr2–/– group than in controls (Figure 4E), although there was no further increase in right ventricular hypertrophy (RVH) (Figure 4F) or in vascular changes, including muscularization of distal arteries (Figure 4G, vessels marked by arrows, and Figure 4H) and loss of peripheral arteries (Figure 4I). Administration of low-dose FK506 prevented chronic hypoxic PAH in both EC-Bmpr2–/– mice and in littermate controls, as judged by lack of elevated RVSP, RVH, and reduced vascular changes. Cardiac output decreased slightly in both genotypes after chronic hypoxia independent of FK506 treatment (Supplemental Table 1). A 3-week treatment with low-dose FK506 did not increase the systemic blood pressure (Supplemental Figure 3). Despite its immunosuppressive properties, FK506, in the dose used, did not reduce total white blood cells, neutrophils, or lymphocytes (Supplemental Table 1). To address whether FK506 rescued EC dysfunction related to loss of BMPR2, we exposed PAECs from the EC-Bmpr2–/– mice to 1% O2 for 24 hours and treated the cells with low-dose FK506 (0.2 ng/ml) or BMP4 (10 ng/ml) for another 8 hours under the same conditions. An increase in apelin mRNA, an EC gene normally regulated by BMP signaling, and in endothelial nitric oxide synthase (eNOS), a gene regulated by apelin (43) and a marker of EC homeostasis were observed with FK506, but not with BMP4 (Figure 4, J and K). The same was true for cells under normoxic conditions (data not shown).
Low-dose FK506 reverses monocrotaline-induced PAH in rats, inhibits proliferation, and induces apoptosis in pulmonary artery SMCs.
As patients with PAH present late in the course of their disease, we assessed whether FK506 could also reverse severe PAH and vascular pathology in an experimental model without a deletion in BMPR2. PAH was induced in adult male Sprague Dawley rats, weighing between 180 and 220 g, with a single s.c. dose of monocrotaline (60 mg). PAH was verified in a subset of animals after day 21, and a 3-week treatment with vehicle or low-dose FK506 (0.05 mg/kg/d) was initiated via s.c. mini-osmotic pump. While vehicle-treated rats showed severe PAH at day 42, FK506 treatment significantly reduced the PAH, measured by the RVSP (Figure 5A) as well as RVH (Figure 5B). The monocrotaline animal model of PAH is characterized by increased muscularity, and FK506-treated animals showed reduced muscularization compared with vehicle-treated animals (Figure 5C).
Figure 5. Low-dose FK506 reverses monocrotaline-induced PAH in rats, inhibits proliferation, and induces apoptosis in PASMCs.

(A) RVSP (mmHg) in rats 21 days after monocrotaline (MCT) treatment (60 mg s.c. once), with or without 3-week treatment with vehicle (VEH) or low-dose FK506 (n = 6–8; **P < 0.01 compared with VEH treated; #P < 0.05 compared with day 21 monocrotaline; §§§P < 0.001 compared with day 42 monocrotaline vehicle, 1-way ANOVA, Bonferroni post test). (B) RVH (Fulton index) in same group as above (***P < 0.001 compared with vehicle treated; §P < 0.05 compared with day 42 monocrotaline vehicle, 1-way ANOVA, Bonferroni post test). (C) Represented lung histology (Movat stain) (scale bar: 100 μm). (D) MTT proliferation assay with PASMCs in the presence or absence of PDGF (20 ng/ml) 72 hours after stimulation with vehicle (CON), BMP4 (10 ng/ml), or FK506 (0.2 and 2.0 ng/ml) (n = 5; **P < 0.01, ***P < 0.001, BMP4 vs. CON [in the presence of PDGF], 1-way ANOVA, Dunnett’s). (E) Assessment of apoptosis with measurement of caspase-3/7 luminescence under 24-hour serum starvation or treatment with BMP4 (10 ng/ml) or FK506 (0.2, 1.0, 2.0, and 15 ng/ml) (n = 5; *P < 0.05 1-way ANOVA, Dunnett’s post test). (F) Luciferase NFAT reporter activation assay in PASMCs stimulated with PDGF (30 ng/ml) and doses of FK506 between 0.2 and 15 ng/ml. (##P < 0.01 and ###P < 0.001, vs. stimulation by PDGF alone, 1-way ANOVA with Bonferroni’s multiple pairs test). (G) Il2/Gapdh mRNA expression in whole lungs of rats 21 days after monocrotaline treatment (60 mg s.c. once), with or without 3-week treatment with vehicle or low-dose FK506 (n = 6–8; nonsignificant, 2-way ANOVA). Mean ± SEM.
We had previously shown that FK506 could increase apelin in pulmonary artery SMCs (PASMCs). Apelin has beneficial autocrine effects on PAEC function and, by a paracrine mechanism, suppresses PASMC proliferation and can induce apoptosis (19). However, we also showed a direct effect of FK506 in inhibiting PDGF-induced PASMC proliferation (Figure 5D) and in inducing apoptosis, as judged by the caspase luminescence assay (Figure 5E). Interestingly, the latter effect of FK506 on SMCs was lost at higher immunosuppressive doses of 15 ng/ml. The mechanism appears to be pSMAD1/5 and ID1 independent (Supplemental Figure 4, A and B) and suggests an alternative pathway, possibly via PPARγ and ApoE (44) or via suppression of NF-ATc, as has been previously shown (31). Using an NFAT reporter assay in PASMCs, we confirmed mild NFAT inhibition at low doses of FK506 (0.2–0.5 ng/ml) and a more pronounced effect at doses clinically used to induce immunosuppression (5–15 ng/ml) (Figure 5F). However, FK506 showed only a trend in reducing expression of IL-2, a major downstream target of NFAT signaling, when assessed in whole lung tissue of vehicle- and monocrotaline-treated rats (Figure 5G), and no effect on IFN-γ, another NFAT target (Supplemental Figure 4C). This suggests that inhibition of NFAT activation is not the major mechanism of action by which low-dose FK506 reverses PAH.
Low-dose FK506 reverses PAH related to neointima formation.
Given that the monocrotaline rat model does not replicate the pathology in humans with PAH (45), we also assessed whether FK506 could reverse severe obliterative pulmonary vascular lesions characterized by neointima formation in distal pulmonary arteries. This pathology is seen in rats treated with the VEGF receptor blocker SUGEN 5416 and exposed to chronic hypoxia (46, 47). Adult male Sprague Dawley rats at 8 weeks of age, weighing between 180 and 220 g, were given a single s.c. dose of SUGEN 5416 (20 mg/kg), exposed to chronic hypoxia for 3 weeks, and then kept in normoxia for another 5 weeks (Figure 6A). This experimental protocol induces severe and progressive PAH, with extensive loss and obliteration of distal arteries associated with neointimal lesions (46). Beginning at 8 weeks, we initiated a 3-week treatment with either vehicle or low-dose FK506, by continuous s.c. infusion via mini-osmotic pump (0.05 mg/kg/d). Vehicle-treated rats showed severe PAH, but FK506 reversed the elevation in RVSP and RVH, resulting in values not significantly different from those of controls (Figure 6B). Cardiac output was lower after administration of SUGEN 5416 and hypoxia and increased with FK506 treatment (Supplemental Table 2). The percentage of vessels with a neointima was reduced from 60% in vehicle-treated rats to less than 20% in those administered FK506 (Figure 6C). Consistent with previous studies, we documented mild emphysema in rats treated with SUGEN 5416 and hypoxia (48, 49), reflected in a 25% decrease in the number of alveoli and a comparable reduction in the number of arteries (Figure 6D). FK506 preferentially restored arterial number so the ratio of arteries per 100 alveoli increased (Figure 6E). As in the murine chronic hypoxia experiments, FK506 also restored endothelial function, as judged by an increase in apelin (Figure 6F) in whole lung tissue. Although we cannot exclude a contribution of NFAT inhibition in FK506-mediated reversal of PAH, we found no significant reduction in targets of NFAT activation, i.e., IL-2 (Figure 6G) and IFN-γ (Supplemental Figure 5).
Figure 6. Low-dose FK506 reverses established PAH and neointima formation in rats.

(A) Timeline of experiments. (B) RVSP and RVH (Fulton index) in rats in normoxia with or without vehicle (VEH) or FK506 and in rats after SUGEN with 3 weeks hypoxia and 5 weeks normoxia, sacrificed at 8 weeks and at 11 weeks following treatment with vehicle or FK506 (n = 8; *P < 0.05; ***P < 0.001 vs. same condition in normoxia, 2-way ANOVA, Bonferroni post test). (C) Representative images (scale bar: 100 μm) and percentage vessels with neointima in hypoxia/SUGEN/normoxia rats at 8 and 11 weeks treated with vehicle or FK506 (n = 8; **P < 0.01 vs. vehicle, 1-way ANOVA, Bonferroni post test). (D) Number of alveoli per mm2 in normoxia (with or without vehicle or FK506) and in rats after hypoxia/SUGEN/normoxia, sacrificed at 11 weeks (with or without 3-week treatment of vehicle or FK506) (n = 8; *P < 0.05 vs. untreated normoxia condition, 2-way ANOVA, Dunnett’s post test). (E) Mean number of arteries per 100 alveoli (5 fields ×20) in same groups as in D (n = 8, #P < 0.05, 1-way ANOVA, selected pairs post test, vehicle vs. FK506). (F) Apelin/β-actin mRNA expression in total lung lysate in same group as in D (n = 8; *P < 0.05, 1-way ANOVA, Dunnett’s post test). (G) Il2/Gapdh mRNA expression in whole lungs of rats in normoxia (with or without vehicle or FK506) and in rats after hypoxia/SUGEN/normoxia, sacrificed at 11 weeks (with or without 3-week treatment of vehicle or FK506) (n = 8; *P < 0.05 vs. vehicle normoxia, #P < 0.05 vs. FK normoxia, nonsignificant trend in decrease of IL-2 with FK506 treatment, 2-way ANOVA, Bonferroni multiple-comparison test). Mean ± SEM.
Discussion
We report the first high-throughput screen of known pharmaceuticals to determine whether any of these agents would increase BMPR2 signaling. Our study design permitted evaluation of strong activators (without exogenous BMP ligand), weaker activators (with exogenous BMP ligand), and potential inhibitors of BMP signaling. The top 3 activators of BMP-mediated signaling and ID1 target gene expression, FK506 (tacrolimus), FK520 (ascomycin), and rapamycin, are potent immunosuppressants that prevent T cell proliferation by interacting with the immunophilin FKBP12 (50). FK506 appears superior to rapamycin in potentiating BMP signaling because it also inhibits the phosphatase calcineurin (51). In addition, FK506 interacts with FKBP12 associated with all 3 BMPR type 1 receptors (ALK1, ALK2, and ALK3), including those preferred by BMPR2 (ALK1 and ALK3), whereas rapamycin only interacts with ALK2. Cyclosporine shares the calcineurin inhibitory properties of FK506 (52), but as it does not bind FKBP12, it is a weak activator of BMP signaling in PAECs.
Also on the list of BMPR2 signaling activators is the TGF-β receptor blocker, SB431542, which could antagonize the enhanced TGF-β signaling that occurs when BMPR2 is deficient (53). Our libraries contained pharmaceuticals currently used to treat PAH, including bosentan, iloprost, and imatinib. None activated BRE-luciferase, although iloprost can induce ID1 by a pSMAD1/5-independent, cAMP-mediated mechanism (54). Recent data suggest that other medications approved and effective for PAH, such as the vasodilator and phosphodiesterase inhibitor sildenafil, can partially restore BMPR2 defective signaling (55), underscoring the importance of this pathway in PAH.
Both at immunosuppressive and at low doses, FK506 not only mimics BMP4 signaling in PAECs but also promotes PAEC survival and increases tube formation, functions essential for preventing loss of vessels and inducing vessel regeneration in PAH (56). In addition, we have evidence of both paracrine (via apelin) (57) and autocrine effects of FK506 in reducing proliferation and mediating apoptosis in SMCs.
TGF-β type I family receptors (including ALK3) share a common glycine-serine–rich sequence, identified as the site of phosphorylation by type 2 receptors as well as the FKBP12 binding site (58). FKBP12 has multiple functions, including Ca++ channel gating and protein folding (37). By serving as a docking protein for the phosphatase calcineurin (39), FKBP12 also modulates the phosphorylation status of TGF-β type I and other receptors, e.g., EGF-R and inositol trisphosphate receptors (50), providing a safeguard against leaky constitutive signaling (37). We showed that FK506 interacts with FKBP12 and is able to dissociate it from type I receptors, e.g., ALK1, ALK2, and ALK3 that form heterodimers with BMPR2. This is a novel finding, although the concept that FK506 can release bound FKBP12 from TGF-β type I receptors or EGF-R (59) has been described previously. Furthermore FK506, even at low doses, inhibits calcineurin activity, which, in concert with removing FKBP12, results in phosphorylation of ALK1, ALK2, and ALK3 and in downstream pSMAD1/5-dependent and -independent signaling. Phosphorylation of the type I receptor induced by FK506 requires kinase activity from a type 2 BMP receptor, and the type I receptor likely co-opts ActivinR2A when BMPR2 is reduced or dysfunctional. Thus, signaling is transduced in mice with deletion of BMPR2 in ECs and in human PAECs with mutant BMPR2.
FK506 also increases EC expression of ALK1 and endoglin (60). As loss-of-function mutations in these genes are observed in PAH associated with hemorrhagic telangiectasia (HHT), FK506 could also be of potential benefit in patients with this disorder. In fact, FK506 was given following liver transplant in a patient with HHT who had multiple arteriovenous malformations, and it was noted that internal and external telangiectases, epistaxes, and anemia disappeared, suggesting that the mechanism of action of FK506 involved partial correction of endoglin and ALK1 haploinsufficiency (60).
Binding and removing FKBP12 from the type I receptors by FK506 is dynamic and varies with different FK506 concentrations at different time points (data not shown), so the effect, particularly, of low-dose FK506 is unlikely to induce the same constitutive activation of BMP signaling that is observed in fibrodysplasia ossificans progressiva. This is a condition characterized by a mutation in ALK2 that permanently reduces binding of FKBP12 (61).
Low-dose FK506 reverses the impaired ability of PAECs from patients with IPAH to form tubular networks (41, 62), consistent with its induction of apelin, a pSMAD-independent target of BMPR2 signaling (19). Apelin (63), a ligand for the G protein–coupled receptor APJ, is highly expressed in endothelial but not in other vascular cells (64). It is proangiogenic (65) and induces eNOS (57). Apelin-null mice develop more severe hypoxia-induced PAH than controls. Patients with IPAH have reduced apelin plasma levels (43), and their PAECs have decreased apelin mRNA and protein (19). Furthermore, the role of apelin in raising levels of a miRNA that represses FGF production is thought to be responsible for the paracrine effect of apelin in suppressing SMC proliferation and inducing SMC apoptosis. This may be the mechanism causing reversal of PAH in both the monocrotaline and the SUGEN hypoxia rat models (66), but we also showed that low-dose FK506 represses cultured PASMC proliferation and induces apoptosis independent of endothelial apelin.
Our studies show efficacy of low-dose FK506 in activating the BMPR2 pathway. Interestingly, it was this dose in contrast to a higher dose that inhibited atherosclerotic plaque development and progression in Apoe–/– mice (33, 34). Moreover, high doses of FK506 (>10 fold of those used in our study) caused systemic hypertension and transplant vasculopathy in animal models, presumably mediated by a paradoxical decrease in eNOS and increase in endothelin production (67). In contrast, the low-dose FK506 used in our studies did not induce systemic hypertension, even after a 3-week treatment.
We chose mice with deletion of BMPR2 in ECs to determine whether FK506 could prevent hypoxia-induced PAH in the absence of endothelial BMPR2 and found that it was effective in both EC-Bmpr2–/– and wild-type genotypes. The latter may be related to reversal of hypoxia-mediated suppression of BMPR2 signaling (68).
Our study is one of few to show that an agent can reverse severe established PAH and neointima formation in the SUGEN5416/hypoxia rat model (46, 66). We suggest that FK506 was able to do so both by upregulating apelin and, to a lesser extent, by suppressing NF-ATc–mediated gene regulation. The effect of inhibiting NFAT activation, in addition to increasing BMPR2 signaling, would be beneficial in treating PAH. Previous studies have shown that inhibition of NF-ATc with rapamycin reverses monocrotaline-induced PAH (31) by restoring Kv1.5 channel expression, decreasing proliferation, and inducing apoptosis of SMCs.
Treatment with FK506 increases cardiac output in the SUGEN5416/hypoxia rat model, perhaps because the interaction of FK506 with FKBPs modulates calcium cycling in the heart. In a mouse model of left heart failure, characterized by increased expression of FKBP12.6 in the heart, FK506 restored contractile function of isolated cardiomyocytes by inhibiting the association with FKBP12.6 and the ryanodine receptor (69).
In conclusion, we used a high-throughput screen to identify a compound, FK506, that is able to activate BMPR2-mediated signaling and endothelial-specific gene regulation (e.g., apelin), even in the absence of exogenous ligand and BMPR2. The efficacy of low-dose FK506 in improving BMP signaling could be valuable in treating PAH as well as other vascular and nonvascular diseases, including cancer, in which BMP signaling is reduced, and bone grafting or spinal fusion surgery, in which activation of BMP is required for a successful result (70, 71).
Methods
High-throughput screen.
Libraries of FDA-approved drugs and bioactive compounds, containing 3,756 unique compounds (NIH-CC, LOPAC, Biomol ICCB Known Bioactives, Microsource spectrum, Biomol FDA-approved drug library), were available from the High-Throughput Bioscience Center at Stanford University. The C2C12 mouse myoblast cell line that stably expresses a BMP response element from the ID1 promoter linked to luciferase (BRE-luc) (25) was used as a reporter cell line for BMP activation. The bioassay can detect luciferase activity after stimulation with a dose as low as 3 pM BMP4 (our ligand of choice, due to the excellent assay sensitivity) (see Supplemental Figure for details).
Reagents.
FK506 (Cayman Chemicals), rapamycin (Cayman Chemicals), cyclosporine (Sigma-Aldrich), shield-1 (a gift from T.J. Wandless, Stanford University), SUGEN5416 (Cayman Chemicals), BMP4 (Sigma-Aldrich), and monocrotaline (Sigma-Aldrich) were used.
Plasmids.
FKBP12-Flag plasmid was a gift from Takeshi Imamura (JFCR Cancer Institute, Tokyo, Japan). HA-ALK1, HA-ALK2, HA-ALK3 have been previously described (72).
Transcription reporter assays.
C2C12 cells were seeded in 24-well plates and then transfected with Polyethylenimine (PEI, Fermentas) with the indicated plasmids. For CAGA-Luc, the luciferase activity was analyzed 24 hours after stimulation with TGF-β3 (5 ng/ml) or FK506 with indicated concentrations. For BRE-luc, the transfected cells were pretreated with LDN-193189 (120 nM) for 30 minutes before stimulation with FK506 (1 μg/ml) for 24 hours. Each experiment was performed in triplicate at least, with normalization to β-galactosidase activity.
Cell culture.
C2C12 mouse myoblastoma cell line was stably transfected with a construct in which the BMP/SMAD elements of the ID1 promoter were linked to luciferase (BRE-luc) (a gift from Peter ten Dijke). C2C12 and 293T were maintained in DMEM supplemented with 10% FBS, penicillin/streptomycin, and Glutamine (Gibco). Human large PAECs (ScienCell) and human and mouse mvPAECs were grown in commercial EC media (ScienCell). Cells were subcultured at a 1:3 ratio in gelatin-coated dishes. Human PASMCs (Lonza) were grown in commercial SMC media (Lonza) and subcultured at a 1:5 ratio. Only passages 3–8 were used.
Immunoprecipitation and immunoblotting.
293T cells were transfected with indicated plasmids, medium was changed 12 hours after transfection, and, 48 hours after transfection, cells were stimulated with FK506 (100 ng/ml) for 30 minutes (see the Supplemental Methods for details).
Isolation of human and mvPAECs.
Human and murine mvPAECs were isolated from digested whole lung tissue using CD31 antibody–coated magnetic beads (Dynabeads; Invitrogen) as previously described (19).
mRNA expression by qRT-PCR.
Total RNA was extracted and purified from whole lung tissue and cells with the RNAeasy Plus Kit (Qiagen) and then reverse transcribed using SuperScript III (Invitrogen) per the manufacturer’s instructions. Expression levels of selected genes were quantified using preverified Assays-on-Demand TaqMan primer/probe sets (Applied Biosystems) and normalized to ribosomal RNA 18S and B2M (mice and humans) and β-actin and Gapdh (rats). The housekeeping gene that was chosen was most consistent throughout the experimental conditions.
Western immunoblotting.
Western immunoblotting was done as previously described (41).
Apoptosis assay and proliferation assays.
PAEC, mvPAEC, and PASMC survival and proliferation were assessed by cell counts and MTT assays. Caspase-3/7 (Promega) was used to detect apoptosis as previously shown (19, 41).
RNAi.
siRNAs for BMPR2 (Dharmacon) were transfected into PAECs using RNAi Max (Invitrogen) as described previously (73). The knockdown efficiency for BMPR2 was determined by Western immunoblotting. siRNAs for BMPR2, ActivinR2A, Activin R2B, ALK1, ALK2, ALK3, and FKBP12 were transfected into C2C12 BRE-luc cells using DharmaFect3, and knockdown efficiency was determined by qRT-PCR. Nontargeting siRNA (Dharmacon) was used as a control.
Tube formation assay.
PAECs were starved for 12 hours in 0.2% FBS, and 10,000 cells were seeded on Matrigel (Trevigen) in serum-free medium with or without stimuli. The number of tubes and tube length were assessed after 8 hours in large PAECs and after 4 hours in mvPAECs.
Creation of cell-specific inducible EC-Bmpr2–/– mice.
Endothelial SCL-CRE ERT mice, with a 5′-endothelial SCL enhancer (–7 to 0.9 kb) that directs expression to endothelium (74), were crossed with Rosa26R mice and crossed with Bmpr2fl/fl mice created previously in our laboratory. Tamoxifen was given at 2 mg per day i.p. for 10 days; after another 14 days, CRE activation led to a deletion in BMPR2 in ECs in lungs as well as all other vascularized tissues. Littermates were used as controls.
Hypoxia/SUGEN/normoxia and monocrotaline rat models.
Adult male Sprague Dawley rats at 8 weeks of age, weighing between 180 and 220 g, were given a single s.c. dose of SUGEN5416 (20 mg/kg) and then placed in chronic hypoxia for 3 weeks, followed by a period of 5 weeks normoxia, as previously described (46). The monocrotaline experiments in rats were carried out as previously described (75).
Statistics.
All data are presented as mean ± SEM. We performed the statistical analyses using 1-way ANOVA when more than 2 groups of data were compared and 2-way ANOVA when 2 conditions were involved. We considered differences to be statistically significant at P < 0.05.
Study approval.
All animal experiments were reviewed and approved by the Stanford University IACUC in accordance with the guidelines of the NIH. The studies on human primary cells were approved by the appropriate Institutional Review Board on human subjects and IACUC at Stanford University.
Supplementary Material
Acknowledgments
We would like to thank Michal Bental Roof for her invaluable help in manuscript editing and the preparation of the figures. This work was supported by NIH grant 1K08HL107450-01 (to E. Spiekerkoetter), a supplemental grant from the Pulmonary Hypertension Association (to E. Spiekerkoetter), NIH grant K12 RFA-HL-07-004 (to E. Spiekerkoetter), American Heart Association Summer Undergraduate Research Fellowship (to R. Haghighat and L. Haghighat), NIH R01 HL074186 (to M. Rabinovitch), and an endowment (to M. Rabinovitch) from the Vera Moulton Wall Center for Pulmonary Vascular Disease at Stanford. We acknowledge the support from the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organisation for Health Research and Development, and the Royal Netherlands Academy of Sciences as well as the LeDucq foundation (to P. ten Dijke).
Footnotes
Conflict of interest: A patent application has been filed for the use of FK506 in pulmonary hypertension (Edda Spiekerkoetter, Marlene Rabinovitch, Philip A. Beachy, David E. Solow-Cordero). Nesrine El-Bizri is a current employee of Gilead Sciences. Thomas J. Wandless owns stock at Theravance and served as a paid expert witness for Fougera.
Citation for this article: J Clin Invest. 2013;123(8):3600–3613. doi:10.1172/JCI65592.
Hirofumi Sawada’s present address is: Department of Pediatrics, Mie University Graduate School of Medicine, Mie, Japan.
Nesrine El-Bizri’s present address is: Gilead Sciences, Foster City, California, USA.
References
- 1.Humbert M, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 suppl S):13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Chin KM, Kim NH, Rubin LJ. The right ventricle in pulmonary hypertension. Coron Artery Dis. 2005;16(1):13–18. doi: 10.1097/00019501-200502000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Stacher E, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(3):261–272. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng Z, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomson JR, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37(10):741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldred MA, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(9):1153–1160. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamid R, et al. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30(4):649–654. doi: 10.1002/humu.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atkinson C, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–1678. doi: 10.1161/01.CIR.0000012754.72951.3D. [DOI] [PubMed] [Google Scholar]
- 9.Austin ED, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ. 2012;3(1):6. doi: 10.1186/2042-6410-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brock M, et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104(10):1184–1191. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 11.Hong KH, et al. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118(7):722–730. doi: 10.1161/CIRCULATIONAHA.107.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.West J, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–1114. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 13.Long L, et al. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98(6):818–827. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 14.Morty RE, et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2007;27(5):1072–1078. doi: 10.1161/ATVBAHA.107.141200. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi H, et al. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;290(3):L450–L458. doi: 10.1152/ajplung.00206.2005. [DOI] [PubMed] [Google Scholar]
- 16.Reynolds AM, et al. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1182–L1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds AM, Holmes MD, Danilov SM, Reynolds PN. Targeted gene delivery of BMPR2 attenuates pulmonary hypertension. Eur Respir J. 2012;39(2):329–343. doi: 10.1183/09031936.00187310. [DOI] [PubMed] [Google Scholar]
- 18.Tamosiuniene R, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109(8):867–879. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alastalo TP, et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest. 2011;121(9):3735–3746. doi: 10.1172/JCI43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, et al. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ Res. 2008;102(10):1212–1221. doi: 10.1161/CIRCRESAHA.108.173567. [DOI] [PubMed] [Google Scholar]
- 21.Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13(8):410–418. doi: 10.1016/S0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 22.Berse M, Bounpheng M, Huang X, Christy B, Pollmann C, Dubiel W. Ubiquitin-dependent degradation of Id1 and Id3 is mediated by the COP9 signalosome. J Mol Biol. 2004;343(2):361–370. doi: 10.1016/j.jmb.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 23.Lee NY, Kirkbride KC, Sheu RD, Blobe GC. The transforming growth factor-beta type III receptor mediates distinct subcellular trafficking and downstream signaling of activin-like kinase (ALK)3 and ALK6 receptors. Mol Biol Cell. 2009;20(20):4362–4370. doi: 10.1091/mbc.E09-07-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, et al. Upregulation of ID protein by growth and differentiation factor 5 (GDF5) through a smad-dependent and MAPK-independent pathway in HUVSMC. J Mol Cell Cardiol. 2006;41(1):26–33. doi: 10.1016/j.yjmcc.2006.03.421. [DOI] [PubMed] [Google Scholar]
- 25.Zilberberg L, ten Dijke P, Sakai LY, Rifkin DB. A rapid and sensitive bioassay to measure bone morphogenetic protein activity. BMC Cell Biol. 2007;8:41. doi: 10.1186/1471-2121-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valdimarsdottir G, et al. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002;106(17):2263–2270. doi: 10.1161/01.CIR.0000033830.36431.46. [DOI] [PubMed] [Google Scholar]
- 27.Bonger KM, Chen LC, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat Chem Biol. 2011;7(8):531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majka S, et al. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir Res. 2011;12:84. doi: 10.1186/1465-9921-12-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paddenberg R, et al. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res. 2007;8:15. doi: 10.1186/1465-9921-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMurtry MS, Bonnet S, Michelakis ED, Haromy A, Archer SL. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamcyin-atorvastatin-simvastatin study. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L933–L940. doi: 10.1152/ajplung.00310.2006. [DOI] [PubMed] [Google Scholar]
- 31.Bonnet S, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104(27):11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Passiatore G, Gentilella A, Rom S, Pacifici M, Bergonzini V, Peruzzi F. Induction of Id-1 by FGF-2 involves activity of EGR-1 and sensitizes neuroblastoma cells to cell death. J Cell Physiol. 2011;226(7):1763–1770. doi: 10.1002/jcp.22505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donners MM, et al. Low-dose FK506 blocks collar-induced atherosclerotic plaque development and stabilizes plaques in ApoE–/– mice. . Am J Transplant. 2005;5(6):1204–1215. doi: 10.1111/j.1600-6143.2005.00821.x. [DOI] [PubMed] [Google Scholar]
- 34.Bai L, et al. Low- but not high-dose FK506 treatment confers atheroprotection due to alternative macrophage activation and unaffected cholesterol levels. Thromb Haemost. 2010;104(1):143–150. doi: 10.1160/TH09-07-0502. [DOI] [PubMed] [Google Scholar]
- 35.Wrana JL. Crossing Smads. Sci STKE. 2000;2000(23):re1. doi: 10.1126/stke.2000.23.re1. [DOI] [PubMed] [Google Scholar]
- 36.Kawabata M, Imamura T, Miyazono K. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998;9(1):49–61. doi: 10.1016/S1359-6101(97)00036-1. [DOI] [PubMed] [Google Scholar]
- 37.Chen YG, Liu F, Massague J. Mechanism of TGFβ receptor inhibition by FKBP12. EMBO J. 1997;16(13):3866–3876. doi: 10.1093/emboj/16.13.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGFβ receptor in complex with FKBP12. Cell. 1999;96(3):425–436. doi: 10.1016/S0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 39.Wang T, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell. 1996;86(3):435–444. doi: 10.1016/S0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 40.de Jesus Perez VA, et al. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184(1):83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez VADJ, et al. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184(1):83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller AA, Hislop AA, Vallance PJ, Haworth SG. Deletion of the eNOS gene has a greater impact on the pulmonary circulation of male than female mice. Am J Physiol Lung Cell Mol Physiol. 2005;289(2):L299–L306. doi: 10.1152/ajplung.00022.2005. [DOI] [PubMed] [Google Scholar]
- 43.Chandra SM, et al. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2011;31(4):814–820. doi: 10.1161/ATVBAHA.110.219980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansmann G, et al. An antiproliferative BMP-2/PPAR gamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118(5):1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 46.Abe K, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121(25):2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 47.Taraseviciene-Stewart L, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15(2):427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 48.Demura Y, Taraseviciene-Stewart L, Scerbavicius R, Tuder RM, Voelkel NF. N-acetylcysteine treatment protects against VEGF-receptor blockade-related emphysema. COPD. 2004;1(1):25–32. doi: 10.1081/COPD-120030412. [DOI] [PubMed] [Google Scholar]
- 49.Voelkel N, Taraseviciene-Stewart L. Emphysema: an autoimmune vascular disease? Proc Am Thorac Soc. 2005;2(1):23–25. doi: 10.1513/pats.200405-033MS. [DOI] [PubMed] [Google Scholar]
- 50.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals. 2008;16(4):318–325. doi: 10.1159/000123041. [DOI] [PubMed] [Google Scholar]
- 51.Wu X, et al. Creating diverse target-binding surfaces on FKBP12: synthesis and evaluation of a rapamycin analogue library. ACS Comb Sci. 2011;13(5):486–495. doi: 10.1021/co200057n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6(7):226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nasim MT, et al. BMPR-II deficiency elicits pro-proliferative anti-apoptotic responses through the activation of TGFbeta-TAK1-MAPK pathways in PAH. Hum Mol Genet. 2012;21(11):2548–2458. doi: 10.1093/hmg/dds073. [DOI] [PubMed] [Google Scholar]
- 54.Yang J, et al. Smad-dependent and smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ Res. 2010;107(2):252–262. doi: 10.1161/CIRCRESAHA.109.209940. [DOI] [PubMed] [Google Scholar]
- 55.Yang J, et al. Sildenafil potentiates bone morphogenetic protein signaling in pulmonary arterial smooth muscle cells and in experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2013;33(1):34–42. doi: 10.1161/ATVBAHA.112.300121. [DOI] [PubMed] [Google Scholar]
- 56.Rabinovitch M. Pathobiology of pulmonary hypertension. Annu Rev Pathol. 2007;2:369–99. doi: 10.1146/annurev.pathol.2.010506.092033. [DOI] [PubMed] [Google Scholar]
- 57.Chun HJ, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118(10):3343–3354. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 59.Mathea S, Li S, Schierhorn A, Jahreis G, Schiene-Fischer C. Suppression of EGFR autophosphorylation by FKBP12. Biochemistry. 2011;50(50):10844–10850. doi: 10.1021/bi2013855. [DOI] [PubMed] [Google Scholar]
- 60.Albinana V, Sanz-Rodriguez F, Recio-Poveda L, Bernabeu C, Botella LM. Immunosuppressor FK506 increases endoglin and activin receptor-like kinase 1 expression and modulates transforming growth factor-beta1 signaling in endothelial cells. Mol Pharmacol. 2011;79(5):833–843. doi: 10.1124/mol.110.067447. [DOI] [PubMed] [Google Scholar]
- 61.Chaikuad A, et al. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J Biol Chem. 2012;287(44):36990–36998. doi: 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masri FA, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L548–L554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 63.Lee DK, et al. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74(1):34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- 64.Sheikh AY, et al. In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol Heart Circ Physiol. 2008;294(1):H88–H98. doi: 10.1152/ajpheart.00935.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kasai A, et al. Apelin is a crucial factor for hypoxia-induced retinal angiogenesis. Arterioscler Thromb Vasc Biol. 2010;30(11):2182–2187. doi: 10.1161/ATVBAHA.110.209775. [DOI] [PubMed] [Google Scholar]
- 66.Kim J, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med. 2013;19(1):74–82. doi: 10.1038/nm.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takeda Y, Miyamori I, Furukawa K, Inaba S, Mabuchi H. Mechanisms of FK 506-induced hypertension in the rat. Hypertension. 1999;33(1):130–136. doi: 10.1161/01.HYP.33.1.130. [DOI] [PubMed] [Google Scholar]
- 68.Wu X, Chang MS, Mitsialis SA, Kourembanas S. Hypoxia regulates bone morphogenetic protein signaling through C-terminal-binding protein 1. Circ Res. 2006;99(3):240–247. doi: 10.1161/01.RES.0000237021.65103.24. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, et al. Transcription factor CHF1/Hey2 regulates EC coupling and heart failure in mice through regulation of FKBP12.6. Am J Physiol Heart Circ Physiol. 2012;302(9):H1860–H1870. doi: 10.1152/ajpheart.00702.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Owens P, et al. Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proc Natl Acad Sci U S A. 2012;109(8):2814–2819. doi: 10.1073/pnas.1101139108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Glassman SD, Howard J, Dimar J, Sweet A, Wilson G, Carreon L. Complications with recombinant human bone morphogenic protein-2 in posterolateral spine fusion: a consecutive series of 1037 cases. Spine (Phila Pa 1976). 2011;36(22):1849–1854. doi: 10.1097/BRS.0b013e3181d133d0. [DOI] [PubMed] [Google Scholar]
- 72.Nakao A, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16(17):5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spiekerkoetter E, et al. S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ Res. 2009;105(7):639–647. doi: 10.1161/CIRCRESAHA.109.205120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sinclair AM, et al. Distinct 5′ SCL enhancers direct transcription to developing brain, spinal cord, and endothelium: neural expression is mediated by GATA factor binding sites. Dev Biol. 1999;209(1):128–142. doi: 10.1006/dbio.1999.9236. [DOI] [PubMed] [Google Scholar]
- 75.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112(3):423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.