

Abstract
For most lysosomal storage diseases (LSDs) affecting the CNS, there is currently no cure. The BBB, which limits the bioavailability of drugs administered systemically, and the short half-life of lysosomal enzymes, hamper the development of effective therapies. Mucopolysaccharidosis type IIIA (MPS IIIA) is an autosomic recessive LSD caused by a deficiency in sulfamidase, a sulfatase involved in the stepwise degradation of glycosaminoglycan (GAG) heparan sulfate. Here, we demonstrate that intracerebrospinal fluid (intra-CSF) administration of serotype 9 adenoassociated viral vectors (AAV9s) encoding sulfamidase corrects both CNS and somatic pathology in MPS IIIA mice. Following vector administration, enzymatic activity increased throughout the brain and in serum, leading to whole body correction of GAG accumulation and lysosomal pathology, normalization of behavioral deficits, and prolonged survival. To test this strategy in a larger animal, we treated beagle dogs using intracisternal or intracerebroventricular delivery. Administration of sulfamidase-encoding AAV9 resulted in transgenic expression throughout the CNS and liver and increased sulfamidase activity in CSF. High-titer serum antibodies against AAV9 only partially blocked CSF-mediated gene transfer to the brains of dogs. Consistently, anti-AAV antibody titers were lower in CSF than in serum collected from healthy and MPS IIIA–affected children. These results support the clinical translation of this approach for the treatment of MPS IIIA and other LSDs with CNS involvement.
Introduction
Most lysosomal storage diseases (LSDs), particularly those affecting the CNS lack effective therapies beyond supportive treatment, resulting, in most cases, in the death of patients at an early age. The ultimate goal in the treatment of disorders that affect diffuse areas of the CNS is to achieve global distribution of the therapeutic agent, while minimizing the delivery-associated risks. The BBB, however, constitutes a major obstacle to attaining this goal, as it prevents efficient delivery to the CNS of most systemically dispensed molecules (1).
Mucopolysaccharidosis type IIIA (MPS IIIA), or Sanfilippo syndrome IIIA, is an autosomic recessive LSD caused by the deficiency of sulfamidase (SGSH), a sulfatase involved in the stepwise degradation of glycosaminoglycan (GAG) heparan sulfate (HS) (2). HS accumulation in the lysosomes of cells is associated with progressive, severe neurodegeneration as well as somatic organ pathology (2, 3). MPS IIIA patients are first diagnosed at 1 to 4 years of age, when delayed psychomotor development and behavioral problems become obvious to their parents. This is followed by a rapid, progressive loss of cognitive and motor skills (2, 3), with death occurring usually by the mid-to-late teenage years (2–4). There is no cure for MSP IIIA. A mouse model of the disease derived from a spontaneous mutation in the catalytic domain of sulfamidase (5) exists and closely resembles the human disease in terms of neurodegeneration, neuroinflammation, hepatosplenomegaly, and shortened lifespan (6–8).
As with most LSDs, the design of therapeutic strategies for MPS IIIA relies on the possibility of cross-correction, based on the fact that soluble lysosomal enzymes present in the extracellular compartment can be taken up by mannose-6-phospate receptor–mediated (M6PR-mediated) endocytosis into affected cells (1). To overcome the limitations imposed by the BBB and efficiently provide the brain with enzyme replacement therapy (ERT) — the main therapeutic option currently available for most LSDs — strategies have required the implantation of delivery devices that allow for repeated administration of the therapeutic proteins to the CNS (9). Direct delivery of ERT to the CNS is currently being tested in MPS IIIA patients (NCT01155778 and NCT01299727, clinicaltrials.gov), but the use of permanent intrathecal delivery devices is associated with substantial risks and shortcomings. Despite its limitations, haploidentical HSC transplantation is another potential therapeutic strategy for the treatment of LSDs (10, 11), although no clinical success has been reported for MPS IIIA yet. Lentiviral vector–mediated transduction of autologous CD34+ HSCs has shown therapeutic efficacy in adrenoleukodystrophy (12), and similar studies currently underway for other LSDs will further establish the safety and efficacy of this approach, which has recently proven successful in treating MPS IIIA mice (13).
In vivo gene therapy offers the possibility of a one-time treatment for MPS IIIA and other inherited diseases, with the prospect of lifelong beneficial effects (14). Adenoassociated virus (AAV) vector–mediated gene transfer, in particular, has shown promising results as an in vivo gene transfer tool, showing long-term production of therapeutic proteins in animal models and in humans (14–16). Extensive gene delivery to the CNS using AAV vectors has been achieved by multiple direct injections into the brain parenchyma (17, 18) or by delivery to the cerebrospinal fluid (CSF) (19–21). Direct delivery of AAV vectors to the brain parenchyma is being clinically explored in MPS IIIA. A phase I/II clinical study (NCT01474343, clinicaltrials.gov) was recently initiated in which AAVrh.10 vectors carrying the sulfamidase and sulfatase–modifying 1 (SUMF1) genes are administered to the brain through burr holes, a procedure previously established for other neurodegenerative disorders (22, 23). Limited vector diffusion from the point of injection required vector administration at several sites, making its delivery technically challenging and requiring the development of specific surgical procedures (24). Alternatively, AAV9, a serotype recently isolated from human tissue (25), has been shown to have broad tropism and to cross the BBB after systemic administration (26, 27), reverting the neurological phenotype of MPS IIIB (28) and MPS IIIA (29) mice. Terminal galactose has been identified as the primary receptor for AAV9 (30), and the glycan-binding avidity of these vectors determines their in vivo fate (31). Additionally, AAV9 vectors delivered into the bloodstream efficiently transduce several peripheral tissues, such as liver, heart, and skeletal muscle, demonstrating a broad distribution profile (27, 32).
Here, we tested the efficacy of AAV9 vectors directly administered to the CSF in reversing the MPS IIIA phenotype in affected mice. We hypothesized that the broad biodistribution profile of this vector serotype, together with CSF-mediated distribution of vectors, would favor widespread transduction of the CNS and correction of the disease. Intracisternal administration of AAV9 vectors carrying the sulfamidase transgene led to increased enzymatic activity throughout the brain and liver, resulting in the reversal of lysosomal pathology in the CNS and somatic tissues, the full correction of behavioral deficits, and a significant extension of lifespan. Similar widespread distribution of transgene expression was observed in a large animal model, in which the efficacy of transduction after CSF delivery was greatly impacted but not completely blocked by high-titer, preexisting anti-AAV antibodies in serum. Our results provide a strong rationale for the future clinical translation of AAV9 vector–mediated sulfamidase gene transfer to MPS IIIA patients by delivering the therapeutic vector through the CSF.
Results
Restoration of enzymatic activity and correction of GAG accumulation in the brain of MPS IIIA mice after delivery of AAV9 sulfamidase into the CSF.
Intracisternal delivery to mice of an AAV9 vector encoding GFP at a dose of 5 × 1010 vector genomes (vg) resulted in widespread brain transduction at higher levels compared with other AAV serotypes, as indicated by the fluorescence intensity throughout the brains of injected mice (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI66778DS1), with detection of vg in the CNS and liver, and low gene copy numbers in other somatic organs (Supplemental Figure 2).
To test the therapeutic efficacy of intra-CSF AAV9 administration, a dose-response study was initiated in which 2 doses (5 × 109 and 5 × 1010 vg per mouse) of an AAV9 vector encoding for murine sulfamidase under the control of the CAG ubiquitous promoter (AAV9-Sgsh) were delivered via intracisternal (i.c.) injection into 2-month-old MPS IIIA mice, an age at which CNS pathology is already established (7) and peripheral accumulation of GAGs is also evident (33). Animals were sacrificed after 4 months of follow up. At the highest dose tested, statistically significant increases in sulfamidase activity were observed in all brain regions analyzed, reaching 11% to 36% of normal values in males (Figure 1A) and 7% to 39% of normal values in females (Supplemental Figure 3A), with the highest levels of Sgsh transgene activity recorded in the most rostral parts of the encephalon (sections I–III, Figure 1A and Supplemental Figure 3A), distal to the point of AAV9-Sgsh administration. Transgene expression resulted in efficient correction of GAG accumulation in several parts of the brain of MPS IIIA mice of both genders to levels indistinguishable from those of WT littermates, with partial reductions achieved in the most caudal portions of the encephalon (sections IV and V, Figure 1B and Supplemental Figure 3B). A partial, but statistically significant, reduction in GAG accumulation was also observed in some, but not all, brain areas tested at the low dose of the AAV9-Sgsh vector (section I in Figure 1B and sections I and II in Supplemental Figure 3B), while no significant increase in sulfamidase transgene levels or decrease in GAG accumulation was observed in MPS IIIA mice injected with AAV9-null vector (noncoding AAV9 vector used as a negative control). Ten to 12 months after a single administration of AAV9-Sgsh vector, brain GAG content remained normal in the treated male and female MPS IIIA mice (Supplemental Figure 4, A and B). Moreover, GAG accumulation was completely corrected in brain sections IV and V, which had previously shown partial reductions when evaluated 4 months after treatment (Figure 1B and Supplemental Figure 3B), suggesting that sustained sulfamidase production over a prolonged period of time is necessary to achieve full correction. In agreement with the correction in GAG storage obtained at the high vector dose, lysosomal integral membrane protein 2 (LIMP2) staining of lysosomes demonstrated a statistically significant reduction in lysosomal signal intensity in all brain regions analyzed (Figure 2). A series of colocalization studies with double immunostaining for LIMP2 and specific markers for neurons (neuronal nuclei, NeuN), astrocytes (glial fibrilar acidic protein, GFAP), and microglia (MAC2 antigen) (Supplemental Figure 5) showed that 73% of LIMP2+ cells in untreated MPS IIIA mice were neurons. Ultrastructural analysis of the frontal cortex by transmission electron microscopy confirmed the reduction in lysosomal pathology, which was very evident in perineuronal glial cells that appeared distended and full of large storage vesicles in null-treated MPS IIIA males and females, while they had a normal appearance in animals treated with the high dose of AAV9-Sgsh (Figure 3 and Supplemental Figure 3C).
Figure 1. Correction of CNS storage pathology after i.c. delivery of AAV9-Sgsh to male MPS IIIA mice.

MPS IIIA male mice were treated with either 5 × 109 (low dose, LD) or 5 × 1010 (high dose, HD) vg of AAV9 vector encoding for murine sulfamidase (Sgsh) under the control of a ubiquitous promoter (CAG) or with 5 × 1010 vg of a noncoding (MPS IIIA–null) vector as a control. Mice were treated at 2 months of age and analyzed 4 months later against age-matched WT and untreated MPS IIIA mice. (A) Percentage of WT sulfamidase activity and (B) GAG content in 5 brain regions analyzed (sections I–V illustrated in the diagrams above the plot). Results are shown as the mean ± SEM; n = 4–6 animals per group. WT sulfamidase activity was set to 100%, which corresponds to 3.51 ± 0.06, 4.40 ± 0.54, 5.03 ± 0.17, 4.31 ± 0.23, and 4.87 ± 0.20 nmol/17 hours/mg of protein for sections I–V, respectively. *P < 0.05; **P < 0.01, and ***P < 0.001 versus MPS IIIA–null.
Figure 2. Normalization of CNS lysosomal compartment size after intra-CSF delivery of AAV9-Sgsh.

(A) LIMP2 immunostaining of lysosomes in different brain regions of WT, null, and HD-treated MPS IIIA male mice. Arrows indicate cells with neuronal morphology. Scale bars: 200 μm and 50 μm (insets). (B) Histograms depict the corresponding signal quantification in all treatment groups. Results are shown as the mean ± SEM. n = 4–5 animals per group. *P < 0.05 and ***P < 0.001 versus MPS IIIA–null.
Figure 3. Frontal cortex ultrastructure.
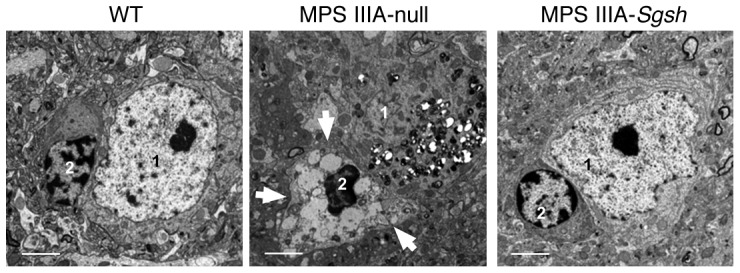
Transmission electron microscopy illustrating the reduction of lysosomal pathology in cells of the frontal cortex following treatment with HD AAV9-Sgsh. Correction was very evident in the perineuronal glial cells, in which large electrolucent vacuoles (arrows) were detected in null-injected MPS IIIA mice but not in healthy (WT) or AAV9-Sgsh–treated male mice. 1, neuron; 2, perineuronal glial cell. Scale bars: 2 μm.
Correction of neuroinflammation in MPS IIIA mice following AAV9-Sgsh gene delivery.
Secondary to the lysosomal pathology in neurons and glial cells, the brains of MPS IIIA mice are characterized by the presence of extensive neuroinflammation (8, 20), a common feature of MPS III in humans (34, 35). To evaluate the effect of AAV9-Sgsh gene transfer on brain inflammation, MPS IIIA mice injected with AAV9-Sgsh or AAV9-null vector were analyzed in parallel to WT mice 4 months after vector delivery. Immunohistochemical analysis of tissue sections from several brain areas was performed using specific stainings for astrocytes and microglia. Immunostaining for GFAP showed a statistically significant reversal of astrocytosis in the brain of the high dose–treated MPS IIIA mice (Figure 4A). Only partial improvement was observed, however, in mice receiving a low dose of vector expressing sulfamidase (data not shown). Similarly, staining with BSI-B4 lectin, which recognizes membrane-associated glycoconjugates containing terminal α-D-galactose residues present on resting and activated microglia but not on other glial cell types (36, 37), showed an absence of microglial infiltration in the brains of WT animals or MPS IIIA mice treated with a high dose of AAV9-Sgsh, but not in the brains of MPS IIIA mice treated with AAV9-null vector (Figure 4B). Also for BSI-B4 staining, partial correction of the disease phenotype was observed at a low dose of AAV9-Sgsh (data not shown).
Figure 4. Correction of neuroinflammation following AAV9-Sgsh treatment.

(A and B) Immunostaining for the astrocyte marker GFAP (A) and for the microglial marker BSI-B4 (B) in sections of frontal, parietal, and occipital cortex, superior colliculus, and thalamus from healthy (WT) male mice, and null- and HD-treated MPS IIIA male mice analyzed 4 months after gene delivery. Scale bars: 50 μm and 20 μm (insets). Histograms represent the signal intensity in each area for each treatment group. Results are shown as the mean ± SEM. n = 4–5 animals per group. **P < 0.01; and ***P < 0.001 versus MPS IIIA–null.
Correction of somatic pathology in affected animals after administration of AAV9-Sgsh via the CSF.
In agreement with the vector biodistribution results for AAV9 (Supplemental Figure 2), sulfamidase activity was detected in liver, reaching levels of approximately 190% of the levels observed in WT animals in the males (Figure 5A) and 20% of the levels observed in WT animals in the females (Supplemental Figure 6A). Sulfamidase activity in serum correlated well with the activity in the liver of males (Figure 5B) and females (Supplemental Figure 6B), suggesting that the liver was the main source of circulating enzymes. Gender differences in the efficiency of liver transduction by AAV vectors, including AAV9, are in agreement with previous reports (29, 33, 38).
Figure 5. Correction of MPS IIIA somatic pathology after CNS-directed gene therapy.

(A) Percentage of normal liver sulfamidase activity in male MPS IIIA mice 4 months after intra-CSF delivery of 5 × 109 (LD) or 5 × 1010 (HD) vg of AAV9-Sgsh or 5 × 1010 vg of a noncoding vector (MPS IIIA–null) as a control. WT sulfamidase activity was set to 100% and equaled 12.77 ± 0.85 nmol/17 hours/mg of protein. (B) Sulfamidase activity was monitored periodically in serum from all experimental groups during the 4-month follow-up period. Activity averaged 160% of that observed in WT mice (27.66 ± 1.47 nmol/17 hours/ml of serum) at the highest dose tested and was practically undetectable in untreated, null-treated, and LD-treated MPS IIIA animals. (C) Quantification of GAG content in liver, heart, lung, spleen, kidney, and urinary bladder in all experimental groups. Results are shown as the mean ± SEM; n = 5–8 animals per group. *P < 0.05 and ***P < 0.001 versus MPS IIIA–null. (D) LAMP1 immunostaining of lysosomes in liver, heart, lung, and urinary bladder demonstrating enlargement of the lysosomal compartment in affected animals (MPS IIIA–null) and a reduction in lysosomal pathology in animals treated with HD AAV9-Sgsh vector. Scale bars: 100 μm and 20 μm (insets). p.i., post injection.
We evaluated the effect of AAV9-Sgsh i.c. administration on somatic organ accumulation of GAGs. Following gene transfer, we observed a significant reduction in GAG accumulation in most organs of both male and female mice (Figure 5C and Supplemental Figure 6C). At the highest dose tested, MPS IIIA male mice showed at least an 85% decrease in GAG content in all tissues analyzed, with complete correction (100% reduction) achieved in most cases (Figure 5C). In female mice, a decrease in GAGs greater than 70% was observed in all tissues except for kidney (Supplemental Figure 6C). We confirmed a correction of GAG accumulation in most somatic organs in animals of both genders sacrificed 10–12 months after administration of a single i.c. dose of AAV9-Sgsh (Supplemental Figure 7, A and B). The correction of GAG accumulation resulted in a reduction in the size of the lysosomal compartment in both genders as soon as 4 months after treatment (Figure 5D and Supplemental Figure 6D). These results suggest that, although lower than in males, serum levels of sulfamidase activity achieved in females animals treated with the high dose of vector were sufficient to correct somatic disease. At the low dose of AAV9-Sgsh, we observed a statistically significant partial reduction in GAG accumulation in most somatic organs tested (Figure 5C and Supplemental Figure 6C). As expected from the results in the high vector dose cohort, we found a lesser extent of disease correction at the low dose in females than in males.
Functional correction of MPS IIIA disease by CSF administration of AAV9-Sgsh.
We assessed the impact of intra-CSF AAV9-Sgsh vector administration on behavior with the open field test, which evaluates the general locomotor and exploratory activity of mice in unknown surroundings. AAV9-Sgsh–treated MPS IIIA mice and controls were evaluated 4 months after gene delivery together with age-matched WT mice. Untreated and AAV9-null–treated MPS IIIA mice displayed reduced exploratory activity compared with healthy mice in terms of total distance traveled, number of lines crossed, and number of rearings on their hind legs (Figure 6, A–C). Intra-CSF delivery of 5 × 1010 vg of AAV9-Sgsh to MPS IIIA mice completely corrected the behavioral deficits, while no therapeutic effect was observed at the low vector dose, suggesting that improvement of CNS function requires considerable correction of lysosomal pathology. Furthermore, MPS IIIA animals receiving the high vector dose lived significantly longer than untreated MPS IIIA mice. Treated males showed a median survival of 24 months, compared with a 16.2-month median survival for MPS IIIA untreated males; P = 0.0132 (Figure 6D). Similarly, untreated MPS IIIA female mice had a median survival of 12.3 months versus 20-month survival for MPS IIIA females treated with AAV9-Sgsh vector (P = 0.0085) (Supplemental Figure 8). Median survival for healthy male and female littermates was 26.4 and 27 months, respectively. The normalization of behavioral responses and the greater survival of treated MPS IIIA mice further demonstrate the therapeutic efficacy of intra-CSF AAV9-Sgsh delivery and support the translation of this approach to the clinic.
Figure 6. Correction of behavioral deficits and prolonged survival following intra-CSF AAV9-Sgsh delivery.

Naive-tested WT, untreated, and treated MPS IIIA male mice were evaluated by the open field test 4 months after i.c. delivery of 5 × 109 (LD) or 5 × 1010 (HD) vg of AAV9-Sgsh or 5 × 1010 vg of AAV9-null vectors. Data correspond to the locomotor and exploratory activity recorded during the first 3 minutes and are represented as the mean ± SEM of 10 to 15 animals per group. (A) Total number of lines crossed, (B) total distance traveled, and (C) total number of rearings on the hind limbs. *P < 0.05, **P < 0.01, and ***P < 0.001 versus MPS IIIA–null. (D) Kaplan-Meier survival analysis in WT (n = 23), MPS IIIA– (n = 12), and HD-treated (n = 8) MPS IIIA male mice. P = 0.013 for untreated versus AAV9-treated MPS IIIA mice.
Widespread vector biodistribution following CSF delivery of AAV9 vectors in a large animal model.
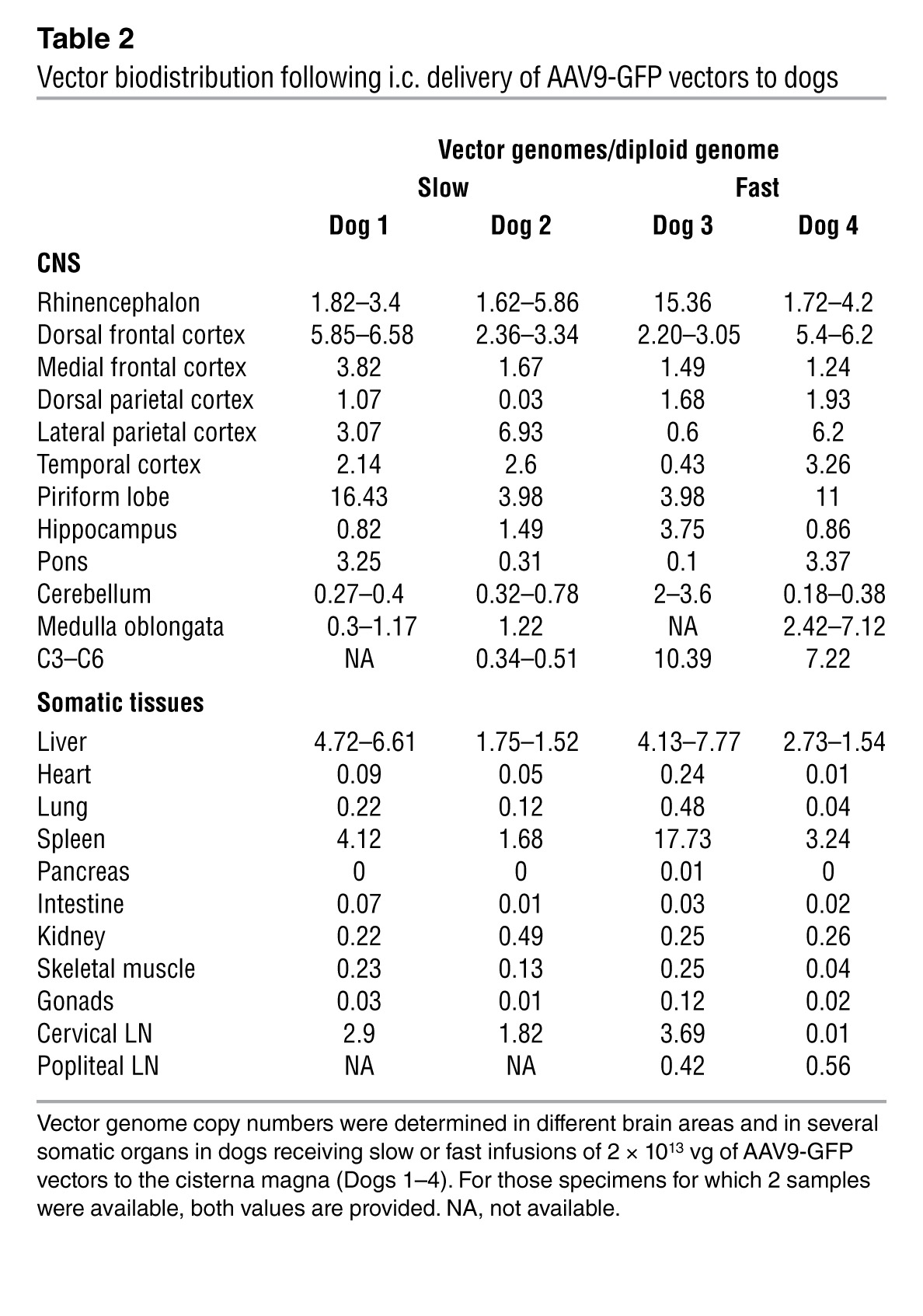
The validation in large animal species of approaches proven successful in rodents is instrumental for moving gene therapies forward into humans. To evaluate vector biodistribution in an animal species with a clinically relevant brain size, we delivered 2 × 1013 vg of AAV9-GFP into the CSF of healthy beagle dogs via cisterna magna injection. Vector was delivered in a total injected volume of 1 ml; Dogs 1 and 2 received the vector through an infusion pump set at 100 μl per minute over a period of 10 minutes and Dogs 3 and 4 as a bolus (injection in under 15 seconds) (Table 1). Intracisternal vector delivery was well tolerated, without alterations in hematological or biochemical parameters (data not shown). Total white cell counts and protein content in CSF samples also remained within normal reference values (Supplemental Table 1). GFP immunodetection in brain sections demonstrated expression of the transgene in widespread areas of the CNS (Figure 7A), which was confirmed by detection of vector genomes in all brain areas analyzed in the 4 dogs (Table 2). Vector transduction was detected in regions close to the cisterna magna, such as the medulla oblongata and the pons, but also in areas distant to the injection site, such as the frontal cortex (Figure 7A and Table 2). In several brain regions, the number of GFP+ cells was greater in areas closer to the brain surface, showing a diffuse horizontal pattern of distribution with respect to the leptomeninges, an observation consistent with CSF-mediated diffusion of vectors from the surface toward deeper areas (Figure 7A). A multifocal pattern of GFP+ cells was also observed in regions of the cerebral cortices, with groups of transduced cells occupying the whole gray matter and reaching the deepest laminae (see occipital and frontal cortices in Figure 7A), and their positive axons were observed in the adjacent white matter (not shown). The morphology of most transduced cells was typical of neuronal cells (arrows, Figure 7A). Immunostaining with specific cell markers confirmed the majority of transduced cells as being neurons, with transgene expression in scattered astrocytes and no detectable transduction of microglia (Supplemental Figure 9). No differences were observed in transduction efficiency (Figure 7B) or vector distribution (Table 2) in relation to the infusion rate. As observed in mice, administration of AAV9 into the CSF resulted in the detection of vector genomes in the liver (Table 2) and transduction of 3% to 5% of hepatocytes (Figure 7C) and low gene copy numbers in other organs except for the spleen (Table 2).
Table 1.
Summary of the experimental treatment and time of follow up for each dog
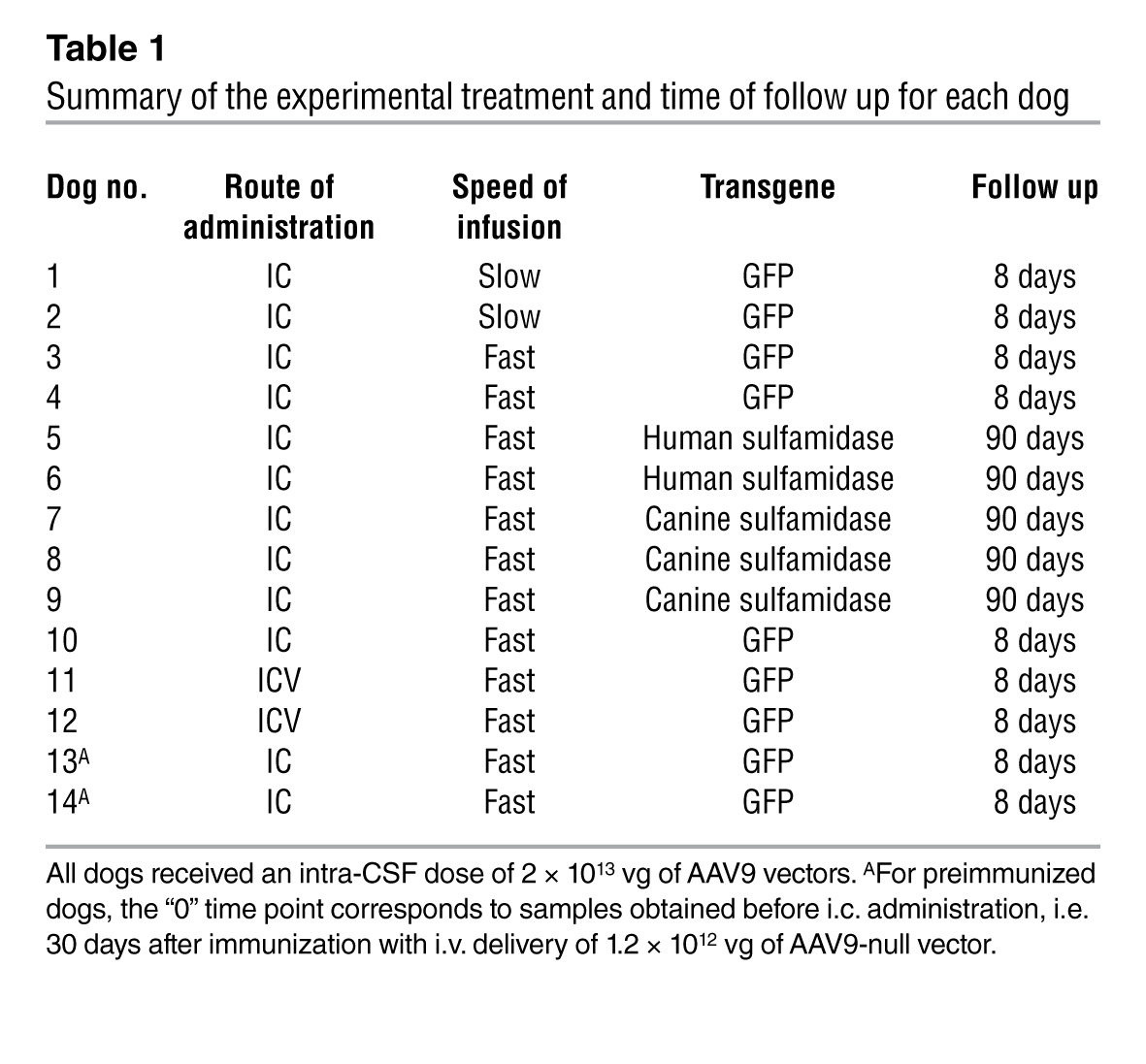
Figure 7. Widespread CNS and liver transduction after i.c. delivery of AAV9 vectors to dogs.

Healthy adult beagle dogs (Dogs 1–4) received a dose of 2 × 1013 vg of AAV9 vectors encoding for the GFP marker, and transgene expression was analyzed by immunohistochemistry 8 days after administration. (A) Widespread CNS transgene expression in Dog 1 following i.c. delivery of AAV9 vectors. Sections correspond to regions scattered throughout the CNS (see diagram). FC, frontal cortex; PC, parietal cortex; OC, occipital cortex; Cer, cerebellum; Hc, hippocampus; Rin, rhinencephalon; Ht, hypothalamus; P, pons; MO, medulla oblongata; SC, spinal cord. (B) Comparison of the effect of the i.c. infusion rate on vector distribution. Vectors were delivered using an infusion pump over the course of 10 minutes (Dogs 1 and 2, slow) or in a bolus (Dogs 3 and 4, fast), and transduction in different CNS areas was compared through quantification of signal intensity in 2 images per area per dog in brain sections immunostained for GFP. Color code corresponds to μm2 of positive area. (C) Liver transduction after i.c. delivery of AAV9 vectors. As a consequence of vector leakage into the circulation, 3%–5% of hepatocytes expressed GFP. Scale bars: 500 μm for FC, PC, OC, Rin, Ht, P, MO, SC, and liver (100 μm for insets), and 100 μm for Cer and Hc.
Table 2.
Vector biodistribution following i.c. delivery of AAV9-GFP vectors to dogs
Increased sulfamidase activity in the CSF of dogs following delivery of sulfamidase-encoding AAV9 vectors.
A therapeutic option for MPS IIIA currently under clinical testing consists of the direct delivery of recombinant sulfamidase to the CSF for its distribution throughout the CNS (NCT01155778 and NCT01299727, clinicaltrials.gov). The approach demands the implantation of a permanent intrathecal delivery device to allow for repeated administrations of the enzyme, a requirement imposed by the short half-life of sulfamidase (39). In contrast, our approach aims at providing a continuous endogenous source of sulfamidase upon a single administration. We delivered an AAV9 vector encoding for human sulfamidase (AAV9-SGSH) to the cisterna magna of healthy beagle dogs (Dogs 5 and 6), scaling up the therapeutic dose used in mice to 2 × 1013 vg, based on the body weight of mice compared with dogs (Table 1). Sulfamidase activity measured in the CSF rose above baseline levels 1 week after administration, peaking around 2 to 3 weeks and decreasing thereafter (Figure 8A). The increase in sulfamidase activity observed following AAV9-SGSH vector delivery coincided with the detection of mononuclear cells, predominantly lymphocytic, in the CSF (Figure 8A and Supplemental Table 1), a phenomenon previously documented in dogs when nonspecies-specific transgenes were used (18). The number of inflammatory cells progressively dropped concomitantly with the loss of sulfamidase activity (Figure 8A). Despite the inflammatory reaction, treated dogs remained clinically well and, in agreement with the fact that 3 months after delivery CSF sulfamidase activity remained above baseline levels in both animals (Figure 8A), the expression of human sulfamidase was detectable throughout different areas of the CNS, as well as in the peripheral nervous system (PNS) and somatic tissues (Supplemental Figure 10A).
Figure 8. Detection of sulfamidase activity in the CSF following intra-CSF delivery of AAV9 vectors.

(A and B) Follow up of sulfamidase activity in the CSF of healthy adult beagle dogs following i.c. administration of 2 × 1013 vg of AAV9 vectors encoding for human (Dogs 5 and 6) (A) or canine (Dogs 7–9) (B) transgenes. Sulfamidase activity (filled symbols) and white cell counts (open symbols) are depicted in the same graph. Basal CSF sulfamidase activity was quantified in CSF samples from 10 untreated, healthy beagle dogs and fell below detection limits. ACT, activity.
Next, we administered 2 × 1013 vg of an AAV9 vector encoding for canine sulfamidase (AAV9-SGSH) to the cisterna magna of 3 additional dogs (dogs 7–9, Table 1). A clear increase in sulfamidase activity was detectable in the CSF of all 3 dogs as soon as 1 week after vector administration (Figure 8B). Similarly to the kinetics of transgene expression observed in dogs injected with the human transgene, activity peaked 2–4 weeks after vector delivery, but contrary to what was observed in animals receiving vectors expressing human sulfamidase, transgene expression was maintained at high levels throughout the 3-month follow-up (observation ongoing). Moreover, white blood cells counts and CSF protein levels remained within normal range at all times (Supplemental Table 1), excluding the presence of an inflammatory process in the CNS of any of the dogs. Clearly, these results demonstrate that the loss of transgene expression observed in Dogs 5 and 6 was due to the use of a nonspecies-specific transgene and that achieving long-term, stable levels of sulfamidase in the CSF of dogs is feasible.
Comparison of i.c. and i.c.v. CSF delivery of AAV9 vectors.
The cisterna magna is easily accessible in mice and dogs, and cisternal puncture is a routine procedure in veterinary medicine. Intracisternal administration is not, however, standard in clinical practice in pediatric patients. We therefore performed a comparative study in which AAV9-GFP vectors were delivered into the CSF by either i.c. injection (Dog 10) or unilateral i.c.v. administration (Dogs 11 and 12) (Table 1) by a technique commonly used in pediatric neurosurgery (40). For i.c.v. vector delivery, a catheter was placed in the largest lateral ventricle, and 1 ml of vector solution was delivered within a few seconds (Figure 9A). The procedure was well tolerated in both animals dosed through this technique, and total white cell counts and protein content in CSF samples remained within normal range (Supplemental Table 1). In i.c.v.-injected dogs, the choroid plexus epithelium and ventricle lining ependimocytes were efficiently transduced in the injected hemisphere (Figure 9B), but positivity for GFP was also observed in the contralateral ventricle (data not shown). In both i.c. and i.c.v.-injected dogs, leptomeningeal cells showed strong immunoreactivity against GFP (Figure 7A and Figure 9B), with an estimated 75%–100% of leptomeningeal cells transduced in the cerebellar and caudal cerebral areas, decreasing slightly toward the most rostral areas of the brain. In the cerebral cortex, positive cells showed the same diffuse horizontal and multifocal patterns of distribution observed after i.c. delivery and spanned through all cortical layers (Figure 9B). Side-by-side analysis of vector distribution and transgene expression in the CNS and somatic organs were not suggestive of major differences in the vector transduction profile with either route of delivery (Figure 10). Efficient transduction of the trigeminal and dorsal root ganglia of the PNS was also observed (Figure 10B). Together, these results show that it is possible to achieve widespread AAV9 vector distribution and transgene expression following i.c.v. delivery to the CNS (Supplemental Figure 11) and PNS of large animals using a clinically approved surgical procedure, thus making the i.c.v. route suitable for the clinical translation of our approach. These results also show that the efficiency of CNS transduction after i.c.v. and i.c. vector delivery in a large animal model is comparable.
Figure 9. Intracerebroventricular administration of AAV9 vectors to dogs.

Healthy adult beagle dogs (Dogs 11 and 12) received a unilateral i.c.v. injection of 2 × 1013 vg of AAV9-GFP vectors. (A) Representative software screenplay depicting the neuronavigator-guided trajectory used by the neurosurgeon to reach the lateral ventricle from the surface of the skull. (B) Transgene expression in Dog 12 was analyzed by immunohistochemistry 8 days after administration. Expression was detected throughout the CNS (see diagram) and PNS (indicated by transduction of dorsal root ganglia) following i.c.v. delivery of AAV9 vectors to the CSF. LV, lateral ventricle; IVv, fourth ventricle; TC, temporal cortex; PL, piriform lobe; DRG, dorsal root ganglia. Scale bars: 250 μm.
Figure 10. Comparison of i.c. and i.c.v. routes of AAV9 delivery.

(A and B) Vector gene copy number and GFP mRNA expression were compared in 3 dogs following CSF delivery of 2 × 1013 vg of AAV9-GFP vectors through either an i.c. (Dog 10) or i.c.v. (Dogs 11 and 12) route. Analysis of samples representative of the whole CNS and PNS (A) and somatic tissues (B). md, medial; dr, dorsal; lt, lateral; vn, ventral; cn, central; R, right; L, left; cv-an, cervical anterior; cv-cd, cervicocaudal; I, intumescence; cv-th, cervicothoracic; th-an, thoracic anterior; th-md, thoracic medial; th-cd, thoracic caudal; ls, lumbosacral; ceq, cauda equina; drg, dorsal root ganglia; lm, lumbar; th, thoracic; cv, cervical. vg/dg, vector genome/diploid genome; IC, intracisternal; ICV, intracerebroventricular.
Asymmetrical distribution of AAV-neutralizing antibodies across the BBB.
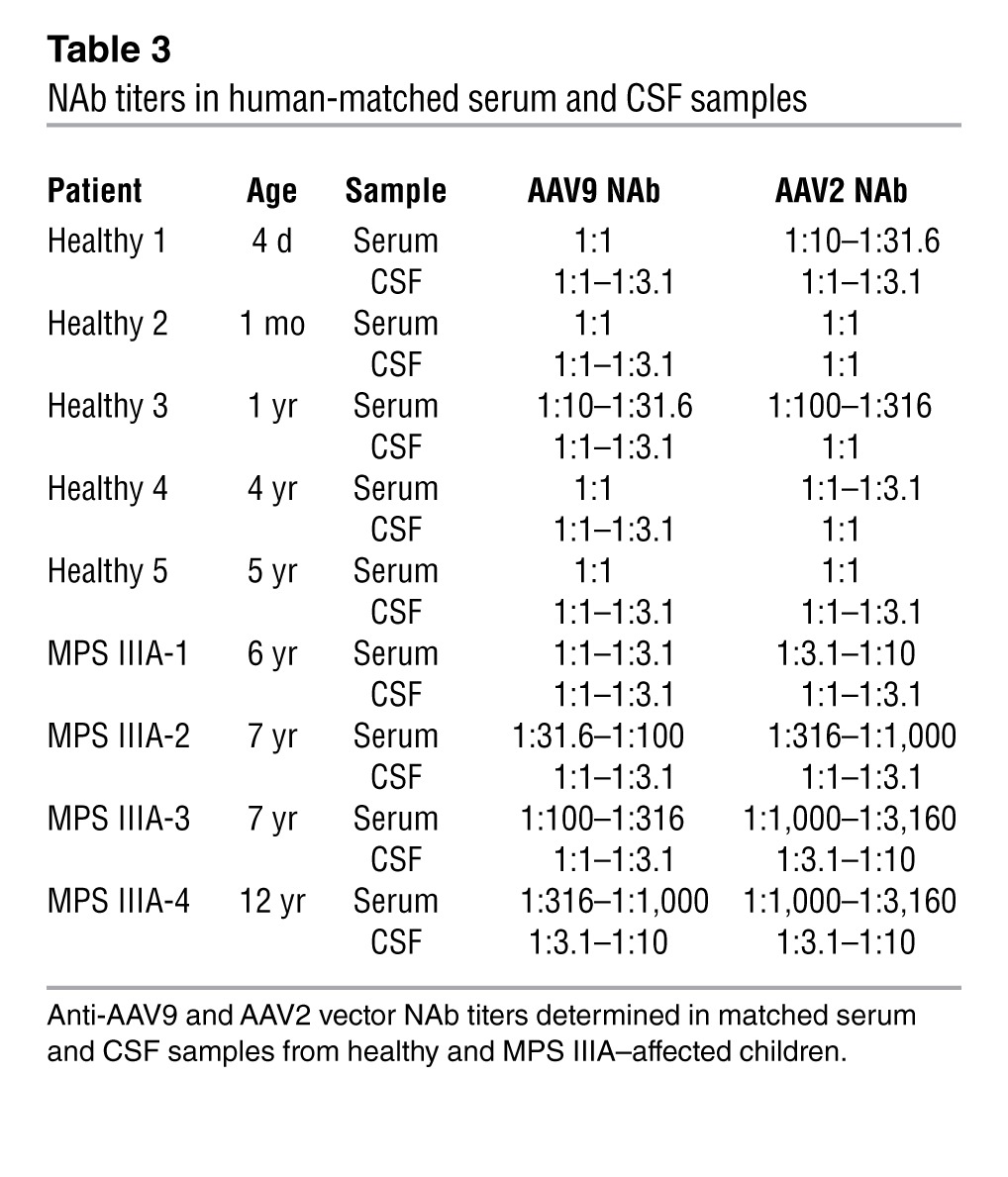
Serum- neutralizing antibodies (NAbs) against AAVs are highly prevalent in humans (41). NAbs can profoundly impact the efficiency of vector transduction after systemic administration (42–44), and several approaches are currently under investigation to overcome this limitation (45, 46). Due to the presence of the BBB, CSF concentrations of immunoglobulins are lower than those in plasma. There is evidence, however, suggesting that the function of the BBB is altered in some LSDs (47), although no data are available on MPS IIIA individuals. To evaluate the prevalence of NAbs against AAV in the CSF of healthy subjects and MPS IIIA–affected individuals, matched serum and CSF samples were collected from subjects of pediatric age (reflecting the age for a therapeutic intervention) and were tested using an in vitro assay previously described (42). Anti-AAV2 and anti-AAV9 NAbs were present in serum at a wide range of titers (Table 3), with anti-AAV2 titers being higher than anti-AAV9 titers, which is consistent with previous studies on the prevalence of antibodies reacting against these AAV serotypes (41). In CSF samples, NAbs were also detected, but titers were consistently lower than those of matched serum samples. Similarly, in MPS-IIIA–affected subjects, NAbs in serum were higher than in the CSF, suggesting that the disease did not alter BBB permeability for circulating immunoglobulins. With the limitation of the small number of matched serum-CSF pairs available, NAb titers seemed to increase with age (48, 49).
Table 3.
NAb titers in human-matched serum and CSF samples
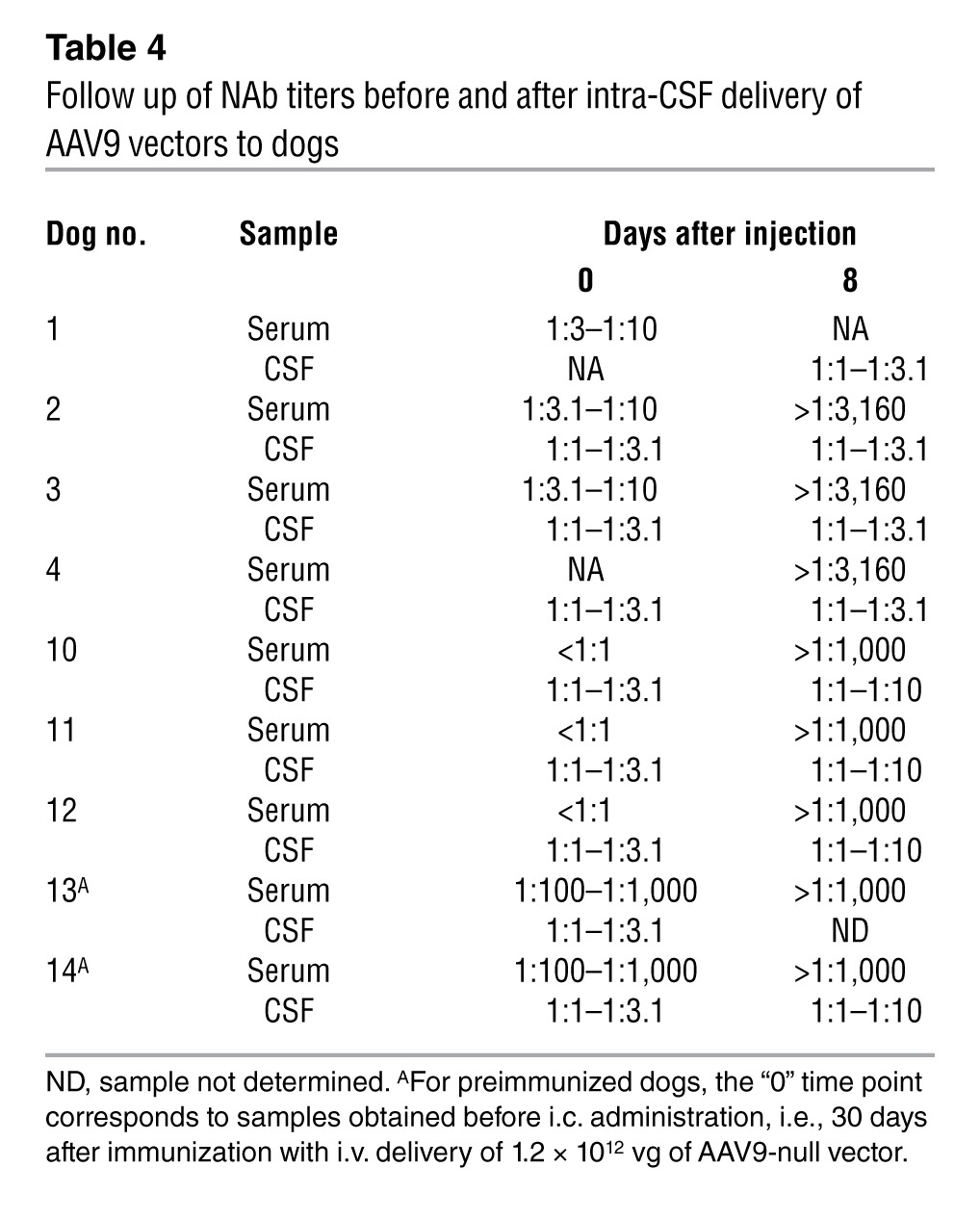
In healthy dogs, the administration of AAV9 vectors into the CSF resulted in robust systemic humoral responses against the AAV9 capsid (Table 4). All dogs presented undetectable levels of anti-AAV9 NAbs in serum and CSF at baseline. As expected, serum titers rose quickly after exposure to vector, and 8 days after administration, all animals had NAb titers greater than 1:1,000. However, in the absence of inflammation, matched CSF samples uncontaminated with blood were negative or had titers less than 1:10, confirming the asymmetrical distribution of NAbs across the BBB. In Dogs 5 and 6, which had signs of inflammation of the CNS, NAb titers in the CSF reached 1:100–1:1,000 (Supplemental Figure 10B).
Table 4.
Follow up of NAb titers before and after intra-CSF delivery of AAV9 vectors to dogs
Impact of humoral immunity on the efficiency of CNS transduction through CSF vector delivery.
Finally, we investigated to what extent the presence of preexisting immunity against AAV influenced the efficiency of intra-CSF AAV9-mediated gene transfer to the CNS. Two beagle dogs (Dogs 13 and 14) were immunized against AAV9 by i.v. injection of 1.2 × 1012 vg of AAV9-null vector. Thirty days following immunization, the animals received 2 × 1013 vg of AAV9-GFP via i.c. delivery (Table 1). The animals were sacrificed 8 days after vector delivery, and vector transduction efficiency was evaluated. Following immunization, serum anti-AAV9 NAb titers rose to greater than 1:1,000 in both dogs; in the CSF, NAb titers were considerably lower, remaining around 1:1 to 1:3.1 (Dogs 13 and 14, day 0; Table 4), an NAb titer sufficient to block vector transduction following systemic delivery (43, 44). Despite the high stringency of the model we used to assess the impact of NAbs on CNS transduction (i.e., serum NAb titers greater than 1:1,000) and given that only a small proportion of humans will present with very high anti-AAV9 antibody titers (41), GFP expression was detected throughout the CNS, particularly in the leptomeninges in both injected animals (Figure 11, A and B), though at what seemed to be slightly lower levels than in naive animals. Quantification of vector copy numbers and GFP mRNA showed vector genomes and transgenic expression in 70% to 95% of CNS samples collected from preimmunized dogs (Figure 11B). In addition, the PNS was also efficiently transduced in preimmunized animals following CSF delivery (Figure 11B), while the transduction of peripheral tissues was completely blocked (Supplemental Figure 12), as expected, due to the high NAb antibody titers in the serum of these animals (Table 4). These data suggest that humoral immunity against AAV may lower the efficacy of AAV gene transfer upon delivery of vector into the CSF, however, significant NAb titers in the CSF and a decrease in CNS transduction would only be observed when circulating NAb titers are very high.
Figure 11. Impact of preexisting humoral immunity on CNS transduction following CSF vector delivery.

(A) Immunodetection of GFP-expressing cells in the CNS following i.c. administration of 2 × 1013 vg of AAV9-GFP vectors to healthy adult beagle dogs with preimmunity against AAV9 (Dogs 13 and 14). FC-Lepto, frontal cortex leptomeninges; OC-Lepto, occipital cortex leptomeninges; OC, occipital cortex; IVv-CPl, fourth ventricle choroid plexus; IVv-Epen, fourth ventricle ependimocytes; Pin, pineal gland; SC-Ceq, spinal cord cauda equina. Scale bars: 200 μm and 100 μm (insets). (B) Vector genome copy number and GFP mRNA expression following intra-CSF delivery of 2 × 1013 vg of AAV9-GFP vectors to naive (Dog 10) or preimmunized (Dogs 13 and 14) dogs. Samples are representative of the whole CNS.
Discussion
Here, we report the efficacy of what we believe to be a novel gene therapy approach based on direct CSF administration of AAV9 sulfamidase for the treatment of MPS IIIA. The combination of this route of delivery, considerably less invasive than the direct injection of AAV vectors into the brain parenchyma, with the widespread transduction of AAV serotype 9 vectors mediates full histopathological and functional correction of not only central, but also somatic, pathology in MPS IIIA mice. A single administration of vector directly into the CSF of MPS IIIA mice resulted in widespread transduction of the CNS, with leakage of vector into the bloodstream and correction of somatic disease in both male and female animals. This translated into a marked improvement in brain function and survival of the animals that was indistinguishable from their healthy littermates. Our study represents what we believe to be the first report of the combined treatment of CNS and somatic disease in MPS IIIA with a single administration of vector to the CNS.
The use of AAV9 vectors delivered i.v. to target the brain has been previously described (26–28). AAV9 is among the few serotypes of AAV able to cross the BBB (26), making it an ideal tool for targeting the brain. However, the approach has several limitations. First, there is the need for high vector doses to achieve correction of CNS pathology (27, 28), doses that must be considerably higher (7.5–20–fold, depending on the study) than those tested here; second, AAV vectors delivered systemically may be neutralized by anti-AAV antibodies (42–44), which are highly prevalent in humans (41); finally, systemic exposure to high doses of AAV vectors may trigger the activation of CD8+ T cell responses directed against the viral capsid in a dose-dependent manner (16, 42, 50, 51). Direct delivery of AAV vectors into the CSF can potentially overcome these limitations, allowing for a lower therapeutic vector dose, limited exposure of vector to serum anti–AAV NAbs (vide infra), and potentially achieving therapeutic efficacy at vector doses below those associated with unwanted anticapsid cytotoxic T cell responses.
Different AAV vector serotypes have been tested after intra-CSF delivery in LSD animal models with variable outcomes. In most cases, widespread CNS vector distribution was reported (19–21), with the exception of i.c.v. administration of AAV4 that resulted in the exclusive transduction of ependyma (52). In our hands, however, AAV9 clearly outperformed all other AAV serotypes tested in transduction efficiency throughout the CNS. Most importantly, none of those studies reported a correction of peripheral disease after CNS-targeted delivery. The profile of AAV9 distribution observed in both species analyzed, with significant brain and liver transduction after CSF delivery, would result from the combination of CSF-mediated vector distribution throughout the CNS and the unique properties of AAV9, a vector with well-documented ability to cross vascular barriers (26, 27).
A noteworthy observation of our study was the existence of gender differences in response to the intra-CSF delivery of AAV9-Sgsh. While vector genome copy numbers, sulfamidase activity levels, and long-term disease correction (assessed 10–12 months after delivery) were very similar in the brains of treated MPS IIIA males and females, clear differences were observed in the periphery. Most strikingly, the levels of sulfamidase activity achieved in the liver and serum of females of the high-dose cohort were lower than those observed in males dosed with an equal amount of vector. We and others have previously reported gender differences in liver transduction by AAV vectors of serotypes 2, 5, 8, and 9 in mice and dogs (33, 38, 53), which are independent of the promoter or mouse strain used (29, 33, 38) and which do not affect the transduction of nonhepatic tissues (29, 38) and seem to be androgen driven (38). Despite the lower level of transduction achieved in treated MPS IIIA females receiving the high dose of vector, sulfamidase activity levels in serum averaged 20% of those found in WT mice during a 4-month follow-up period, and this was sufficient to achieve long-term correction of pathological accumulation of GAGs in most somatic organs and considerably prolonged survival. There seems to be a very low threshold of activity in LSDs above which lysosomal enzymes are capable of coping with substrate influx and avoiding accumulation. Preclinical studies of ERT or gene therapy approaches in other forms of MPS have demonstrated that low levels of circulating enzyme (as low as 5% of normal) can mediate the correction of GAG accumulation in most somatic tissues (54, 55). This low enzymatic activity requirement for disease correction is further supported by clinical evidence; a clear correlation between MPS IIIA clinical manifestations and the patients’ underlying mutations have recently been demonstrated in a large cohort of MPS IIIA–affected individuals (2). This report identified carriers of mutations associated with a certain degree of residual enzymatic activity (3) that showed a relatively mild disease phenotype, suggesting that low, constant levels of activity are enough to change the clinical course of the disease. Overall, our results emphasize the importance of including female cohorts when assessing the efficacy of AAV-based gene therapy approaches for inherited diseases that affect both genders.
In an effort to translate MPS IIIA results to the clinic, we assessed the feasibility and tolerability of our approach in a large animal model. We first demonstrated widespread CNS transduction after i.c. administration of AAV9-GFP vectors in naive, healthy dogs. Vector doses were scaled up from the doses used in mice and resulted in vector dissemination to the CNS, but also to the liver, confirming the results obtained in the murine model of MPS IIIA. We next used an i.c.-delivered AAV9 vector encoding for human sulfamidase, currently under clinical development. Following vector delivery, an increase in sulfamidase activity was measured in the CSF. In this experiment, transgenic expression was short lived due to the development of what appeared to be an immune response against the nonspecies-specific human transgene, which was not observed in mice when the species-specific transgene was used. This finding can be explained based on the differences in the amino acid sequence of the human versus canine sulfamidase protein (94% homology). Immune responses against the nonspecies-specific transgene were also observed in rhesus macaques undergoing hepatic gene transfer with AAV vectors encoding for human coagulation factor IX (FIX) (56–58) and in MPSI and IIIB dogs following CNS gene transfer with human transgenes (18). Importantly, at least in the context of hemophilia B, immune responses against the human transgene observed in nonhuman primates did not predict the outcome in humans, as no humoral or cellular immunity against the FIX transgene was documented in clinical trials, even in patients carrying null mutations in the endogenous FIX gene, who are generally at higher risk of mounting an immune responses against the WT protein (16, 42). It is worth mentioning that greater than 90% of the mutations causing MPS IIIA described so far are missense mutations and that recent studies on a large cohort of MPS IIIA patients showed that greater than 98% of subjects were carriers of at least 1 allele corresponding to a missense mutation and were therefore expected to have some degree of tolerance to sulfamidase (2). The confirmation that the loss of transgenic expression observed in Dogs 5 and 6 was due to the use of a nonspecies-specific transgene comes from the dogs that were administered vectors encoding for canine sulfamidase. In these animals, the initial kinetics of transgenic expression and the levels of sulfamidase activity in the CSF were similar to those observed in dogs injected with the human transgene. However, in dogs expressing the canine transgene, activity remained at high levels, and no increase in white blood cell counts or CSF protein levels was observed throughout the 3-month follow-up period, excluding the presence of CNS inflammation in these dogs. Hence, this study demonstrates that achieving long-term, stable levels of sulfamidase production following CSF delivery of AAV9 vectors is feasible, as shown here for what we believe to be the first time in a large animal. It also highlights the importance of carefully evaluating the choice of the use of a transgene in preclinical studies in order to avoid overestimating potential immunogenicity of the therapeutic transgene itself. Preclinical studies of intrathecal delivery of a recombinant enzyme (39, 59–61), now being translated to humans (NCT01155778 and NCT01299727, clinicaltrials.gov), confirm that an increase in sulfamidase levels in CSF can be effective in counteracting the pathological changes associated with MPS IIIA and further support the rationale of our study.
While we showed safety and efficacy in MPS IIIA mice and evidence of widespread transgenic expression in dogs using i.c. delivery of AAV9 vectors, the injection procedure used is uncommon in the clinic due to the risk of complications derived from the proximity of the cisterna magna to vital centers. We therefore focused on the i.c.v. route of administration, as it is a safer alternative for the delivery of AAV9-SGSH to the CSF in humans. Ventricular access to the CSF through the lateral ventricle, or ventriculostomy, is an important diagnostic and therapeutic tool for neurosurgeons, with more than 40,000 procedures performed per year in the United States (62). Using the i.c.v route for vector delivery, we achieved a very similar pattern of vector distribution throughout the CNS, the PNS, and other body organs and systems to that achieved in the 7 dogs previously dosed via the i.c. route and in which transgenic expression was measured. For both the i.c. and i.c.v. routes, efficient transduction was detected from the most rostral part of the encephalon (frontal cortex) to the end of the spinal cord (cauda equina), and the pattern suggested CSF-driven penetration of vector from the CNS surface to deeper areas.
An alternative route of CSF delivery is via lumbar puncture. Although this is a common clinical procedure, limited distribution of products in widespread CNS areas is achieved through this route. Preclinical data in pigs show that AAV9-derived gene expression following local lumbar intrathecal administration remains restricted to the areas of injection and that widespread spinal cord transduction requires several administrations of vector at the cervical, thoracic, and lumbar regions, suggesting that vector penetration occurs mostly in the vicinity of the catheter tip used for delivery (63). Global CNS gene delivery following intrathecal AAV administration has been documented in cynomolgus macaques, a nonhuman primate species of small size (~3–7 kg), using self-complementary AAV vectors diluted in a hyperosmotic buffer (64); while the study provides evidence that the approach is potentially feasible, detailed confirmatory studies in larger animal models are needed. In humans, a comparison of aminoglycoside therapies after CSF administration indicates that lumbar delivery results in high aminoglycoside concentration in lumbar CSF, but rather low concentrations in ventricular CSF, whereas instillation in the lateral ventricles results in high ventricular and lumbar CSF concentrations (65). The results of these studies confirm the concept of unidirectional flow of CSF and suggest that i.c.v. administration of products may result in the most widespread distribution of drugs within CSF-containing compartments (63, 65).
Finally, our study also examined the prevalence of anti-AAV NAbs in the serum and CSF of humans and their possible impact on vector-mediated CNS transduction following delivery to the CSF. To the best of our knowledge, we showed, for the first time, that MPS IIIA subjects carry anti-AAV antibodies in serum and CSF at levels similar to those in healthy individuals, indicating that despite the pathological accumulation of GAGs, the BBB is still intact, a result consistent with the lack of CNS correction through systemic ERT in this disease. Importantly, NAb titers were consistently lower in the CSF than in serum, both in humans and in dogs immunized through i.v. AAV vector administration. Despite being detectable, NAb titers in the CSF did not completely block AAV vector transduction in the brain, a result in contrast to previously published results (66) and in agreement with a recent study that explored CNS transduction following intra-CSF delivery in nonhuman primates with low serum NAb titers (64). Altogether, our study demonstrates that CNS gene transfer is possible in the presence of preexisting anticapsid antibodies. This observation, in conjunction with the low prevalence of NAbs against AAV in healthy and MPS IIIA–affected children, suggest that most patients would be eligible for enrollment in AAV vector gene transfer trials targeting the CNS via i.c.v. vector delivery. Conversely, as demonstrated here, NAbs will likely block peripheral organ transduction, possibly requiring ERT in combination with gene transfer to achieve whole body correction of MPS IIIA.
One final question remains regarding whether brain function can be restored simply by providing the missing enzyme, taking into account that for most LSDs, clinical signs appear once some degree of tissue damage has already occurred. We and others have determined that by 2 months of age, MPS IIIA mice have established pathology in brain and somatic organs, with significant accumulation of GAGs and lysosomal distention (7, 33). In MPS IIIA mice 6 to 18 weeks of age, repeated intra-CSF ERT achieved partial, but not complete, reversion of the disease phenotype, which was attributed to the late initiation of treatment (61). In our study, 4 months after i.c. administration of AAV9-Sgsh vectors to 2-month-old mice, i.e. mice at 6 months of age, the animals showed normal CNS GAG content and no signs of lysosomal distention or neuroinflammation, suggesting that constant local production of the enzyme might be more efficient in reversing established disease. Moreover, at 12 to 14 months of age, i.e., an age at which untreated MPS IIIA mice are dying, brain GAG content remained normal in treated MPS IIIA male and female mice that received a single administration of vector 10–12 months beforehand. The observation, in both genders, of complete GAG normalization in caudal sections of the brain that had shown partial reductions when evaluated 4 months after treatment, suggests that these areas require sustained sulfamidase production over a prolonged period of time to be fully corrected, a result never described in prior studies and that has important implications for the design of future clinical trials for both gene transfer and ERT approaches to treating MPS IIIA and other lysosomal storage disorders. Normalization of GAG content was associated with normal behavior and lifespan, indicative of full phenotypic reversal, while untreated MPS IIIA mice showed behavioral deficits as soon as 10 weeks of age (67).
In summary, intra-CSF delivery of AAV9 vectors led to transgenic expression in widespread areas of the CNS, PNS, and liver, with reversal of central and somatic pathology in MPS IIIA mice leading to full correction of behavioral deficits and substantially extended lifespan. Long-term expression of the therapeutic gene is fundamental to achieving full correction of the pathological accumulation of GAGs. These results are fully translatable to a large animal model, showing long-term expression of the species-specific transgene and similar transgene biodistribution to mice when vector was delivered using a procedure commonly used in pediatric neurosurgery. Our results provide strong evidence supporting the clinical translation of this approach to treat MPS IIIA and other LSDs affecting the CNS.
Methods
Animals.
Congenic C57Bl/6 mice carrying an inactivating amino acid change (D31N) in the sulfamidase gene were used (5). Affected MPS IIIA and healthy littermates were bred from heterozygous founders (The Jackson Laboratory). Genotyping was performed as previously described (5). Mice were fed a standard diet ad libitum (Harlan Teklad) and were maintained under a 12-hour light-dark cycle. Healthy male beagle dogs were purchased from Isoquimen or Marshall BioResources. Animals were fed individually once daily at 9:00 am with 30 g/kg of body weight of standard dry food (Elite Nutrition; Nestle).
Recombinant AAV vectors.
AAV expression cassettes were generated by cloning the cDNA of murine sulfamidase (clone ID: D330015N16; RIKEN), optimized human sulfamidase (GeneArt; Life Technologies), or GFP under the control of the ubiquitous CAG promoter (hybrid of the chicken β-actin promoter and CMV enhancer) into single-stranded AAV backbone plasmids. The GFP constructs also contained the WPRE element to enhance expression. AAV vectors were produced by helper virus-free transfection of HEK293 cells and purified using an optimized cesium chloride gradient–based purification protocol that renders vector preps of high purity and devoid of empty capsids (68).
Vector administration.
For i.c. delivery in mice, animals were anesthetized by i.p. injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), and the skin of the posterior part of the head, from behind the ears to approximately between the scapulae, was shaved. Mice were held in a prone position, with the head at a slightly downward inclination. A 2-mm rostro-caudal incision was made to introduce a Hamilton syringe at an angle of 45° to 55° into the cisterna magna, between the occiput and the C1 vertebra, and 5 μl of vector dilution was injected. Given that the CNS was the main target compartment for vector delivery, the mice were dosed with the same number of vector genomes per mouse, irrespective of gender or body weight. In dogs, anesthesia started with premedication with 0.02 mg/kg of i.v. buprenorphine hydrochloride (Buprex; Schering-Plough). Thirty minutes later, induction was performed with i.v. administration of 4 mg/kg of propofol (PropoVet; B. Braun Medical) and 0.5 mg/kg of diazepam. After endotracheal intubation, anesthesia was maintained by inhalation of 2% isoflurane (IsoVet; B. Braun) in 100% oxygen. With the dog positioned in lateral recumbency, a 22-gauge needle was introduced into the cisterna magna between the occipital bone and the C1 vertebra. A syringe connected to a fluid extension catheter tube with a 3-way stopcock was attached to the needle for delivery of 1 ml of total volume over a period of 15 seconds (fast) or 10 minutes (slow), with the assistance of an infusion pump (Harvard Apparatus). For i.c.v. injection, anesthetized dogs were subjected to presurgical magnetic resonance imaging to establish brain coordinates. An image-based neuronavigation system (Stealthstation AxiEM; Medtronic) was used to guide the placement of a 2.5-mm-diameter Teflon catheter into the lateral ventricle via a small burr hole made through the parietal bone. Throughout all surgical procedures, temperature, cardiac and respiratory frequency, capnography, arterial pressure, pulse, and electrocardiography were monitored using a multifunctional patient Vet Care monitor (B. Braun).
Sulfamidase activity and GAG quantification.
Four months after injection, the mice were anesthetized with ketamine and xylazine, blood was extracted by cardiac puncture, and the animals were intracardially perfused with 10 ml of PBS to completely clear blood from the tissues. Brain and tissue samples were sonicated in 500 μl of water, and sulfamidase activity was assayed in supernatants with a 2-step protocol using a 4-methylumbelliferone–derived fluorogenic substrate (4-MU; Moscerdam Substrates), as previously described (69). Briefly, the first step consisted of the incubation of 30 μg of tissue protein extract or 10 μl of serum or CSF, with 10 mmol per liter of 4-MU-αGIcNS for 17 hours at 47°C. The second incubation was carried out in the presence of 10 U/ml of α-glucosidase (Sigma-Aldrich) in 0.2% BSA for 24 hours at 37°C. After stopping the reaction, the released fluorescence was measured. When indicated, sulfamidase activity was normalized against total protein content quantified by a Bradford assay (Bio-Rad). For GAG quantification, samples were weighed and digested with proteinase K, and extracts were clarified by centrifugation and filtration (Ultrafree MC; Millipore). A Blyscan sulfated glycosaminoglycan kit (Biocolor Life Science Assays) was used for GAG measurement using chondroitin 4-sulfate as a standard. Results were normalized to wet tissue weight.
Histology and electron microscopy.
Formalin-fixed, paraffin-embedded tissue sections were incubated overnight at 4°C with rat anti-LAMP1 (SC-1992; Santa Cruz Biotechnology); rabbit anti-LIMP2 (NB400; Novus Biologicals); rabbit anti-GFAP (Z0334; Dako Cytomation); goat anti-GFP (Ab6673; Abcam); chicken anti-GFP (Ab13970; Abcam); mouse anti-NeuN (MAB377; Millipore); goat anti-IBA1 (Ab5076; Abcam); and mouse anti-MAC2 (CL8942AP; Cedarlane) antibodies; and BSI-B4 lectin (L5391; Sigma-Aldrich). The secondary antibodies were: biotinylated rabbit anti-rat IgG (E0467; Dako) or biotinylated goat anti-rabbit IgG (31820; Vector Laboratories). Bright-field sections were stained with 3.3-diaminobenzidine (Sigma-Aldrich) and counterstained with hematoxylin. Images were obtained with an Eclipse E800 optical microscope (Nikon). For immunofluorescence, streptavidin-conjugated Alexa 488 (S-11223; Molecular Probes) or streptavidin-Alexa 568 (S-11226; Molecular Probes) was used for visualization. TO-PRO-3 (Invitrogen) was used to counterstain nuclei. Signal intensity was quantified with images obtained using a confocal microscope (Leica Microsystems). NIS Elements Advanced Research 2.20 software was used to quantify GFP, LIMP2, GFAP, and BSI-B4 signals in 3 to 5 images (original magnification, ×20) per animal of each brain region, using the same signal threshold settings for all animals. Then, the percentage of positive area was calculated, i.e., the area, in pixels, with a positive signal over the total tissue area in the image. For transmission electron microscopy, samples were processed as previously described (33) and examined with a Hitachi H-7000 transmission electron microscope (Hitachi).
Open field test.
The open field test was performed in 6-month-old mice between 9:00 am and 1:00 pm. Animals were placed in the center of a brightly lit chamber (41 × 41 × 30 cm) crossed by 2 bundles of photobeams (LE 8811; Panlab) that detect horizontal and vertical movements. Three squared concentric regions were delimited in the arena: center (14 × 14 cm), periphery (27 × 27 cm), and border (41 × 41 cm). Motor and exploratory activities were evaluated during the first 3 minutes. The total number of rearings and total number of lines crossed were recorded by photobeam disruption (SesaCom32; Panlab). The total distance covered was evaluated using a video tracking system (SMART Junior; Panlab).
Vector genome copy number and transgenic expression.
Total DNA was isolated with a MasterPureDNA Purification Kit (Epicentre) from overnight tissue digestions in proteinase K (0.2 mg/ml). The vector genome copy number in 20 or 100 ng of total DNA was determined by quantitative PCR with primers and probe specific for the WPRE element in the AAV-GFP vector. Forward primer: 5′-CGGCTGTTGGGCACTGA-3′; reverse primer: 5′-GGAAGGTCCGTCGAATTGA-3′; probe: 5′-ATGGCTGCTCGCCTGTGTTGCC-3′. The final values were determined by comparison with a reference standard curve built from serial dilutions of the linearized plasmids bearing the GFP expression cassette spiked into 20 ng or 100 ng of nontransduced genomic DNA. Total RNA was purified from tissues homogenized in TriPure Isolation Reagent (Roche) using an RNeasy Mini kit (QIAGEN). cDNA was synthesized with a Transcriptor First Strand cDNA Synthesis kit (Roche). Quantitative PCR was performed using a Light Cycler 480 Probes Master (Roche) with GFP-specific primers and probe. Forward primer: 5′-AGCTGGACGGCGACGTAA-3′; reverse primer: 5′-GTGCAGATGAACTTCAGGGTCA-3′; probe: 5′-CGTGTCCGGCGAGGGCGA-3′ (Invitrogen). These were normalized to the expression of the dog RPLP0 gene: forward primer: 5′-GCTGCTGGCCAATAAGGTT-3′; reverse primer: GGGCTGGCACAGTGACTT-3′.
Anti-AAV antibody determination.
Anti-AAV2 and AAV9 NAb titers were determined in human and dog CSF and serum samples as previously described (42) using an in vitro neutralization assay. An AAV self-complementary vector expressing the Renilla luciferase reporter gene under the control of the chicken β-actin promoter (scAAV-Luc) was used in the assay. To increase the efficiency of AAV vector transduction in vitro, 2V6.11 cells (ATCC) were used, which expressed the adenoviral gene E4 under the control of an inducible promoter. Cells were seeded in a 96-well plate at a density of 1.25 × 104 cells per well, and a 1:1,000 dilution of ponasterone A (Invitrogen) was added to the medium to induce E4 expression.On the day of assay, serial dilutions of heat-inactivated test serum were mixed with medium containing vector. Residual activity of the reporter transgene was measured using a luminometer.
Statistics.
All results are expressed as the mean ± SEM. Statistical comparisons were made using 1-way analysis of variance (ANOVA), and multiple comparisons between control and treatment groups were made using Dunnett’s post test. A P value less than 0.05 was considered statistically significant. The Kaplan–Meier method was used to analyze survival, and the log-rank test was used for comparisons.
Study approval.
All experimental procedures were approved by the Ethics Committee for Animal and Human Experimentation of the Universitat Autònoma de Barcelona.
Supplementary Material
Acknowledgments
We thank Xavier León, Marta Moya, Ester Blasco, ρngel Vázquez, and David Ramos for technical assistance. We would like to give special thanks to Xavier Codony for his advice on the evaluation of behavioral deficits. Funding was received from the Ministerio de Ciencia e Innovación, Plan Nacional I+D+I (SAF2008-00962, SAF2011-24698); the MPS España Foundation and Laboratorios ESTEVE, S.A.; and the European Community (CLINIGENE LSHB-CT-2006-018933). A. Ribera and A. Ruzo received fellowships from the Ministerio de Ciencia e Innovación, S. Marcó from the Generalitat de Catalunya, and P. Villacampa from the Fundación Ramón Areces.
Footnotes
Conflict of interest: Albert Ruzo, Eduard Ayuso, and Fatima Bosch are coinventors on a patent application for the use of AAV vectors for the treatment of MPS IIIA.
Citation for this article: J Clin Invest. 2013;123(8):3254–3271. doi:10.1172/JCI66778.
References
- 1.Enns GM, Huhn SL. Central nervous system therapy for lysosomal storage disorders. Neurosurg Focus. 2008;24(3–4):E12. doi: 10.3171/FOC/2008/24/3-4/E11. [DOI] [PubMed] [Google Scholar]
- 2.Valstar MJ, et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010;68(6):876–887. doi: 10.1002/ana.22092. [DOI] [PubMed] [Google Scholar]
- 3.Meyer A, et al. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics. 2007;120(5):e1255–e1261. doi: 10.1542/peds.2007-0282. [DOI] [PubMed] [Google Scholar]
- 4.Ruijter GJ, et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab. 2008;93(2):104–111. doi: 10.1016/j.ymgme.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharyya R, Gliddon B, Beccari T, Hopwood JJ, Stanley P. A novel missense mutation in lysosomal sulfamidase is the basis of MPS III A in a spontaneous mouse mutant. Glycobiology. 2001;11(1):99–103. doi: 10.1093/glycob/11.1.99. [DOI] [PubMed] [Google Scholar]
- 6.Bhaumik M, et al. A mouse model for mucopolysaccharidosis type III A (Sanfilippo syndrome). Glycobiology. 1999;9(12):1389–1396. doi: 10.1093/glycob/9.12.1389. [DOI] [PubMed] [Google Scholar]
- 7.Crawley AC, et al. Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res. 2006;1104(1):1–17. doi: 10.1016/j.brainres.2006.05.079. [DOI] [PubMed] [Google Scholar]
- 8.Wilkinson FL, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7(4):e35787. doi: 10.1371/journal.pone.0035787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felice BR, et al. Safety evaluation of chronic intrathecal administration of idursulfase-IT in cynomolgus monkeys. Toxicol Pathol. 2011;39(5):879–892. doi: 10.1177/0192623311409595. [DOI] [PubMed] [Google Scholar]
- 10.Staba SL, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350(19):1960–1969. doi: 10.1056/NEJMoa032613. [DOI] [PubMed] [Google Scholar]
- 11.Krivit W, Peters C, Shapiro EG. Bone marrow transplantation as effective treatment of central nervous system disease in globoid cell leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy, mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III. Curr Opin Neurol. 1999;12(2):167–176. doi: 10.1097/00019052-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Cartier N, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326(5954):818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 13.Langford-Smith A, et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol Ther. 2012;20(8):1610–1621. doi: 10.1038/mt.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12(5):341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 15.Buchlis G, et al. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood. 2012;119(13):3038–3041. doi: 10.1182/blood-2011-09-382317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathwani AC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciron C, et al. Human alpha-iduronidase gene transfer mediated by adeno-associated virus types 1, 2, and 5 in the brain of nonhuman primates: vector diffusion and biodistribution. Hum Gene Ther. 2009;20(4):350–360. doi: 10.1089/hum.2008.155. [DOI] [PubMed] [Google Scholar]
- 18.Ellinwood NM, et al. Safe, efficient, and reproducible gene therapy of the brain in the dog models of Sanfilippo and Hurler syndromes. Mol Ther. 2011;19(2):251–259. doi: 10.1038/mt.2010.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Passini MA, Watson DJ, Vite CH, Landsburg DJ, Feigenbaum AL, Wolfe JH. Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J Virol. 2003;77(12):7034–7040. doi: 10.1128/JVI.77.12.7034-7040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraldi A, et al. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet. 2007;16(22):2693–2702. doi: 10.1093/hmg/ddm223. [DOI] [PubMed] [Google Scholar]
- 21.Fu H, Dirosario J, Kang L, Muenzer J, McCarty DM. Restoration of central nervous system alpha-N-acetylglucosaminidase activity and therapeutic benefits in mucopolysaccharidosis IIIB mice by a single intracisternal recombinant adeno-associated viral type 2 vector delivery. J Gene Med. 2010;12(7):624–633. doi: 10.1002/jgm.1480. [DOI] [PubMed] [Google Scholar]
- 22.Worgall S, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19(5):463–474. doi: 10.1089/hum.2008.022. [DOI] [PubMed] [Google Scholar]
- 23.Janson C, et al. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther. 2002;13(11):1391–1412. doi: 10.1089/104303402760128612. [DOI] [PubMed] [Google Scholar]
- 24.Souweidane MM, et al. Gene therapy for late infantile neuronal ceroid lipofuscinosis: neurosurgical considerations. J Neurosurg Pediatr. 2010;6(2):115–122. doi: 10.3171/2010.4.PEDS09507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao G, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78(12):6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duque S, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17(7):1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol Ther. 2011;19(6):1025–1033. doi: 10.1038/mt.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruzo A, et al. Correction of pathological accumulation of glycosaminoglycans in CNS and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum Gene Ther. 2012;23(12):1237–1246. doi: 10.1089/hum.2012.029. [DOI] [PubMed] [Google Scholar]
- 30.Bell CL, et al. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J Clin Invest. 2011;121(6):2427–2435. doi: 10.1172/JCI57367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen S, et al. Glycan binding avidity determines the systemic fate of adeno-associated virus type 9. J Virol. 2012;86(19):10408–10417. doi: 10.1128/JVI.01155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inagaki K, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruzo A, et al. Liver production of sulfamidase reverses peripheral and ameliorates CNS pathology in mucopolysaccharidosis IIIA mice. Mol Ther. 2012;20(2):254–266. doi: 10.1038/mt.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurihara M, Kumagai K, Yagishita S. Sanfilippo syndrome type C: a clinicopathological autopsy study of a long-term survivor. Pediatr Neurol. 1996;14(4):317–321. doi: 10.1016/0887-8994(96)00083-5. [DOI] [PubMed] [Google Scholar]
- 35.Tamagawa K, Morimatsu Y, Fujisawa K, Hara A, Taketomi T. Neuropathological study and chemico-pathological correlation in sibling cases of Sanfilippo syndrome type B. Brain Dev. 1985;7(6):599–609. doi: 10.1016/S0387-7604(85)80008-5. [DOI] [PubMed] [Google Scholar]
- 36.Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4). J Histochem Cytochem. 1990;38(11):1683–1686. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- 37.Streit WJ, Kreutzberg GW. Lectin binding by resting and reactive microglia. J Neurocytol. 1987;16(2):249–260. doi: 10.1007/BF01795308. [DOI] [PubMed] [Google Scholar]
- 38.Davidoff AM, Ng CY, Zhou J, Spence Y, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102(2):480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- 39.Crawley AC, et al. Enzyme replacement reduces neuropathology in MPS IIIA dogs. Neurobiol Dis. 2011;43(2):422–434. doi: 10.1016/j.nbd.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Rengachary SS, Wilkins RH. Neurosurgical Operative Atlas. Vol. 1. Baltimore, Maryland, USA: Williams and Wilkins; 1991. [Google Scholar]
- 41.Boutin S, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 42.Manno CS, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 43.Jiang H, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108(10):3321–3328. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scallan CD, et al. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood. 2006;107(5):1810–1817. doi: 10.1182/blood-2005-08-3229. [DOI] [PubMed] [Google Scholar]
- 45.Mingozzi F, et al. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther. 2013;20(4):417–424. doi: 10.1038/gt.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monteilhet V, et al. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther. 2011;19(11):2084–2091. doi: 10.1038/mt.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Begley DJ, Pontikis CC, Scarpa M. Lysosomal storage diseases and the blood-brain barrier. Curr Pharm Des. 2008;14(16):1566–1580. doi: 10.2174/138161208784705504. [DOI] [PubMed] [Google Scholar]
- 48.Calcedo R, et al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin Vaccine Immunol. 2011;18(9):1586–1588. doi: 10.1128/CVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li C, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2012;19(3):288–294. doi: 10.1038/gt.2011.90. [DOI] [PubMed] [Google Scholar]
- 50.Mingozzi F, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13(4):419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 51.Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2011;11(4):321–330. doi: 10.2174/156652311796150354. [DOI] [PubMed] [Google Scholar]
- 52.Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005;25(41):9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, et al. Sustained correction of disease in naive and AAV2-pretreated hemophilia B dogs: AAV2/8-mediated, liver-directed gene therapy. Blood. 2005;105(8):3079–3086. doi: 10.1182/blood-2004-10-3867. [DOI] [PubMed] [Google Scholar]
- 54.Donsante A, Levy B, Vogler C, Sands MS. Clinical response to persistent, low-level beta-glucuronidase expression in the murine model of mucopolysaccharidosis type VII. J Inherit Metab Dis. 2007;30(2):227–238. doi: 10.1007/s10545-007-0483-4. [DOI] [PubMed] [Google Scholar]
- 55.Garcia AR, DaCosta JM, Pan J, Muenzer J, Lamsa JC. Preclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS II. Mol Genet Metab. 2007;91(2):183–190. doi: 10.1016/j.ymgme.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 56.Mingozzi F, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110(7):2334–2341. doi: 10.1182/blood-2007-03-080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nathwani AC, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107(7):2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nathwani AC, et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther. 2011;19(5):876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hemsley KM, Hopwood JJ. Delivery of recombinant proteins via the cerebrospinal fluid as a therapy option for neurodegenerative lysosomal storage diseases. Int J Clin Pharmacol Ther. 2009;47(suppl 1):S118–S123. doi: 10.5414/cpp47118. [DOI] [PubMed] [Google Scholar]
- 60.Hemsley KM, et al. Effect of cisternal sulfamidase delivery in MPS IIIA Huntaway dogs — a proof of principle study. Mol Genet Metab. 2009;98(4):383–392. doi: 10.1016/j.ymgme.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 61.Hemsley KM, Beard H, King BM, Hopwood JJ. Effect of high dose, repeated intra-CSF injection of sulphamidase on neuropathology in MPS IIIA mice. Genes Brain Behav. 2008;7(7):740–753. doi: 10.1111/j.1601-183X.2008.00413.x. [DOI] [PubMed] [Google Scholar]
- 62.American Association of Neurological Surgeons. National Neurosurgical Procedural Statistics 2006 Survey. Meadows, Illinois, USA: American Association of Neurological Surgeons; 2006. [Google Scholar]
- 63.Federici T, et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2012;19(8):852–859. doi: 10.1038/gt.2011.130. [DOI] [PubMed] [Google Scholar]
- 64.Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20(4):450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook AM, Mieure KD, Owen RD, Pesaturo AB, Hatton J. Intracerebroventricular administration of drugs. Pharmacotherapy. 2009;29(7):832–845. doi: 10.1592/phco.29.7.832. [DOI] [PubMed] [Google Scholar]
- 66.Samaranch L, et al. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. 2012;23(4):382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lau AA, Crawley AC, Hopwood JJ, Hemsley KM. Open field locomotor activity and anxiety-related behaviors in mucopolysaccharidosis type IIIA mice. Behav Brain Res. 2008;191(1):130–136. doi: 10.1016/j.bbr.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 68.Ayuso E, et al. High AAV vector purity results in serotype- and issue-independent enhancement of transduction efficiency. Gene Ther. 2010;17(4):503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 69.Karpova EA, et al. A fluorimetric enzyme assay for the diagnosis of Sanfilippo disease type A (MPS IIIA). J Inherit Metab Dis. 1996;19(3):278–285. doi: 10.1007/BF01799255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.