

Abstract
Aging is a major risk factor for a large number of disorders and functional impairments. Therapeutic targeting of the aging process may therefore represent an innovative strategy in the quest for novel and broadly effective treatments against age-related diseases. The recent report of lifespan extension in mice treated with the FDA-approved mTOR inhibitor rapamycin represented the first demonstration of pharmacological extension of maximal lifespan in mammals. Longevity effects of rapamycin may, however, be due to rapamycin’s effects on specific life-limiting pathologies, such as cancers, and it remains unclear if this compound actually slows the rate of aging in mammals. Here, we present results from a comprehensive, large-scale assessment of a wide range of structural and functional aging phenotypes, which we performed to determine whether rapamycin slows the rate of aging in male C57BL/6J mice. While rapamycin did extend lifespan, it ameliorated few studied aging phenotypes. A subset of aging traits appeared to be rescued by rapamycin. Rapamycin, however, had similar effects on many of these traits in young animals, indicating that these effects were not due to a modulation of aging, but rather related to aging-independent drug effects. Therefore, our data largely dissociate rapamycin’s longevity effects from effects on aging itself.
Introduction
Aging is an important risk factor for a number of disorders and functional impairments. Therapeutic targeting of the aging process may therefore represent an innovative strategy in the quest for novel and potentially broadly effective treatments against diseases associated with old age, including neurodegenerative diseases, cardiovascular diseases, cancers, and type 2 diabetes mellitus (1).
The vast majority of current research that aims to identify novel anti-aging interventions uses lifespan measures as a primary readout to detect anti-aging effects. Mechanisms involved in cell growth and metabolism are commonly implicated players in lifespan regulation (2). Specifically, the target of rapamycin (TOR) signaling pathway is an important and evolutionarily conserved player in longevity regulation, with effects in flies (3), worms (4, 5), yeast (6, 7), and mice (8–11). The recent report of lifespan extension in mice treated with the FDA-approved mammalian TOR (mTOR) inhibitor rapamycin (9) represented the first demonstration of pharmacological extension of maximal lifespan in mammals; additional studies have since confirmed this important finding (8, 10, 11).
Extension of lifespan does not necessarily indicate effects on aging. Lifespan extension could be caused by isolated suppression of specific life-limiting pathologies, such as cancers. This is a plausible scenario, because rapamycin has known antineoplastic properties (12–14), and cancers are the leading cause of death in many mouse strains, including C57BL/6J and all strains in which rapamycin was shown to have longevity effects (9, 15). An alternative scenario is that rapamycin’s anticancer effects represent one aspect of a more general effect of this compound on aging.
Whether mTOR inhibition actually decelerates mammalian aging rates, then, remains unknown. Addressing this question requires determining whether rapamycin delays a broad range of age-dependent functional and structural alterations in a number of different cell types, tissues, and organ systems. While some recent reports suggest that rapamycin may indeed prevent aging phenotypes in addition to age-related mortality and cancer (8, 16, 17), these reports are highly limited in scope, examining only a paucity of aging phenotypes and not accounting for potential aging-independent drug effects.
Here, we present results from a comprehensive, large-scale assessment of a wide range of structural and functional aging phenotypes (>150 molecular, cellular, histopathological, and functional aging phenotypes across >25 different tissues), which we performed to test whether rapamycin promotes healthy aging in male C57BL/6J mice.
Results
Rapamycin was previously reported to extend lifespan, even if treatment is initiated late in life (9). To determine whether age of treatment onset is an important variable affecting the possible healthspan effects of the drug, we included 3 different cohorts in our study, in which rapamycin or vehicle control treatment commenced in young adulthood (i.e., at 4 months), in midlife (i.e., at 13 months) or late in life (i.e., at 20–22 months). Animals were treated with rapamycin for 1 year before analyses commenced (i.e., analysis at 16, 25, or 32–34 months, respectively), unless otherwise stated.
Rapamycin-induced lifespan extension is known to occur in mice of both genders (with larger effect sizes in females in a genetically heterogeneous stock of mice) (9, 10). We therefore focused our study on one sex. We chose to analyze male mice because a proper analysis of females would have required taking into account phenotypic variation associated with the estrus cycle.
Rapamycin was detected in whole blood of treated animals and showed extensive tissue distribution (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI67674DS1), consistent with the known pharmacokinetic properties of the drug (18). Survival analysis applied to all treated cohorts, and lifespan extension by rapamycin was demonstrated relative to corresponding vehicle controls (Figure 1), confirming previously reported longevity effects (8–11).
Figure 1. Rapamycin extended lifespan.

Survival curves were calculated for rapamycin- and vehicle-treated mice (all cohorts; n = 68 per group). Statistical analysis using a Cox proportional hazards model with the data stratified by cohort revealed a significant effect of rapamycin treatment (P = 0.0326).
To determine whether rapamycin slows aging, we assessed a large number of structural and functional aging phenotypes in multiple cell types, tissues, and organs systems in our aging mouse cohorts. Based on their modulation by aging and/or rapamycin treatment, the resulting phenotypes could be categorized as: (a) aging phenotypes rescued by rapamycin (Table 1); (b) aging phenotypes not altered by rapamycin (Table 2); and (c) aging phenotypes worsened by rapamycin and/or influenced by rapamycin only (Supplemental Table 2). Traits in the first category (aging phenotypes rescued by rapamycin) may be related to retarded development of aging phenotypes under chronic rapamycin treatment, which would support the hypothesis that rapamycin slows aging. Alternatively, rapamycin may influence these traits in a direct way, without actually modulating aging. To test for aging-independent drug effects, we examined whether rapamycin affects the respective phenotypes in young mice (Table 1). Traits affected similarly in young animals and aging mice are influenced by rapamycin in a direct way, not via a modulation of aging itself.
Table 1.
Aging phenotypes rescued by rapamycin treatment
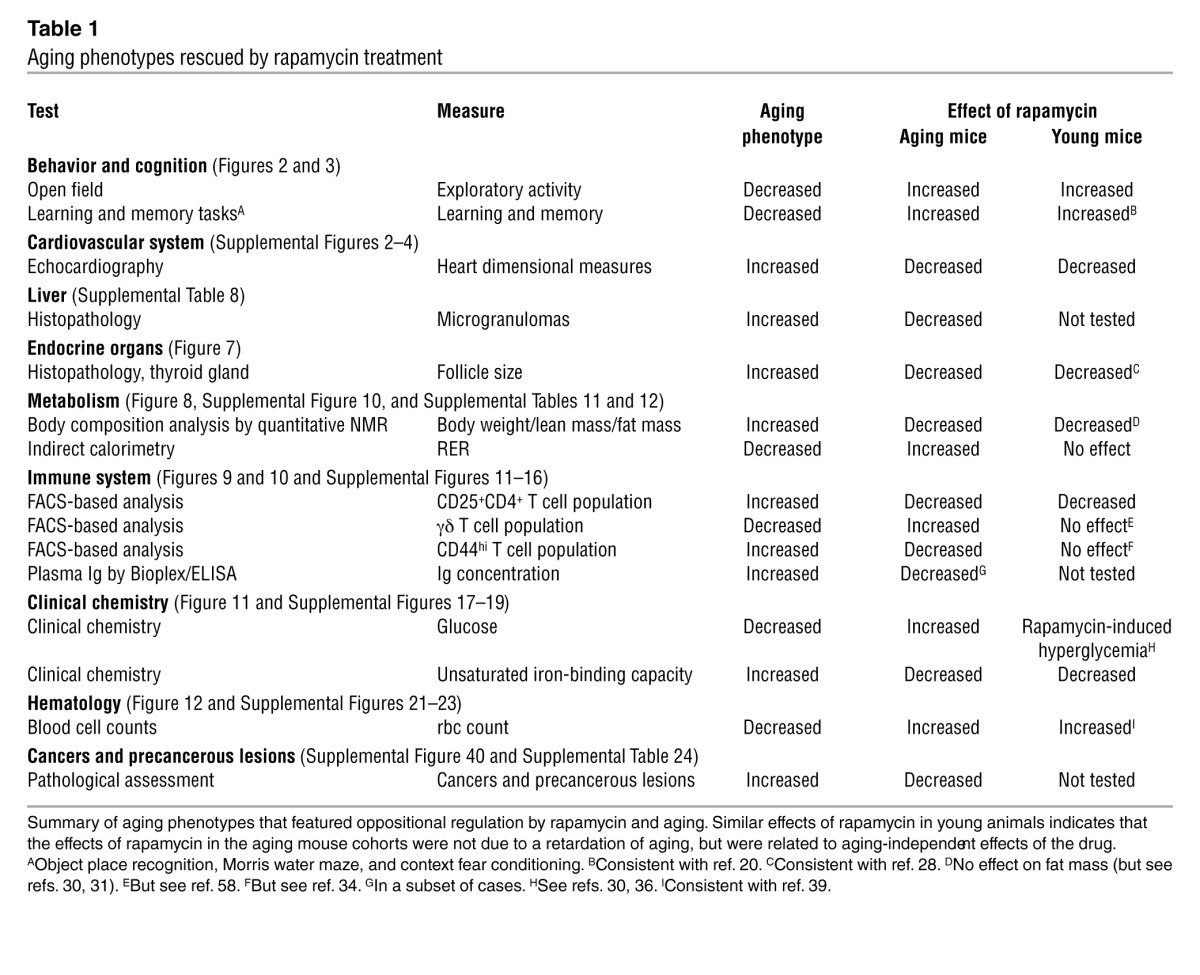
Table 2.
Aging phenotypes not measurably altered by rapamycin

Rapamycin treatment resulted in aging-independent improvements of some age-associated neurobehavioral phenotypes.
We started our assessment of age-related health parameters by testing neurological and behavioral functions in rapamycin- and vehicle-treated aged mice and young controls. A modified SHIRPA screen was used to examine general health, autonomous functions, reflexes, posture, movement, and spontaneous behavior. A subset of SHIRPA measures (presence of tremor and gait abnormalities) showed clear age-related changes that were not prevented by rapamycin treatment (Supplemental Table 3).
Motor coordination and balance was examined with the accelerating rotarod, which revealed age-related impairments that were not improved by rapamycin treatment (Figure 2, A and B). Nociceptive functions were examined using the hot plate test, which demonstrated that aging increased reaction latencies on the hot plate, and these were not affected by rapamycin treatment (Figure 2, C and D).
Figure 2. Rapamycin treatment had limited effects on aging-associated neurobehavioral phenotypes.

Basic neurological functions were assessed in the 16-month (young control, n = 10; vehicle, n = 18; rapamycin, n = 20) and 25-month (young control, n = 10; vehicle, n = 11; rapamycin, n = 16) cohorts. (A and B) Latency to fall in the context of a motor coordination test on the accelerating rotarod. (C and D) Latency to first and second reaction on a hot plate test performed to assess nociceptive functions. (E and F) Exploratory activity, as examined in an open field assay. (E) Distance traveled. (F) Number of rearings. (G and H) Effects of rapamycin on exploratory activity in young mice (vehicle, n = 9; rapamycin, n = 8). Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font.
Exploratory activity was examined in an open field assay, in which mice were allowed to explore freely a novel environment. Mice of the 16-month cohort showed the expected strongly reduced levels of exploration, as evidenced by reduced distance-traveled measures and reduced number of rearings, and these reductions were improved by rapamycin treatment (Figure 2, E and F). These results could indicate either that chronic rapamycin slowed the age-related decline in activity levels, or, alternatively, that chronic rapamycin could result in aging-independent effects on exploratory activity in mice. To distinguish between these possibilities, we tested open field exploratory activity in young adult male C57BL/6J mice that had been chronically treated with rapamycin or vehicle (see Methods). Rapamycin increased the number of rearings and the total distance traveled in these young mice (Figure 2, G and H), which indicated that rapamycin increased exploratory activity in an aging-independent fashion.
Aging is associated with declining cognitive functions, including impairments in learning and memory formation (19). We examined learning and memory in the animals using 3 different hippocampus-dependent memory paradigms: the Morris water maze, context fear conditioning, and an object place recognition task (Figure 3).
Figure 3. Chronic rapamycin treatment resulted in aging-independent improvements of learning and memory.

Learning and memory was examined in our aging cohorts using an object place recognition paradigm (A), the Morris water maze (B–D) and a context fear conditioning paradigm (E) (11-month vehicle, n = 20; 20-month vehicle, n = 16; 11-month rapamycin, n = 20; 20-month rapamycin, n = 19). (A) Exploration times of the objects in the novel and familiar location, respectively, during the test of the object place recognition task. (B) Escape latencies during training on a hidden version of the Morris water maze task. (C) Quadrant occupancy and (D) target crossing measures during the probe trial given after completion of training in the Morris water maze. TQ, target quadrant; OQ, other quadrants. (E) Activity suppression ratios during a context test given 1 day after associative training in a context fear conditioning paradigm. (F–H) To test for aging-independent effects of rapamycin on learning and memory, we assessed young mice chronically treated with rapamycin or vehicle (n = 15 per group) in the Morris water maze. (F) Escape latencies during training. (G) Quadrant occupancy and (H) target crossings during a probe trial delivered after completion of training. All graphs show mean ± SEM.
In the object place recognition task (Figure 3A), mice were allowed to explore an environment that contained 2 identical objects in defined locations of the arena. During the test phase, the position of one of the objects was altered, while the other object remained in its previous location. Analysis of exploration times during the test revealed significant object/age interaction (P = 0.0293, 3-way ANOVA with age and treatment as between-subjects factors and object as within-subjects factors), indicative of preferential exploration of the object in the novel location by the younger animals and a lack of such preferential exploration in 20-month-old mice. Our analyses did not show a significant object/treatment interaction (P = 0.4889), which indicated that rapamycin treatment had no clear effect on object position memory in this task.
In the Morris water maze (Figure 3, B–D), mice were trained to find an escape platform hidden underneath the water surface in a constant location of the pool. Analysis of escape latencies (Figure 3B) during training indicated significant effects of aging and, possibly, rapamycin, such that escape latencies were increased in aged mice and were decreased by rapamycin treatment (P = 0.0021, P = 0.0548, and P = 0.2419 for age, treatment, and age/treatment interaction, respectively, 3-way ANOVA with age and treatment as between-subjects factors and training day as within-subjects factor). To test how accurately animals had learned the platform location, we gave a probe trial after completion of training, during which we removed the platform from the pool and analyzed the swim pattern of the animals. Analysis of the probe trial measures — quadrant occupancy and target crossings (Figure 3, C and D) — indicated that spatial learning and memory was significantly enhanced by chronic rapamycin treatment, regardless of age group (quadrant occupancy, P = 0.0021, treatment/quadrant interaction; target crossings, P = 0.4412, treatment/quadrant interaction; 3-way ANOVA with age and treatment as between-subjects factors and quadrant as within-subjects factor).
Associative learning and memory was analyzed in a context fear conditioning paradigm (Figure 3E). Conditioned fear was assessed 1 day after conditioning in the training context. Whereas age effects were not significant, rapamycin treatment significantly improved fear conditioning (P = 0.4623, P = 0.0018, and P = 0.1601 for age, treatment, and age/treatment interaction, respectively; 2-way ANOVA with age and treatment as between-subjects factors).
These findings indicated that chronic rapamycin treatment enhanced learning and memory on different tasks in our aged cohorts. To test whether rapamycin exerts effects on aging, or, alternatively, whether aging-independent drug effects accounted for the above results, we tested spatial learning and memory in the Morris water maze in the young adult male C57BL/6J mice chronically treated with rapamycin or vehicle. Rapamycin significantly reduced escape latencies during the training trials (P = 0.0190, 2-way ANOVA with treatment as between-subjects factor and training day as within-subjects factor; Figure 3F). Analysis of the probe trial measures — quadrant occupancy and target crossings (Figure 3, G and H) — showed a significant treatment/quadrant interaction for the target crossings measure (P = 0.0411 and P = 0.2456 for target crossings and quadrant occupancy, respectively; 2-way ANOVA with treatment as between-subjects factor and quadrant as within-subjects factor), indicating that young rapamycin-treated animals searched in a more selective fashion during the probe trial than vehicle controls. Consistent with a previous report (20), these data demonstrated that learning and memory in the Morris water maze is enhanced by chronic rapamycin treatment in an aging-independent manner.
There is a known strong age-dependent decline in adult hippocampal neurogenesis (21). Our quantitative assessment of doublecortin (DCX) immunoreactivity in the dentate gyrus showed a loss of DCX in aged animals (Supplemental Figure 1), consistent with a substantial decrease in the neurogenic capacity of the aged hippocampus. Chronic rapamycin treatment tended to increase dentate gyrus DCX immunoreactivity in aged animals, but this effect was far from reaching statistical significance.
Rapamycin had no effect on age-related reductions in grip strength and cross-sectional muscle fiber area.
Aging is accompanied by progressive loss of muscle mass and muscle force (22). We recorded 2-paw (i.e., forepaw) and 4-paw grip strength measurements in aged rapamycin- and vehicle-treated mice as well as young controls (16-, 25-, and 34-month cohorts; Figure 4, A–F). We observed the expected grip strength decline in all measures across all groups of aged mice. Rapamycin had no apparent protective effect on aging-associated grip strength loss. We also measured cross-sectional muscle fiber areas in quadriceps femoris muscle of rapamycin- or vehicle-treated aged mice and corresponding young controls (16- and 25-month cohorts; Figure 4, G and H), which showed the expected aging-associated decrease in cross-sectional muscle fiber area. Rapamycin treatment did not ameliorate this aging trait, but rather led to a further decrement in muscle fiber area, at least in one of the cohorts.
Figure 4. Rapamycin did not improve age-related reductions in grip strength and cross-sectional muscle fiber area.

(A–F) Aging-related decline in muscle strength was assessed using 2-paw (A, C, and E) and 4-paw (B, D, and F) grip strength tests in the 16-month (A and B; young control, n = 10; vehicle, n = 18; rapamycin, n = 20), 25-month (C and D; young control, n = 10; vehicle, n = 11; rapamycin, n = 16), and 34-month (E and F; young control, n = 10; vehicle, n = 9; rapamycin, n = 14) cohorts. (G and H) Muscle fibers were automatically identified on H&E-stained sections of quadriceps femoris muscle of the 16-month (G; young control, n = 786 muscle fibers from 5 mice; vehicle, n = 534 muscle fibers from 6 mice; rapamycin, n = 600 muscle fibers from 8 mice) and 25-month (H; young control, n = 330 muscle fibers from 6 mice; vehicle, n = 741 muscle fibers from 4 mice; rapamycin, n = 1,371 muscle fibers from 5 mice) cohorts using image segmentation and analysis software. Shown are examples of histological images and classification maps (generated via automated segmentation of histological images) along with quantification of cross-sectional muscle fiber area. Scale bar: 400 μm. Whisker plots display 25th percentile, median, and 75th percentile as well as minimum-to-maximum (A–F) or 10th to 90th percentile (G and H). P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font.
Rapamycin did not improve age-related ophthalmological impairments.
The most common aging-associated pathology in the anterior part of the eye is cataract formation (23). We used Scheimpflug imaging to measure abnormalities in the anterior segment of the eye, including lens density, in animals of the 16- and 25-month cohorts. Vehicle-treated aged animals showed the expected clear increase in average lens density relative to young control mice, and this deterioration was not ameliorated by rapamycin treatment (Figure 5A). The posterior part of the eye was assessed using in vivo optical coherence tomography and histology, which demonstrated normal retinal thickness and apparently unaltered retinal vasculature in aged mice (Figure 5B and data not shown). To test visual acuity, we subjected animals of the 16- and 25-month cohorts to the virtual drum vision test (Figure 5C). Visual acuity deteriorated with age in both cohorts, and rapamycin treatment failed to improve these age-related impairments.
Figure 5. Rapamycin did not improve age-related ophthalmological impairments.

Summary of findings from ophthalmological assessment of the 16-month (young control, n = 10; vehicle, n = 18; rapamycin, n = 20) and 25-month (young control, n = 10; vehicle, n = 8; rapamycin, n = 14) cohorts. (A) Quantification of lens densities, assessed via Scheimpflug imaging; example images are also shown. (B) Optical coherence tomography was performed for in vivo assessment of the posterior part of the eye. Example images are shown. No age or treatment effects were noted. (C) Visual acuity was examined with the virtual drum test. Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font.
Rapamycin had limited effects on age-dependent functional and histopathological phenotypes in the cardiovascular system.
Aging is associated with substantial changes in heart structure and function (24). To examine cardiac aging phenotypes, we performed echocardiography in rapamycin- and vehicle-treated aged mice and young controls. Echocardiographic studies were performed in 2 different settings. First, basic echocardiography was performed to determine heart dimensional measures in the aforementioned 16- and 25-month cohorts (see Supplemental Results and Supplemental Figure 2). To more fully explore cardiac physiology, we performed additional echocardiographic studies in another cohort of animals: 26-month-old animals in which rapamycin or vehicle treatment was started at 13 months of age, along with 3- to 4-month-old young controls fed vehicle diet. These additional studies included assessment of cardiac output, ejection fraction, and fractional shortening as well as measurements of flow velocities and pressure gradients across the aortic valve, pulmonary valve and mitral valve. Our results showed a number of functional alterations in aged mice, including reduced ejection fraction, reduced fractional shortening, and reduced pressure gradients and blood flow velocities across the aortic and pulmonary valves in aged animals compared with young controls (Figure 6 and Supplemental Figure 3). Rapamycin treatment did not measurably improve these functional cardiac aging phenotypes, but instead reduced some functional cardiac outcome measures even further, such as ejection fraction and fractional shortening, although these differences did not reach statistical significance (Figure 6, A and B).
Figure 6. Aging-associated functional cardiac alterations were not restored by rapamycin treatment.

Key findings of a comprehensive assessment of cardiac function using echocardiography (young control, n = 10; 26-month vehicle, n = 17; 26-month rapamycin, n = 10). (A) Ejection fraction. (B) Fractional shortening. (C) Flow velocity across the aortic valve. (D) Pressure gradient across the aortic valve. (E) Flow velocity across the pulmonary valve. (F) Pressure gradient across the pulmonary valve. Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font.
Next, we performed detailed histopathological assessments of the myocardium and the aortic vascular wall to identify possible effects of rapamycin on cardiovascular pathology associated with normal aging. As expected, aged mice showed significant myocardial pathology, including ventricular dilation, myocardial hypertrophy, myocardial infarction, fibrosis, and thickening of the heart valves (Supplemental Figure 5 and Supplemental Table 4), which did not appear to be improved by rapamycin and may have even been worsened in the 34-month cohort.
A hallmark of arterial aging in humans is atherosclerosis. Although atherosclerosis does not spontaneously develop in C57BL/6J mice, histopathological changes reminiscent of arterial precursor lesions (e.g., accumulation of glycosaminoglycans and chronic inflammatory infiltrates) do occur in mice (25). Additionally, during aging, arteries lose their elasticity due to elastic fiber fragmentation and excessive collagen deposition in the arterial wall (25). We performed histopathological analyses of aortic sections, assessing the arterial wall for the presence of degenerative changes (i.e., division and disorganization of elastic fibers and replacement of smooth muscle cells by mucinous substance). Our analyses revealed that aging was associated with the expected degenerative changes (Supplemental Figure 5 and Supplemental Table 5). We were not able to detect beneficial effects of rapamycin on aortic degeneration in our mouse cohorts (Supplemental Figure 5 and Supplemental Table 5), although larger sample sizes may be required to detect possible effects of rapamycin on aortic degeneration.
Rapamycin had no measurable effects on hepatic fibrosis, but appeared to reduce microgranulomas in the aged liver.
Age-associated changes in the liver include fat accumulation, fibrotic changes, inflammatory/immune alterations, and cancers (26). We performed histopathological assessment of liver-associated aging phenotypes in rapamycin- and vehicle-treated aged mice and young controls, including evaluation of liver steatosis, periportal fibrosis, polyploidy, hepatitis, and hepatic microgranulomas (Supplemental Tables 6–8 and Supplemental Figure 6). Logistic regression analysis indicated that aging was associated with a significant increase in the prevalence of periportal fibrosis and polyploidy (Supplemental Tables 6 and 7). We were unable to discern significant effects of rapamycin on these hepatic aging phenotypes, although larger sample sizes would be needed to detect possible effects of rapamycin, if any, on these traits. However, rapamycin did show a trend toward attenuating the age-associated increase in hepatic microgranuloma prevalence. We did not test for aging-independent effects of rapamycin on hepatic microgranulomas, due to the relatively low prevalence of this pathology in young mice (Supplemental Table 8).
Rapamycin had aging-independent effects on thyroid follicle size.
Aging-related changes in the thyroid include increased follicle size and decreased colloid resorption, reflective of decreased thyroid activity in the aged animal (27). We performed morphometric histopathological analyses to assess follicle size distribution in the thyroid of rapamycin- or vehicle-treated aged mice and young controls. Our analyses showed the expected aging-associated increase in thyroid follicle size, which was significantly decreased by rapamycin treatment (Figure 7A and Supplemental Figure 7). In order to determine whether these effects of rapamycin were related to modulation of aging or aging-independent effects on thyroid follicle size, we analyzed follicle size distribution in the young mice chronically treated with rapamycin or vehicle. Rapamycin similarly decreased thyroid follicle size in young animals (Figure 7B), which is consistent with the important role of PI3K/AKT/mTOR signaling in the regulation of thyroid follicle size (28) and indicates that these effects were aging independent.
Figure 7. Rapamycin decreased thyroid follicle size in an aging-independent manner.

Thyroid follicles were automatically identified on H&E-stained thyroid sections using image segmentation and analysis software. Shown are examples of histological images and classification maps (generated via automated segmentation of histological images) along with quantification of follicle surface area. (A) Follicle size distribution in rapamycin- and vehicle-treated aged mice as well as young controls (young control, n = 1,428 follicles from 12 mice; 16-month vehicle, n = 566 follicles from 8 mice; 25-month vehicle, n = 241 follicles from 3 mice; 34-month vehicle, n = 440 follicles from 5 mice; 16-month rapamycin, n = 1,064 follicles from 10 mice; 25-month rapamycin, n = 277 follicles from 4 mice; 34-month rapamycin, n = 535 follicles from 8 mice). (B) Corresponding data from a comparison of young animals treated with rapamycin (n = 1,182 follicles from 8 mice) or vehicle (n = 1,543 follicles from 9 mice). Scale bars: 500 μm. Whisker plots display 10th percentile, 25th percentile, median, 75th percentile, and 90th percentile. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font.
Age-dependent thyroid pathologies include follicular cell hyperplasia and adenoma formation (29). We assessed the frequency of these pathological lesions in rapamycin- or vehicle-treated aged mice and found an age-dependent increase in the prevalence of follicular cell hyperplasia and thyroid adenoma formation in our mouse cohorts (Supplemental Table 9). We were unable to detect a significant effect of rapamycin treatment on the prevalence of these pathologies; again, larger sample sizes may be needed for the detection of rapamycin’s effects, if any, on this trait. Aging-related histopathological alterations in the adrenal glands include adrenal adenomas, subcapsular cell hyperplasias, and lipofuscin depositions (29). Advanced age was associated with nonsignificant increases in their prevalence (Supplemental Table 10 and Supplemental Figures 8 and 9).
Aging is associated with prominent deposition of lipofuscin within the adrenal gland parenchyma. We used morphometric histopathology to assess quantitatively the presence of lipofuscin deposits in the adrenal glands of rapamycin- or vehicle-treated aged mice and young controls. Statistical analyses of the data showed an increase in adrenal gland lipofuscin deposits in rapamycin-treated animals (Supplemental Figure 9), indicating that rapamycin did not ameliorate this histopathological phenotype.
Rapamycin counteracted a subset of age-related metabolic changes, but had no effect on others.
Aged mice showed significantly increased body weight due to increased fat and/or lean mass (Supplemental Figure 10). Rapamycin treatment tended to decrease body weight as well as fat and/or lean mass, at least in some cases (25-month cohort; Supplemental Figure 10). Studies in the young adult rapamycin- or vehicle-treated mice suggested body weight and lean mass reductions by rapamycin (Supplemental Figure 10), indicating that the effects of rapamycin on these parameters are likely aging independent. Although we were unable to detect any effect of rapamycin on fat mass in young mice (Supplemental Figure 10), previously documented direct effects of rapamycin on fat mass and adipogenesis (30, 31) caution against the conclusion that rapamycin’s effects were aging dependent.
Next, we used indirect calorimetry to assess energy metabolism in rapamycin- or vehicle-treated mice of the 16- and 25-month cohorts as well as young controls. O2 consumption (and thereby energy turnover) was reduced in aged mice and was not improved by rapamycin treatment (Figure 8B and Supplemental Tables 11 and 12). Aged mice showed a reduced respiratory exchange ratio (RER; Figure 8C and Supplemental Tables 11 and 12), indicating a proportional substrate preference toward lipid oxidation in aged mice. Rapamycin treatment restored age-related changes in RER to levels similar to those in young controls, at least in the 25-month cohort. Experiments in a young group of animals indicated no effects of rapamycin on RER in young mice (Figure 8D), suggesting that rapamycin may have influenced RER specifically in aged animals. Body temperature was significantly reduced in the 25-month cohort, and these age-related impairments in thermoregulation (32) were not corrected by rapamycin treatment (Figure 8A and Supplemental Tables 11 and 12). In conclusion, rapamycin may counteract some of the metabolic effects associated with older age (i.e., altered RER), but had no effect on others (i.e., O2 consumption and body temperature).
Figure 8. Rapamycin treatment had no significant effect on age-related changes in maximal O2 consumption and body temperature, but increased RER.

(A–C) Findings from a metabolic assessment (indirect calorimetry) of the 25-month cohort (young control, n = 10; vehicle, n = 10; rapamycin, n = 14). (A) Body temperature. (B) Maximal O2 consumption. (C) Average RER. Data were analyzed by fitting them with a linear model against the factors of age (young vs. old), treatment (rapamycin vs. vehicle), and body weight. (D) Effects of rapamycin on RER in young mice (control, n = 9; rapamycin, n = 8). Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font. See Supplemental Tables 11 and 12 for complete findings.
Rapamycin treatment affected age-related changes in T lymphocytes.
Age-related immune dysfunction increases the prevalence of infection and autoimmune disease in the elderly and may also contribute to increased cancer risk in aged individuals (33). Immune aging phenotypes include reduced thymic output, decreased naive T lymphocyte numbers, expansion of memory-type (CD44hi) T cells, reduced CD4/CD8 ratio, reduced NK cell frequency, increased granulocyte numbers, and increased levels of Igs (33).
To test for alterations in these immune phenotypes, we performed 10-color polychromatic flow cytometry and quantified various immune cell populations in young and old mice (Figure 9 and Supplemental Figures 11–13). We observed a strong aging-related decline in the frequency of CD4+ T lymphocytes (Figure 9A). Aged mice showed the expected increase in the frequency of CD44hi T cells (CD4+ and CD8+; Figure 9D and Supplemental Figures 11–13), indicative of an activated/memory T cell phenotype. Rapamycin treatment did not restore the aging-associated decrease in CD4+ T cells, but reduced the number of CD44hi T cells (CD4+ and CD8+) in the 16-month cohort. Additionally, rapamycin opposed aging-associated changes in the frequency of CD25+CD4+ T cells in the 16-month cohort (Figure 9B and Supplemental Figures 11–13), but not in the 25- and 34-month cohorts, in which rapamycin increased CD25+CD4+ T cell frequency. We observed strong and consistent aging-associated reductions in the frequency of NK cells (Figure 9F), which were not modified by rapamycin.
Figure 9. Rapamycin counteracted a subset of age-related changes in the T lymphocyte compartment.

(A) CD4+ T cell populations in the 16-month cohort. (B and C) CD25+CD4+ T cell populations in the 16-month cohort (B) and in young animals chronically treated with rapamycin or vehicle (C). (D and E) CD44hiCD4+ T cell populations in the 16-month cohort (D) and in young animals chronically treated with rapamycin or vehicle (E). (F) NK cell frequency in the 16-month cohort. Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font. See Supplemental Figures 11–13 for complete datasets.
In summary, our analyses revealed a set of effects of rapamycin that opposed aging phenotypes within the T lymphocyte compartment (i.e., effects on CD44hi T cells, CD25+CD4+ T cells, and γδ T cells). mTOR inhibition is well known to have a variety of effects on T lymphocytes, including a shift in CD44 expression (34), which suggests that the observed effects on T cell aging phenotypes may reflect age-independent drug effects. To test for age-independent drug effects in our experimental setting, we performed flow cytometry in the young mice chronically treated with rapamycin or vehicle. These analyses revealed some of the same effects of rapamycin in young mice as in aging mice (decreased CD25+CD4+ T cells; Figure 9C), whereas other effects were not present (no decrease in CD44hi T cell frequency; Figure 9E). These data therefore indicate that a subset of the effects of rapamycin on T cell phenotypes, in particular the frequency of CD44hi T cells, may have been specific to the 16-month cohort.
We performed Bioplex/ELISA to measure plasma Ig levels (IgM, IgA, IgG1, IgG2a, IgG2b, IgG3, and IgE), which are expected to increase with advanced age (Figure 10 and Supplemental Figures 14–16). We found the expected increase in plasma Ig concentrations in aged mice. Rapamycin decreased Ig concentrations in some cases (Figure 10, A, D, and H). The effects of rapamycin on plasma Ig concentrations in young mice were not assessed.
Figure 10. Rapamycin partially counteracted age-associated increases in plasma Ig concentrations.

Plasma Ig levels in the 16-month (A and B) and 34-month (C–H) cohorts. Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font.
Rapamycin treatment had no apparent effect on most aging-associated changes in clinical chemistry parameters.
We next performed clinical chemistry studies (Figure 11 and Supplemental Figures 17–19). Our main findings were clear aging-related changes in concentrations of plasma sodium, calcium, and chloride (increased), glucose (decreased), and triglycerides (decreased), in line with previously reported aging effects (35). Moreover, old mice showed increased alkaline phosphatase and α-amylase levels in their plasma, in line with previously published data (35). Rapamycin treatment had by and large no measurable effect on aging-related alterations in clinical chemistry measures, with only 2 exceptions in the entire clinical chemistry dataset. First, rapamycin decreased unsaturated iron binding capacity (which was increased in the 34-month cohort). Second, in the 16-month cohort, rapamycin increased plasma glucose levels (which decreased in aged mice). These effects of rapamycin, however, most likely represent aging-independent drug effects: unsaturated iron binding capacity was also decreased by rapamycin in young animals (data not shown). Treatment with rapamycin or other mTOR inhibitors is known to induce insulin resistance and hyperglycemia (36). In agreement with this notion, we observed that rapamycin exacerbated age-related glucose tolerance impairments in rapamycin-treated animals of the 16-month cohort (Supplemental Figure 20).
Figure 11. Rapamycin treatment had no apparent effect on most aging-associated changes on clinical chemistry parameters.

Shown are selected clinical chemistry results from the 16-month cohort (young control, n = 10; vehicle, n = 16; rapamycin, n = 19). (A) Plasma sodium concentration. (B) Plasma calcium concentration. (C) Plasma chloride concentration. (D) Total protein in plasma. (E) Plasma glucose. (F) Plasma α-amylase. Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font. See Supplemental Figures 17–19 for complete clinical chemistry findings from all cohorts.
Because steroid hormone concentrations are known to decline with age (37), we measured plasma corticosterone, androstenedione, and testosterone levels. Aging was associated with decreased androstenedione and/or testosterone plasma levels, which were not corrected by rapamycin treatment (Supplemental Tables 13–15).
Rapamycin increased rbc counts via aging-independent mechanisms and had no effect on elevated leukocyte and platelet numbers in aged mice.
Aging in C57BL/6J mice is known to be associated with altered cellular composition of the peripheral blood (35, 38). We therefore included basic hematological assessments of rapamycin- or vehicle-treated aged mice and young controls. Platelets and leukocytes were elevated in aged animals, without apparent effects of rapamycin on this phenotype (Figure 12, A and B). Our studies also revealed reduced rbc counts, associated with decreased mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH), in aged mice (Figure 12C and Supplemental Figures 21–23). We observed increased rbc distribution width (RDW) in aged animals, indicative of a higher degree of anisocytosis. Our observations are consistent with known aging effects on blood cell counts (35, 38). Notably, rapamycin-treated animals of the 16-month cohort showed erythrocyte counts indistinguishable from those of young controls (Figure 12C and Supplemental Figure 21). However, chronic rapamycin treatment was previously shown to increase rbc counts in young animals (i.e., apart from any aging-related changes) (39). To test for aging-independent effects of rapamycin on rbc counts in our setting, we analyzed the young mice chronically treated with rapamycin or vehicle; rapamycin also tended to increase erythrocyte counts in these animals (Figure 12D). Thus, the effects of rapamycin in aged animals are likely not linked to aging per se, but are instead aging-independent effects on erythrocyte production and/or turnover.
Figure 12. Rapamycin increased rbc counts.

(A–C) Hematology results for the 16-month cohort (young control, n = 10; vehicle, n = 16; rapamycin, n = 19). (A) wbc count. (B) Platelet count. (C) rbc count. (D) Effects of rapamycin on rbc count in young mice (control, n = 9; rapamycin, n = 8). Whisker plots display minimum, 25th percentile, median, 75th percentile, and maximum. P values and fit coefficients with 95% confidence intervals are shown; statistically significant differences (P < 0.05) are denoted by bold font. See Supplemental Figures 21–23 for complete hematology results.
Rapamycin treatment was associated with considerable nephro- and gonadotoxicity.
During assessment of histopathological aging phenotypes in kidneys and the male reproductive tract, we noted signs of considerable nephro- and gonadotoxicity (see Supplemental Results, Supplemental Figures 24–28, and Supplemental Tables 16–18). Additional side effects observed in rapamycin-treated animals were impaired glucose tolerance (Supplemental Figure 20) and hypercholesterolemia (Supplemental Figure 17).
Additional analyses.
Additional analyses revealed only marginal or no effects of aging on the respective measures (Supplemental Results, Supplemental Figures 29–39, and Supplemental Tables 19–23).
Rapamycin suppressed carcinogenesis in aging mice.
Rapamycin is well known to suppress the de novo formation of cancers, as well as corresponding precursor lesions, and also has inhibitory effects on established tumors (12–14). Accordingly, rapamycin has been found to extend lifespan and reduce tumor burden in mouse lines with a range of early-onset cancers (40, 41). Cancers are the leading cause of death in many mouse strains, including C57BL/6J and all strains in which rapamycin was previously shown to have longevity effects (9, 15), which indicates that rapamycin’s antineoplastic properties may underlie its longevity effects in mice. We therefore sought to determine whether rapamycin suppresses carcinogenesis in our aging mouse cohorts. We determined the number of animals presenting with cancers and/or precancerous lesions upon pathological assessment. Young controls were consistently devoid of cancers and precancerous lesions. We detected several cancers in aged mice, including hepatocellular carcinoma, lymphoma, histiocytic sarcoma, bronchoalveolar adenocarcinoma, and follicular adenocarcinoma of the thyroid, as well as precancerous lesions (pancreatic ductal metaplasia) (Supplemental Figure 40 and Supplemental Table 24). Rapamycin significantly reduced the proportion of aged mice presenting with cancers and/or precancerous lesions upon pathological assessment in the 16-month cohort (vehicle, 4 of 10; rapamycin, 0 of 15; P = 0.0166, Fisher exact test), but had no measurable effect in the 25-month cohort (vehicle, 1 of 5; rapamycin, 2 of 8; P = 1.0, Fisher exact test) or the 34-month cohort (vehicle, 1 of 5; rapamycin, 3 of 10; P = 1.0, Fisher exact test). Note that a considerable proportion of animals in the latter 2 cohorts died during the course of the study (of natural causes) and were therefore not available for pathological assessment at conclusion of our study (dropout due to death, 25-month vehicle, 55%; 25-month rapamycin, 38%; 34-month vehicle, 54%; 34-month rapamycin, 44%; 16-month vehicle, 20%; 16-month rapamycin, 5%). The interpretation of the cancer data from 25- and 34-month cohorts is therefore complicated by relatively small sample size and a selection bias toward animals that did not perish prior to pathological assessment (with cancers representing the main causes of death in C57BL/6J mice). Our data are in line with prior reports of delayed cancer-caused death in rapamycin-treated aging mice (9, 10) and support the view that lethal neoplastic disease is delayed in rapamycin-treated animals during the course of normal aging.
Discussion
Rapamycin was recently shown to increase lifespan in mice (9, 10). In principle, lifespan extension could have been related either to isolated effects on specific life-limiting pathologies, in particular cancers, or to cancer-suppressive effects of rapamycin, which are part of broader effects against aging in mice. In the present study, we tested whether rapamycin slows mammalian aging rates by assessing this compound’s effects on a wide range (one of the most comprehensive published to date) of functional and structural aging phenotypes in C57BL/6J mice. We observed a large number of aging phenotypes that were not improved by rapamycin in any apparent and measurable way (Table 2), which indicated that rapamycin did not prevent the signs and symptoms of aging in a universal fashion.
We also identified a number of aging phenotypes that were restored by rapamycin, at least partially (Table 1). It is possible that rapamycin modulates these traits by slowing aging-related processes; alternatively, rapamycin may influence these traits in a direct way, without actually modulating aging (i.e., aging-independent effects). To distinguish between these possibilities, we examined whether rapamycin affects the respective phenotypes in young mice. Traits that were similarly affected in young and aging cohorts would likely be influenced in a direct way, not via modulation of aging itself.
In a number of these cases, we observed similar effects of rapamycin in young and aging animals, which indicated that rapamycin acted in an aging-independent manner. For instance, rapamycin enhanced spatial learning and memory if chronically administered during aging, but it had similar effects in young mice, in line with another recent report that also demonstrated aging-independent enhancements of learning and memory by chronic rapamycin treatment (20). Consistent with a recent report by Miller and colleagues (10), we found rapamycin to increase exploratory activity in aged animals. However, rapamycin also increased exploratory activity in young mice in our study, indicative of another aging-independent effect, rather than an effect on aging per se. Chronic rapamycin treatment has been shown to elevate monoamine levels (dopamine, norepinephrine, and serotonin) in the midbrain, which may contribute to the improvements in learning and memory (20) and may also explain rapamycin’s stimulatory effect on exploratory activity (42).
We observed reduced rbc counts in aged mice that were elevated by rapamycin treatment. Consistent with a prior report of increased rbc counts by rapamycin treatment in young animals (39), our data showed a trend toward elevated rbc counts in treated young mice. Advanced age was associated with an increase in Treg (CD25+CD4+) frequency, a change that was counteracted by rapamycin in the 16-month cohort. However, rapamycin had similar effects on Tregs in young mice, indicative of further aging-independent drug effects.
Few of the tested effects of rapamycin in our dataset were seen only in aged mice, not in young animals (RER, fat mass, γδ T cells, and CD44hi T cells); however, previous reports have shown aging-independent effects of mTOR inhibition on CD44 expression in T lymphocytes and fat mass (30, 31, 34). Together, our present data and the considerations outlined above strongly support the view that the bulk of the rapamycin-modulated aging traits we observed were related to aging-independent effects of the drug and not caused by rapamycin’s effects on aging itself.
Our study has several limitations. We assessed only a single inbred mouse strain and gender (i.e., male C57BL/6J mice); it is possible that rapamycin has more effective healthspan effects in other strains and/or in female. Moreover, some of our analyses, particularly some of the count-based histopathological analyses in the older cohorts, are limited, as sample sizes were too low to draw definitive conclusions about possible effects of rapamycin or the absence thereof. We have therefore reported 95% confidence intervals of fit coefficients and odds ratios throughout in order to help assess the strength of the data. In cases with wide confidence intervals, larger sample sizes may be needed to extract possible effects of rapamycin.
A recent study by Wilkinson et al. (16) addressed the question of whether rapamycin slows aging by studying a number of age-related phenotypes in a population of genetically heterogeneous mice. In their study, rapamycin improved the following age-associated phenotypes: hepatic lipidosis, atypical nuclei in cardiomyocytes, endometrial cystic hyperplasia, adrenal tumors, changes in mechanical tendon properties, and reduced motor activity levels.
Comparing our dataset with that of Wilkinson et al., a few phenotypes assessed by both studies show clear-cut effects. There is agreement on adverse testicular side effects of rapamycin treatment in exposed males, and both studies report increased motor activity in rapamycin-treated aged animals. In contrast to Wilkinson et al., we found no worsening of cataracts under rapamycin treatment. Discrepancies in findings may be related to different genetic backgrounds used — C57BL/6J mice (present study) versus a stock of genetically heterogeneous mice (16) — or technical differences in the assessment of lens densities — Scheimpflug imaging with computer-assisted automated measurements of lens density (present study) versus traditional slit lamp examination with investigator-based rating of lens densities (16).
Rapamycin’s effects on aging traits in the Wilkinson et al. study were not tested for the possible aging-independency of the effects (16). The effects on liver lipidosis, for example, could be explained by aging-independent drug effects, given that mTORC1 signaling plays an important role in lipid biogenesis (43). Aging-independent effects of rapamycin could also explain the inhibition of endometrial hyperplasia: inactivating mutations in Tsc2, an important mTOR repressor, were shown to result in endometrial hyperplasia, which could then be suppressed by pharmacological mTOR inhibition (44). In the present study, we showed that rapamycin had stimulatory effects on exploratory activity, not only if applied for extended periods during aging, but also if applied during young adulthood, which indicates that this effect of rapamycin is aging independent. It will therefore be important to address which of the phenotypes in the Wilkinson et al. study (16) may be related to aging-independent effects of rapamycin.
Genetic deletion of S6k1, a downstream effector in the mTORC1 signaling pathway, was found by Selman et al. to extend lifespan in female C57BL/6J mice (45). Moreover, the authors demonstrated that 600-day-old S6k1–/– mutants showed improvement in several aging-associated phenotypes, such as impaired motor coordination, reduced exploratory activity, reduced bone volume, and elevated CD44+ T cell counts (45). This analysis did not include a comparison of young adult S6k1–/– mutants with the respective littermate controls; thus, it is currently unknown whether the observed effects are due to slowed aging or represent aging-independent consequences of S6k1 deletion. In fact, there is overlap between the age-related phenotypes reported by Selman et al. to be influenced by S6k1 deletion (45) and those we found to be influenced by chronic rapamycin treatment, namely, exploratory activity and CD44hi T cell count. As discussed above, the effect on exploratory activity in our study reflected an aging-independent consequence of rapamycin treatment.
Our data indicated that the longevity effects of rapamycin are not associated with broad modulation of aging. Therefore, the present results best fit a model in which rapamycin has limited effects on mammalian aging itself and extends lifespan via inhibition of specific pathologies that limit life in aged animals. Neoplastic disease is an obvious suspect, because malignant neoplasias are the leading cause of death in many mouse strains, including all strains in which longevity effects have been demonstrated for rapamycin (9, 15). In the recent study by Miller et al. (using genetically heterogeneous mice; ref. 10), 95.6% of control animals and 88.6% of rapamycin-treated animals had neoplastic disease as their likely cause of death. Both vehicle- and rapamycin-treated animals eventually died of cancers, but controls did so at younger ages (10).
mTOR-related cell signaling plays an important role in multiple steps of carcinogenesis, including the establishment of precancerous lesions, their progression to cancer, and subsequent cancer growth (12–14). Accordingly, rapamycin has inhibitory effects on de novo cancer formation and also inhibits the progression of malignant neoplastic disease by suppressing cancer growth and, additionally, modifying host responses to cancer (e.g., angiogenesis) (12–14). The present study demonstrated that cancers and precancerous lesions were less frequent in rapamycin-treated mice of the 16-month cohort than in corresponding age-matched controls. Our data, together with known effects of rapamycin on carcinogenesis and the NIA Interventions Testing Program’s cause-of-death data in aging rapamycin-treated mice mentioned above (10), support a model wherein rapamycin delays the onset and/or slows the progression of eventually lethal neoplastic disease. Possible effects of rapamycin on non-neoplastic causes of death are less likely to have contributed to the drug’s reported longevity effects (having accounted for a small fraction of deaths in these populations; see above). However, future studies should test whether rapamycin also extends lifespan in mouse strains (or other mammalian species) that die predominantly due to non-neoplastic pathology.
In summary, our findings confirmed the extension of mammalian lifespan by rapamycin. However, rapamycin had limited effects on a large number of murine aging phenotypes. Although age-related changes in several traits were opposed by rapamycin, this was often due to aging-independent drug effects (i.e., similar effects of rapamycin were observed in young mice), which indicates that rapamycin influenced these traits in a direct way and not by slowing aging. Our data therefore seem to largely dissociate rapamycin’s longevity effects in mice from effects on aging itself.
Methods
Mice.
Male C57BL/6J Rj mice were obtained from Janvier and randomly assigned to rapamycin or vehicle treatment groups. We had 3 aging treatment cohorts, distinguished by age of treatment onset. Treatment was initiated in young adulthood (approximately 4 months of age; n = 20 per treatment group), in midlife (13 months of age; n = 21 per treatment group), or late in life (20–22 months of age; n = 27 per treatment group). Animals were treated for >12 months before analyses started at 16, 25, and 34 months of age, respectively. Animals were kept on their respective diet for the entirety of the study. In all cases, the experimenter was blinded to the animal group assignments.
Initial behavioral studies were performed at the DZNE/Bonn (object place recognition, Morris water maze, and context fear conditioning). Animals were subsequently shipped to the German Mouse Clinic (46, 47) for additional analyses (16-month vehicle, n = 19; 16-month rapamycin, n = 20; 25-month vehicle, n = 12; 25-month rapamycin, n = 16; 34-month vehicle, n = 11; 34-month rapamycin, n = 16). Cohorts were analyzed separately, each side by side with a group of young adult (3–6 months old) male C57BL/6J Rj mice (Janvier) kept on vehicle control diet (n = 10). The following analyses were performed, in the order described (46, 47): open field, modified SHIRPA, rotarod, hot plate test, indirect calorimetry, NMR-based body composition analysis, awake echocardiography, Scheimpflug imaging, optical coherence tomography, virtual drum vision test, clinical chemistry, hematology, FACS analysis of peripheral blood leukocytes, Bioplex ELISA (Ig concentrations), steroid metabolism, pathology.
Parameters that were altered by aging and that appeared to be rescued by rapamycin (i.e., aging and rapamycin had oppositional effects on the parameters) were further assessed in groups of young adult mice in order to test for aging-independent effects of rapamycin. For the water maze experiment, we used male C57BL/6J Rj mice (n = 15 per group) that had been on rapamycin or vehicle treatment for 6 weeks prior to the experiment (6 months of age at start of treatment). For all other experiments, 12-week-old male C57BL/6J Rj mice were placed on rapamycin (n = 8) or vehicle control diet (n = 9) for a 3-month period prior to analysis.
Mice were housed under SPF conditions in groups of 2–5 per cage. We kept animals on a 12-hour light/12-hour dark cycle. Mice received water and food ad libitum. Local and federal regulations regarding animal welfare were followed.
Rapamycin treatment.
Rapamycin treatment was essentially as previously described (9). In brief, rapamycin was purchased from LC Laboratories and microencapsulated by Southwest Research Institute using a spinning disk atomization coating process, with Eudragit S100 (Röhm Pharma) as the coating material. TestDiet provided Purina 5LG6 food containing encapsulated rapamycin or just the coating material (vehicle control). The encapsulated rapamycin was administered at 14 mg/kg food.
HPLC-based detection of rapamycin.
Rapamycin concentrations were measured in whole blood and tissue lysates prepared from brain, kidney, liver, muscle, heart, and visceral fat of 22-month-old animals that had been on rapamycin or vehicle diet for 5 months. After protein precipitation, rapamycin was separated from matrix components by reversed-phase HPLC on a C8 column using the Agilent 1100 binary pump system. After electrospray ionization, selected ion fragments were detected and quantified in the API4000 triple-quadrupole tandem MSMS (Applied Biosystems/Sciex).
Object place recognition.
After handling and habituation to the empty test arena, mice were subjected to a single 16-minute training session, during which they were allowed to explore freely 2 identical objects (small glass bottles) that were placed in defined locations of the test arena. On the next day, a 6-minute test session was performed, during which the position of one of the objects was changed, while the position of the other remained unaltered. An automated tracking system (Ethovision XT; Noldus) was used to monitor and record the behavior of the animals. The time the animals spent exploring the object in the novel location and the known location during the test was hand-scored by an experienced observer from the videotape.
Hidden version of the Morris water maze.
The water maze was performed essentially as previously described (48). Each animal received 6 daily training trials in the hidden version of the Morris water maze (in blocks of 2 consecutive trials) for 5 consecutive days. Training trials were completed when mice climbed on the escape platform or when 1 minute had elapsed, whichever came first. To evaluate the accuracy with which the animals had learned the position of the escape platform, we performed a probe trial after completion of training. We determined the time that mice spent searching in the target quadrant (which previously contained the escape platform) or the other quadrants during the probe trial. Additionally, we analyzed the number of crossings of the exact target location (i.e., where the platform was during training) and compared it with crossings of analogous positions in the other quadrants.
Context fear conditioning.
A near infrared video fear conditioning system (Med Associates) was used to test context fear conditioning. The training session was 184 seconds total duration; 2-second, 0.75-mA shocks were delivered via the metal grid floor of the chambers after 60 and 122 seconds. A single test session was given on the next day, during which animals were placed in the chamber for 180 seconds to record behavior. To evaluate conditioned fear, we calculated activity suppression ratios for each animal as (activity during test)/(activity during test + activity during baseline).
Open field.
The test apparatus (ActiMot; TSE) was a transparent and infrared light–permeable acrylic test box (45.5 cm × 45.5 cm × 39.5 cm internal measurements) equipped with 2 pairs of light-beam strips. Animals were allowed to freely explore the test arena for 20 minutes. Illumination levels were set to approximately 150 lux in the corners and 200 lux in the middle of the test arena. Parameters analyzed were total distance traveled and number of rearings.
Modified SHIRPA.
We used a modification of the SHIRPA screen (49) to assess 23 parameters related to neurological functions and general health as previously described ( http://www.har.mrc.ac.uk/services/phenotyping/neurology/shirpa.html) (46, 47).
Rotarod.
An accelerating rotarod (Bioseb) was used to measure motor coordination, balance, and motor learning abilities (50). Mice were placed on the rotarod, and the rod rotations were subsequently accelerated from 4 to 40 rpm during the 5-minute trial period. Trials were terminated when animals fell off the rod or showed passive cycling or when 5 minutes had elapsed, whichever came first. Animals were given 3 trials, with intertrial intervals of 15 minutes. Latencies to fall were analyzed across groups using linear mixed-effects models that consider effects of body weight variation.
Hot plate test.
Mice were placed on a metal surface maintained at 52°C ± 0.2°C (hot plate system; TSE). Locomotion on the hot plate was constrained by a circular Plexiglas wall (20 cm high, 18 cm diameter). Mice remained on the plate until they performed 1 of 3 behaviors regarded as indication of a painful sensation: hindpaw licking, hindpaw shake/flutter, or jumping. To avoid tissue injury, a 30-second cutoff time was used.
Grip strength.
A grip strength meter system (Bioseb) was used to assess grip strength in rapamycin- or vehicle-treated aged mice as well as young controls. Mice were allowed to hold on to the grid with either 2 or 4 paws (for 2-paw and 4-paw grip strength measurements, respectively). Each mouse was given 3 trials over the course of 1 minute, and average values were used to represent the grip strength of an individual mouse.
Echocardiography.
Echocardiography on the 16- and 25-month cohorts was performed at the German Mouse Clinic in Munich. Left ventricular function was evaluated by transthoracic echocardiography, using a Vevo 2100 Imaging System (Visual Sonics) with a 30-MHz probe. Examinations in Munich were performed on conscious animals. Left ventricular parasternal short- and long-axis views were obtained in B-mode imaging, and left ventricular parasternal short-axis views were obtained in M-mode imaging at the papillary muscle level. The short-axis M-mode images were used to measure left ventricular end-diastolic internal diameter (LVEDD), left ventricular end-systolic internal diameter (LVESD), diastolic and systolic septal wall thickness (SWT), and diastolic and systolic posterior wall thickness (PWT) in 3 consecutive beats, according to the American Society of Echocardiography leading edge method (51).
Cardiac Doppler-Echocardiography in Münster was performed on a specialized ultrasound system (VEVO 2100; VisualSonics). In Münster, another cohort was assessed: 26-month-old mice in which rapamycin or vehicle treatment had been initiated at 13 months of age, and 3- to 4-month-old young controls. Examinations were performed as previously described (52). In brief, following induction of anesthesia with isoflurane (2.0% isoflurane, 98% O2), examinations were performed with the system’s 30-MHz linear probe. ECG-triggered parasternal short-axis views were obtained together with a parasternal long-axis view. Routinely pulsed-wave Doppler signals of the velocity time integral of the mitral valve inflow, the aortic outflow, and the pulmonic outflow were obtained. Ultrasound image analyses were performed according to the American Society of Echocardiography (ASE) leading edge method (51) using the VEVO 2100 implemented image analyses software.
Scheimpflug imaging.
Images of corneas and lenses were taken with the Pentacam digital camera system (Oculus). Mice were held on a platform such that the vertical light slit (light source: LEDs, 475 nm) was orientated toward the middle of the eyeball. Distance between eye and camera was adjusted with the help of the software provided in order to guarantee optimal focus. Subsequently, measurements were started manually. Mean density across the lens was quantified with the densitometry tool provided. In addition to the quantitative assessment of lens density, we examined qualitatively the morphology of cornea and lens.
Optical coherence tomography.
Eye fundus and retina were analyzed with a Spectralis OCT (Heidelberg Engineering) modified with a 78-diopter double aspheric lens (Volk Optical) fixed directly to the outlet of the device. A contact lens with a focal length of 10 mm (Roland Consult) was applied to the eye of the mouse with a drop of methylcellulose (Methocel 2%; OmniVision). Eyes were treated with 1% atropine to ensure pupil dilation. Images were taken as described previously (53). The parameters evaluated were number of main blood vessels (fundus), retinal thickness, and morphology of retinal layers.
Virtual drum vision test.
Vision tests were performed using a virtual optomotor system (Cerebral Mechanics) as described previously (54). Briefly, a rotating cylinder covered with a vertical sine-wave grating was calculated and drawn in virtual 3-dimensional space on 4 computer monitors facing to form a square. Visually unimpaired mice track the grating with reflexive head and neck movements (head-tracking). Vision threshold was quantified by a randomized simple staircase test. Rotation speed and contrast were set to 12.0 d/s and 100%, respectively.
Blood collection.
Blood samples were taken from isoflurane-anesthetized mice by puncturing the retrobulbar sinus with nonheparinized glass capillaries (Neolab). Blood samples collected from ad libitum–fed mice were divided into 2 portions. The major portion was collected in a heparinized tube (Li-heparin, no. 078028; KABE). Plasma from this portion was used for Ig concentration measurement, clinical chemistry assessment, and steroid level measurement. The cell pellet was used for FACS-based assessment of cellular immune aging phenotypes (i.e., quantitative assessment of leukocyte populations). Another smaller portion was collected (using the same capillary) in an EDTA-coated tube (no. 078035; KABE) and used for hematological assessments.
Clinical chemistry.
Clinical chemistry assessment was performed using a Beckman-Coulter AU 480 autoanalyzer and reagents from Beckman-Coulter (except free fatty acids, which were measured using kit NEFA-HR2 from Wako Chemicals, and glycerol, which was measured using a kit from Randox Laboratories) as previously described (46, 47).
Glucose tolerance test.
For the intraperitoneal glucose tolerance test, food was withdrawn for 16–18 hours overnight. For determining the basal blood glucose level of unfed mice, the tip of the tail was scored using a sterile scalpel blade, and a small drop of blood was analyzed with the Accu-Chek Aviva glucose analyzer (Roche). Mice were then injected intraperitoneally with glucose (2 g/kg body mass) using a sterile 20% glucose solution (B. Braun). 15, 30, 60, and 120 minutes after glucose injection, additional blood samples (1 drop each) were collected and used to determine blood glucose levels.
Hematology.
EDTA-blood was used to measure basic hematological parameters with a blood analyzer (ABC-Blutbild-Analyzer; Scil Animal Care Co.) as previously described (46, 47).
FACS-based analysis of peripheral blood leukocytes.
These analyses were performed as previously described (46, 47). In brief, peripheral blood leukocytes were stained with fluorescence-conjugated monoclonal antibodies (BD Biosciences — Pharmingen), and samples were analyzed using a 3-laser, 10-color flow cytometer (LSRII, BD; Gallios, Beckman Coulter). The following main leukocyte (CD45+) populations were examined: T cells (CD3+), CD4+ T cells, CD8+ T cells, γδ T cells (CD3+γδ_TCR+), Tregs (CD4+CD25+), NK cells (NKp46+NK1.1+CD5–), B cells (CD19+B220+), NK T cells (NKp46+NK1.1+CD5+), monocytes (non-NK, nongranulocyte, CD11b+). Additionally analyzed subsets included T cells coexpressing CD62L or CD44, B cells coexpressing IgD and/or MHCII, and NK cells expressing CD11b.
Ig concentrations.
Plasma levels of IgM, IgG1, IgG2a, IgG2b, IgG3, and IgA were determined simultaneously in the same sample using a bead-based assay with monoclonal anti-mouse antibodies conjugated to beads of different regions (Biorad) and a Bioplex reader (Biorad). Plasma was also analyzed for total IgE, using a classical immunoassay isotype-specific sandwich ELISA.
Steroid metabolism.
Steroids were quantified in plasma using a validated high-throughput LC-MS/MS method that combines HPLC (Shimadzu Prominence) and the use of a Triple Quadrupol Mass Spectrometer (4000 QTrap; ABSciex). Sample preparation and LC-MS/MS measurements were as described previously (55).
Indirect calorimetry and body composition analysis.
Metabolic assessments were performed as described previously (46, 47). In brief, each mouse was placed individually in a metabolic cage for a period of 21 hours with free access to food and water. High-precision CO2 and O2 sensors measured the difference in CO2 and O2 concentrations in air volumes flowing through individual cages. The following parameters were calculated from these measures: O2 consumption, RER (calculated as VCO2/VO2), heat production (HP; calculated as [4.44 + (1.43 × RER)] × VO2, and expressed in mW). Additionally, we determined total daily food intake and rectal body temperature, as well as physical activity (distance travelled and number of rearings) using infrared light beam frames set up around the cages.
Pathology.
Mice were sacrificed with CO2, then analyzed macroscopically and weighed ( http://eulep.pdn.cam.ac.uk/Necropsy_of_the_Mouse/index.php). After complete dissection, all organs were fixed in 4% buffered formalin and embedded in paraffin for histological examination. 4-μm-thick sections from skin, heart, muscle, lung, cerebrum, cerebellum, thymus, spleen, cervical lymph nodes, trachea, thyroid and parathyroid glands, adrenal glands, esophagus, stomach, intestine, liver, gall bladder, pancreas, kidneys, reproductive organs, and urinary bladder were cut and stained with H&E, Periodic acid Schiff stain (PAS), Elastica van Gieson (EvG; Weigert’s stain), Movat pentachrome, and/or Congo Red (for which 8-μm-thick sections were used). Slides were digitalized using a virtual slide system (NanozoomerHT2.0; Hamamatsu). All histopathological sections were evaluated by a board-certified pathologist. In addition, computer-assisted analyses — using automated image segmentation software, such as Definiens Tissue Studio (56) and CellProfiler (57) — were carried out to measure quantitatively specific aspects of age-related tissue changes. For immunohistochemical assessment of DCX expression in the dentate gyrus, we fixed brains in 4% buffered formalin, performed a dehydration step in 30% sucrose, and generated 40-μm coronal section series with a sliding microtome (Leica). DCX expression was detected using a primary antibody against DCX (goat anti-DCX, 1:100; Santa Cruz Biotechnology), a biotinylated secondary antibody (biotinylated donkey anti-goat IgG, 1:500; Dianova), and incubation in avidin-biotin peroxidase complex (ABC Elite; Vector) and diaminobenzidine (DAB; Roche). Anti–phospho–histone H2A.X (Ser139) antibody (clone JBW301, catalog no. 05-636, 1:500; Millipore) was used after heat-induced antigen retrieval with citrat buffer (pH 6) for 20 minutes. Immunoreaction was carried out in the automated immunostainer Discovery XT (Roche-Ventana) using the “Momab” kit to prevent background staining according to the manufacturer’s instructions. DAB as chromogen and counterstaining using Mayer’s acid hemalum (both Roche-Ventana) was also performed in the Discovery autostainer.
Western blot detection of carbonylated proteins.
We measured protein carbonylation in liver lysates using the OxyBlot Protein Oxidation Detection Kit (Millipore) according to the manufacturer’s instructions. In brief, protein carbonyl groups were derivatized by reaction with 2,4-dinitrophenylhydrazine and analyzed by SDS-PAGE followed by immunoblotting for DNP groups and development using chemiluminescence.
ELISA-based detection of 8-hydroxydeoxyguanosine (8-OHdG).
We measured the abundance of 8-OHdG in DNA (isolated from rapamycin- or vehicle-treated 26-month-old mouse liver as well as from young control liver) using the OxiSelect Oxidative DNA Damage ELISA Kit (Cell Biolabs) according to the manufacturer’s instructions.
Statistics.
Unless stated otherwise, data were analyzed by fitting them with a linear fixed-effects model against the factors of age (young vs. old) and treatment (rapamycin vs. vehicle). Results from these analyses are typically reported as P value, fit coefficient, and 95% confidence interval of fit coefficient. For analysis of count-based data (such as much of the histopathological data), we used regular or ordered logistic models as appropriate. Analysis of data from young animals treated with rapamycin or vehicle was by unpaired 1-tailed t test, unless stated otherwise. A P value less than 0.05 was considered statistically significant.
Study approval.
The present study was approved by “Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen” (Recklinghausen, Germany) as well as “Regierung von Oberbayern” (Munich, Germany).
Supplementary Material
Acknowledgments
We thank Reinhard Seeliger, Anke Bettenbrock, Jan Einicke, Ralf Fischer, Elfi Holupirek, Maria Kugler, Jacqueline Müller, Elenore Samson, Florian Schleicher, Waldemar Schneider, Ann-Elisabeth Schwarz, Nadine Senger, Yvonne Sonntag, Bettina Sperling, Alida Theil, Lucie Thurmann, Susanne Wittich, Anja Wohlbier, Christoph Möhl, and Mary Vlassopoulou as well as the GMC and DZNE animal caretaker teams (Manuela Huber, Boris Schön, Heidi Marr, Annica Miedl, Tina Reichelt, Michael Gerstlauer, Renate Huber, Horst Wenig, Jacqueline Agor, and Stefan Hansen). This work has been funded by the German Federal Ministry of Education and Research to the German Center for Diabetes Research (DZD e.V.), to the German Center for Neurodegenerative Diseases (DZNE e.V.), to the GMC (NGFN-Plus grants 01GS0850, 01GS0851, 01GS0852, 01GS0853, 01GS0854, GS0868, 01GS0869; Infrafrontier grant 01KX1012) and to the German Center for Vertigo and Balance Disorders (grant 01 EO 0901), by the Initiative and Networking Fund of the Helmholtz Association in the framework of the Helmholtz Alliance for Mental Research in an Ageing Society (HA-215), by the Helmholtz Alliance ICEMED (Initiative and Network Fund of the Helmholtz Association), by DFG Sonderforschungsbereich 655 and 656, and by IZKF Münster PIX ECHO.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(8):3272–3291. doi:10.1172/JCI67674.
See the related Commentary beginning on page 3204.
References
- 1.Olshansky SJ, Perry D, Miller RA, Butler RN. In pursuit of the longevity dividend. Scientist. 2006;20(3):28–36. [Google Scholar]
- 2.Kapahi P, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11(6):453–465. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14(10):885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426(6967):620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 5.Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131(16):3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- 6.Kaeberlein M, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310(5751):1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 7.Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20(2):174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller RA, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011;66(2):191–201. doi: 10.1093/gerona/glq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anisimov VN, et al. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011;10(24):4230–4236. doi: 10.4161/cc.10.24.18486. [DOI] [PubMed] [Google Scholar]
- 12.Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19(56):6680–6686. doi: 10.1038/sj.onc.1204091. [DOI] [PubMed] [Google Scholar]
- 13.Garber K. Rapamycin’s resurrection: a new way to target the cancer cell cycle. J Natl Cancer Inst. 2001;93(20):1517–1519. doi: 10.1093/jnci/93.20.1517. [DOI] [PubMed] [Google Scholar]
- 14.Kopelovich L, Fay JR, Sigman CC, Crowell JA. The mammalian target of rapamycin pathway as a potential target for cancer chemoprevention. Cancer Epidemiol Biomarkers Prev. 2007;16(7):1330–1340. doi: 10.1158/1055-9965.EPI-07-0045. [DOI] [PubMed] [Google Scholar]
- 15.Blackwell BN, Bucci TJ, Hart RW, Turturro A. Longevity, body weight, and neoplasia in ad libitum-fed and diet-restricted C57BL6 mice fed NIH-31 open formula diet. Toxicol Pathol. 1995;23(5):570–582. doi: 10.1177/019262339502300503. [DOI] [PubMed] [Google Scholar]
- 16.Wilkinson JE, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11(4):675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majumder S, et al. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell. 2012;11(2):326–335. doi: 10.1111/j.1474-9726.2011.00791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Napoli KL, Wang ME, Stepkowski SM, Kahan BD. Distribution of sirolimus in rat tissue. Clin Biochem. 1997;30(2):135–142. doi: 10.1016/S0009-9120(96)00157-9. [DOI] [PubMed] [Google Scholar]
- 19.Murphy GG, Rahnama NP, Silva AJ. Investigation of age-related cognitive decline using mice as a model system: behavioral correlates. Am J Geriatr Psychiatry. 2006;14(12):1004–1011. doi: 10.1097/01.JGP.0000209405.27548.7b. [DOI] [PubMed] [Google Scholar]
- 20.Halloran J, et al. Chronic inhibition of mTOR by rapamycin modulates cognitive non-cognitive components of behavior throughout lifespan in mice. Neuroscience. 2012;223:102–113. doi: 10.1016/j.neuroscience.2012.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kempermann G, Kuhn HG, Gage FH. Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci. 1998;18(9):3206–3212. doi: 10.1523/JNEUROSCI.18-09-03206.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romero-Suarez S, et al. Muscle-specific inositide phosphatase (MIP/MTMR14) is reduced with age and its loss accelerates skeletal muscle aging process by altering calcium homeostasis. Aging (Albany NY). 2010;2(8):504–513. doi: 10.18632/aging.100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harding JJ. Aging of Organs and Systems. 2003. Aging of the eye. In: Aspinall R, ed. pp. 179–200. Dordecht, The Netherlands: Kluwer Academic Publishers; [Google Scholar]
- 24.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19(7):213–220. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulckaen H, et al. Low-dose aspirin prevents age-related endothelial dysfunction in a mouse model of physiological aging. Am J Physiol Heart Circ Physiol. 2008;294(4):H1562–H1570. doi: 10.1152/ajpheart.00241.2007. [DOI] [PubMed] [Google Scholar]
- 26.Zhang YM, et al. Expression of tissue inhibitor of matrix metalloproteinase-1 in aging of transgenic mouse liver. Chin Med J (Engl). 2006;119(6):504–509. [PubMed] [Google Scholar]
- 27.Kmiec Z, Kotlarz G, Smiechowska B, Mysliwski A. The effect of fasting and refeeding on thyroid follicule structure and thyroid hormone levels in young and old rats. Arch Gerontol Geriatr. 1998;26(2):161–175. doi: 10.1016/S0167-4943(97)00040-X. [DOI] [PubMed] [Google Scholar]
- 28.Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007;67(3):959–966. doi: 10.1158/0008-5472.CAN-06-3524. [DOI] [PubMed] [Google Scholar]
- 29.Haines DC, Chattopadhyay S, Ward JM. Pathology of aging B6;129 mice. Toxicol Pathol. 2001;29(6):653–661. doi: 10.1080/019262301753385988. [DOI] [PubMed] [Google Scholar]
- 30.Houde VP, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59(6):1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deblon N, et al. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br J Pharmacol. 2012;165(7):2325–2340. doi: 10.1111/j.1476-5381.2011.01716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds MA, Ingram DK, Talan M. Relationship of body temperature stability to mortality in aging mice. Mech Ageing Dev. 1985;30(2):143–152. doi: 10.1016/0047-6374(85)90003-X. [DOI] [PubMed] [Google Scholar]
- 33.Henson SM, Aspinall R. Aging of Organs and Systems. 2003. Aging and the immune system. In: Aspinall R, ed. pp. 225–242. Dordecht, The Netherlands: Kluwer Academic Publishers; [Google Scholar]
- 34.Zhang S, et al. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood. 2011;117(4):1228–1238. doi: 10.1182/blood-2010-05-287821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frith CH, Suber RL, Umholtz R. Hematologic and clinical chemistry findings in control BALB/c and C57BL/6 mice. Lab Anim Sci. 1980;30(5):835–840. [PubMed] [Google Scholar]
- 36.Larsen JL, et al. Tacrolimus and sirolimus cause insulin resistance in normal sprague dawley rats. Transplantation. 2006;82(4):466–470. doi: 10.1097/01.tp.0000229384.22217.15. [DOI] [PubMed] [Google Scholar]
- 37.Coquelin A, Desjardins C. Luteinizing hormone and testosterone secretion in young and old male mice. Am J Physiol. 1982;243(3):E257–E263. doi: 10.1152/ajpendo.1982.243.3.E257. [DOI] [PubMed] [Google Scholar]
- 38.Leuenberger HG, Kunstyr I. Gerontological data of C57BL/6J mice. II. Changes in blood cell counts in the course of natural aging. J Gerontol. 1976;31(6):648–653. doi: 10.1093/geronj/31.6.648. [DOI] [PubMed] [Google Scholar]
- 39.Diekmann F, et al. mTOR inhibition and erythropoiesis: microcytosis or anaemia? Nephrol Dial Transplant. 2012;27(2):537–541. doi: 10.1093/ndt/gfr318. [DOI] [PubMed] [Google Scholar]
- 40.Anisimov VN, et al. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010;176(5):2092–2097. doi: 10.2353/ajpath.2010.091050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komarova EA, et al. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/– mice. . Aging (Albany NY). 2012;4(10):709–714. doi: 10.18632/aging.100498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fink JS, Smith GP. Mesolimbicocortical dopamine terminal fields are necessary for normal locomotor and investigatory exploration in rats. Brain Res. 1980;199(2):359–384. doi: 10.1016/0006-8993(80)90695-2. [DOI] [PubMed] [Google Scholar]
- 43.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19(22):R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCampbell AS, Harris HA, Crabtree JS, Winneker RC, Walker CL, Broaddus RR. Loss of inhibitory insulin receptor substrate-1 phosphorylation is an early event in mammalian target of rapamycin-dependent endometrial hyperplasia and carcinoma. Cancer Prev Res (Phila). 2010;3(3):290–300. doi: 10.1158/1940-6207.CAPR-09-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selman C, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326(5949):140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuchs H, et al. Mouse phenotyping. Methods. 2011;53(2):120–135. doi: 10.1016/j.ymeth.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 47.Gailus-Durner V, et al. Systemic first-line phenotyping. Methods Mol Biol. 2009;530:463–509. doi: 10.1007/978-1-59745-471-1_25. [DOI] [PubMed] [Google Scholar]
- 48.Heinen M, Hettich MM, Ryan DP, Schnell S, Paesler K, Ehninger D. Adult-onset fluoxetine treatment does not improve behavioral impairments and may have adverse effects on the Ts65Dn mouse model of Down syndrome. Neural Plast. 2012;2012:467251. doi: 10.1155/2012/467251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8(10):711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- 50.Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1968;259(2):211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- 51.Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation. 1978;58(6):1072–1083. doi: 10.1161/01.CIR.58.6.1072. [DOI] [PubMed] [Google Scholar]
- 52.Stypmann J, Engelen MA, Troatz C, Rothenburger M, Eckardt L, Tiemann K. Echocardiographic assessment of global left ventricular function in mice. Lab Anim. 2009;43(2):127–137. doi: 10.1258/la.2007.06001e. [DOI] [PubMed] [Google Scholar]
- 53.Fischer MD, et al. Noninvasive, in vivo assessment of mouse retinal structure using optical coherence tomography. PLoS One. 2009;4(10):e7507. doi: 10.1371/journal.pone.0007507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci. 2004;45(12):4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- 55.Haller F, Prehn C, Adamski J. Nat Protoc. Quantification of steroids in human mouse plasma using online solid phase extraction coupled to liquid chromatography tandem mass spectrometry. doi: 10.1038/nprot.2010.22 . [Google Scholar]
- 56. Sommer C, Straehle C, Köthe U, Hamprecht FA. 8th IEEE International Symposium on Biomedical Imaging. March 30–April 2, 2011; Chicago, Illinois, USA. [Google Scholar]
- 57.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler: free, versatile software for automated biological image analysis. Biotechniques. 2007;42(1):71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 58.Li H, Pauza CD. Rapamycin increases the yield and effector function of human gammadelta T cells stimulated in vitro. Cancer Immunol Immunother. 2011;60(3):361–370. doi: 10.1007/s00262-010-0945-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.