

Abstract
Nephrotic syndrome (NS) is divided into steroid-sensitive (SSNS) and -resistant (SRNS) variants. SRNS causes end-stage kidney disease, which cannot be cured. While the disease mechanisms of NS are not well understood, genetic mapping studies suggest a multitude of unknown single-gene causes. We combined homozygosity mapping with whole-exome resequencing and identified an ARHGDIA mutation that causes SRNS. We demonstrated that ARHGDIA is in a complex with RHO GTPases and is prominently expressed in podocytes of rat glomeruli. ARHGDIA mutations (R120X and G173V) from individuals with SRNS abrogated interaction with RHO GTPases and increased active GTP-bound RAC1 and CDC42, but not RHOA, indicating that RAC1 and CDC42 are more relevant to the pathogenesis of this SRNS variant than RHOA. Moreover, the mutations enhanced migration of cultured human podocytes; however, enhanced migration was reversed by treatment with RAC1 inhibitors. The nephrotic phenotype was recapitulated in arhgdia-deficient zebrafish. RAC1 inhibitors were partially effective in ameliorating arhgdia-associated defects. These findings identify a single-gene cause of NS and reveal that RHO GTPase signaling is a pathogenic mediator of SRNS.
Introduction
Nephrotic syndrome (NS) is caused by malfunction of the kidney glomerular filter, resulting in proteinuria, hypoalbuminemia, and edema. NS is classified by its response to steroid treatment into steroid-sensitive nephrotic syndrome (SSNS) and steroid-resistant nephrotic syndrome (SRNS) categories. SSNS represents one of the most frequent kidney diseases and constitutes 80% of all childhood NS. Histologically, it presents as minimal change nephrotic syndrome (MCNS). Very little is known about its primary causes, disease processes, or mechanisms of treatment. SRNS is mostly refractory to therapy and leads to end-stage kidney disease (ESKD) within a few years of onset, requiring renal replacement therapy for survival. It causes about 15% of all ESKD in children (1), is considered one of the most intractable kidney diseases, and has a 30% recurrence risk in renal transplants, leading again to ESKD. Histologically, SRNS presents as focal segmental glomerulosclerosis (FSGS) or as the early-onset developmental variant, diffuse mesangial sclerosis (DMS) (2). There is clinical overlap between SSNS and SRNS, as some individuals with SSNS and MCNS may later develop SRNS with FSGS histology.
The discovery of single-gene causes of SRNS has generated the first insights into its pathogenesis by revealing that the defective proteins are essential for glomerular podocyte function (3). Podocytes constitute the outer epithelial layer of the 3-layered capillaries in the glomerular filter, which also consists of the glomerular basement membrane (GBM) and the endothelial cell (EC) layer (4). Podocytes are neuron-like cells that extend multiple tubulin-based primary processes, which branch off actin-based foot processes. The foot processes interdigitate with those of neighboring podocytes and form between them the glomerular slit diaphragm, which is critical for the filtering process and the retention of protein in the bloodstream. Integrin molecules in the “sole plate” of the foot processes tether the foot processes to the GBM. Loss of foot processes and slit membrane integrity causes glomerular protein leakage and NS. The products of single-gene causes of SRNS play a role in cell-cell signaling at the podocyte slit membrane (NPHS1, NPHS2, CD2AP, PTPRO/GLEPP1) (5–8) for regulation of the foot process actin network (ACTN4 and INF2) (9, 10) or in foot process-GBM interaction (LAMB2 and ITGA3) (11–14).
While the disease mechanisms of NS are still obscure, features of podocyte cell migration were recently implicated in its pathogenesis. In this concept, a sessile podocyte phenotype represents the physiologic mode, whereas a migratory phenotype represents the nephrotic mode (15). Actin remodeling by members of the RHO family of small GTPases (referred to hereafter as RHO GTPases) regulates the 2 modes, where active RHOA and increased actin remodeling are seen in the nephrotic state (16). Conversely, inactivation of RHO GTPases may cause proteinuria in animal models, e.g., in Cdc42–/– mice (17) as well as in transgenic mice that express a dominant-negative Rhoa mutant (18). Apparently, both increased and decreased RHO GTPase signaling interfere with the state of podocyte mobility, thus causing proteinuria (19). Furthermore, mutations in the nonmuscle class I myosin MYO1E cause proteinuria in humans due to decreased podocyte migratory ability (20).
Many of the genes that cause NS, if mutated, act in a recessive mode (e.g., NPHS1, NPHS2, and PLCE1) (5, 6, 21). Because recessive single-gene mutations directly represent the primary cause of a genetic disease, gene identification offers a powerful approach toward revealing disease mechanisms. Since recessive mutations predominantly convey loss of function, recessive single-gene defects can be directly transferred into animal models to study the related disease mechanisms and to screen for small molecules as possible treatment modalities. However, no causative gene is known for SSNS. Likewise, in more than 80% of all cases of SRNS, the molecular cause is unknown, and treatment options have yet to be discovered (22).
Although the onset of SRNS in the first year of life is mostly a single-gene disorder, i.e., more than two-thirds of cases are caused by a mutation in 1 of only 5 different disease genes (NPHS1, NPHS2, WT1, LAMB2, or PLCE) (21, 23), genetic mapping data strongly suggest that there are a multitude of additional disease genes that cause SRNS later in life (24). The finding that some of the more recently identified recessive genetic causes of NS are exceedingly rare (LAMB2, refs. 12, 25; PLCE1, ref. 21; and COQ6, ref. 26) necessitates the ability to identify novel single-gene causes of NS in families with only a single affected individual. The new method of whole-exome resequencing (WER) theoretically offers a powerful approach to identifying genes in rare recessive diseases. However, its utility is hampered by the large number of genetic variants that are observed with WER in any given individual (27, 28). To overcome this limitation, we developed a strategy that a priori restricts the number of variants by combining WER (28) with homozygosity mapping (24). We applied this approach to families affected by SRNS or SSNS and identified a novel recessive, full-penetrance cause of SRNS. We generated direct evidence in humans that loss of function of ARHGDIA causes NS by interference with RHO GTPase signaling, thereby altering the podocyte migratory state. We recapitulated features of NS in a zebrafish model and demonstrated that RAC1 inhibitors mitigate the NS phenotypes. Our findings may have implications for the development of new therapeutic approaches to NS.
Results
Mutations in ARHGDIA cause NS.
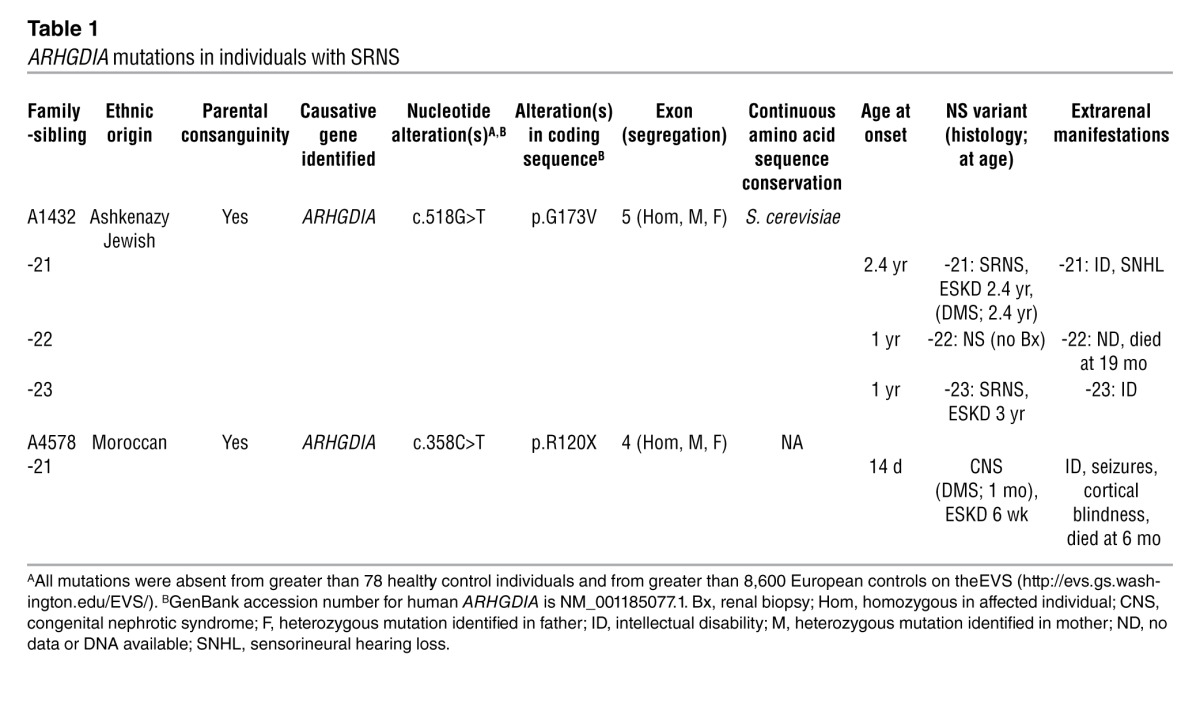
We performed homozygosity mapping (HM) in a family (A1432) of Ashkenazi Jewish origin in whom 2 siblings had early-onset SRNS with renal histology of DMS. HM yielded 5 regions of homozygosity by descent as candidate regions for a recessive SRNS gene (Figure 1A) (24). Using WER, we detected in both siblings a homozygous missense mutation (c.518G>T;p.G173V) of ARHGDIA (RefSeq accession number NM_001185077.1) encoding the RHO GDP dissociation inhibitor α (Figure 1B and Table 1). The altered amino acid residue is conserved, including S. cerevisiae. When we examined 65 additional individuals with DMS and 350 individuals with SRNS, we detected a homozygous mutation (c.358C>T;p.R120X) in an infant (A4578-21) with congenital NS (Figure 1B and Table 1). This infant also exhibited renal histology of DMS (Figure 1C and Table 1). Both mutations were absent from more than 190 ethnically matched healthy control individuals and from more than 8,600 European controls in the Exome Variant Server (EVS) ( http://evs.gs.washington.edu/EVS/). The primary cellular function of ARHGDIA is to interact with RHO GTPases, including RHOA, RAC1, and CDC42, locking them into their cytosolic, inactive, GDP-bound states (29). Thus, ARHGDIA indirectly regulates actin cytoskeleton–dependent cellular functions. Depletion of Arhgdia was shown to cause early-onset NS in mice (30).
Figure 1. Mapping and WER reveal a novel single-gene cause of NS (ARHGDIA).

(A) Homozygosity mapping in family A1432 consisting of 2 siblings with SRNS. NPL scores are plotted across the human genome. The x-axis shows SNP marker positions on human chromosomes concatenated from p-ter (left) to q-ter (right). Genetic distance is given in centimorgans. Maximum NPL peaks indicate the candidate regions of homozygosity by descent (red circles). The gene ARHGDIA is positioned (arrowhead) within one of the mapped candidate regions. (B) Chromatograms of ARHGDIA mutations in SRNS families A1432 and A4578. Gene symbols (underlined), family numbers, mutations, and predicted translational changes are given (see also Table 1). Sequence traces are shown for mutations above normal controls. Mutated nucleotides are indicated by arrowheads. (C) Renal histology of individual A4578-21 with DMS and an ARHGDIA mutation. Right kidney explanted at 2 months of age reveals on H&E staining advanced tubular dilation, atrophy, and casts (left). Original magnification, ×50. PAS stain (right) reveals the characteristic pattern of DMS featuring a small, sclerosed, simplified glomerulus with a corona of vacuolized podocytes (arrowhead). Original magnification, ×630.
Table 1.
ARHGDIA mutations in individuals with SRNS
Arhgdia localizes to podocytes in glomeruli.
Because most gene products that are defective in SRNS are located in glomerular podocytes, we examined adult rat kidney sections and found subcellular localization of ARHGDIA. Indeed, ARHGDIA is enriched in both differentiated and undifferentiated human podocytes (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI69134DS1). In glomeruli, ARHGDIA is prominently expressed in podocytes as identified by the expression of nuclear WT1 (Figure 2A). ARHGDIA partially colocalized with synaptopodin, which colocalizes with ARHGAP24 (31) and regulates RHOA signaling (Figure 2A) (32). ARHGDIA appeared to localize to the nucleus in some podocytes, and nuclear localization of ARHGDIA has been previously reported (33). ARHGDIA also colocalized with the phospholipase C ε 1 (PLCε1) protein, which is defective in SRNS type 3 (21). ARHGDIA partially colocalized with PLCε1 to podocyte cell bodies and primary processes (Figure 2B), suggesting a defect in podocyte function as central to the pathogenesis of ARHGDIA mutations.
Figure 2. ARHGDIA, RAC1, CDC42, and RHOA colocalize and interact in rat glomeruli.

(A) Coimmunofluorescence of ARHGDIA with podocyte marker proteins in rat glomeruli. ARHGDIA is highly expressed in podocytes, as identified by the expression of nuclear WT1. ARHGDIA partially colocalized with synaptopodin, but not with podocalyxin or GLEPP1. (B) ARHGDIA partially colocalized with the SRNS protein PLCε1 in proximal cell bodies and primary processes of podocytes. Direct fluorescent labeling of anti-PLCε1. (C) Coimmunofluorescence of ARHGDIA with the RHO GTPases RAC1, CDC42, and RHOA in adult rat glomeruli. ARHGDIA partially colocalized with RHOA, RAC1, and CDC42 in proximal cell bodies and primary processes, whereas RHOA, RAC1, and CDC42 exhibited a broad glomerular staining pattern in podocyte cell bodies and processes. Scale bars: 10 μm. (D) Coimmunoprecipitation of ARHGDIA in rat renal glomerular lysates. The protein complex precipitated by an anti-ARHDGIA antibody includes the RHO small GTPases RHOA, RAC1, and CDC42. The immunoprecipitation (IP) experiment is representative of more than 3 experiments.
Since ARHGDIA is a regulator of RHO GTPases (29), we examined its colocalization with the RHO GTPases RHOA, RAC1, and CDC42. RHOA, RAC1, and CDC42 exhibited broad glomerular staining in podocyte cell bodies and processes (Figure 2C). ARHGDIA partially colocalized with RHOA, RAC1, and CDC42 in proximal cell bodies and primary processes. Our findings are congruent with the proposed regulatory role of RHO GTPases in these podocyte structures (15, 19).
ARHGDIA and RHO small GTPases form a complex, and mutations abrogate interaction.
To further elucidate the role of ARHGDIA in the functioning of the RHO small GTPases RAC1, CDC42, and RHOA in podocytes, we performed coimmunoprecipitation experiments of ARHGDIA in rat renal glomerular lysates as an endogenous, disease-relevant source of podocyte proteins. We demonstrate that an anti-ARHGDIA antibody coimmunoprecipitated with all 3 RHO small GTPases, RHOA, RAC1, and CDC42 (Figure 2D), strongly suggesting that the pathogenesis of ARHGDIA defects is related to RHO small GTPase function.
We then performed coimmunoprecipitation studies in cultured human podocytes to examine whether the 2 ARHGDIA mutants, which we identified in the family with SRNS, affect protein-protein interaction. Overexpression of the ARHGDIA mutants (R120X and G173V) abrogated interaction with endogenous RHOA, RAC1, and CDC42. (Figure 3A and Supplemental Figure 2A). Comparable results were seen upon a GST pulldown assay in rat renal glomerular lysates (Supplemental Figure 2B).
Figure 3. Effects of disease-causing ARHGDIA mutations on protein-protein interaction, RHO GTPase activity, and podocyte migration.

(A) Interaction of wild-type ARHGDIA and 2 mutants (p.R120X and p.G173V) with RHO GTPases. FLAG-tagged ARHGDIA constructs were transfected into podocytes and were coimmunoprecipitated with endogenous RHO GTPases. Note that the R120X and G173V mutants abrogated interaction with RHOA, RAC1, and CDC42. (B) Active GTP-bound forms of RAC1 and CDC42 precipitated from podocytes expressing FLAG-ARHGDIA (wild-type and mutants) using a GST-PAK1 (CRIB, CDC42, and RAC interactive binding domain) pulldown assay. Five percent input represents the controls for equal loading. Note that, compared with mock cells, podocytes expressing ARHGDIA-WT exhibited a substantial decrease in active RAC1 and CDC42. This decrease was abrogated in the null mutant R120X and is diminished in the G173V mutant. (C) Active GTP-bound RHOA precipitated from podocytes expressing FLAG-ARHGDIA (wild-type and mutants) using a GST-rhotekin (RHO-binding domain [RBD]) pulldown assay. Overexpression of either wild-type or mutant ARHGDIA resulted in a substantial decrease in relative RHOA activity compared with mock cells. PD, pulldown. All IPs and PDs are representative of more than 3 experiments. (D) Effect on podocyte migration of wild-type ARHGDIA and 2 mutants found in patients with SRNS. Migration assay was performed using the xCELLigence system (described in Methods). Overexpression of wild-type ARHGDIA in podocytes inhibited serum-induced migration (green). However, the mutants G173V and R120X failed to inhibit migration (red). Error bars are shown in one direction only for clarity and indicate SDs for more than 3 independent experiments (see also Supplemental Figure 5).
ARHGDIA mutations cause increased levels of active RAC1 and CDC42 and fail to regulate the podocyte migratory phenotype.
As ARHGDIA is known to regulate the inactive GDP-bound versus the active GTP-bound states of RHO GTPases (29), we examined the effect of ARHGDIA mutations on the active states of RHO GTPases. We used purified PAK1 to assay the active GTP-bound states of RAC1 and CDC42 (Figure 3B) and used rhotekin to assay the active state of RHOA (Figure 3C). As expected, wild-type ARHGDIA, when overexpressed in cultured human podocytes, decreased the GTP-bound forms of RAC1, CDC42, and RHOA. In contrast, overexpression of the truncating ARHGDIA mutant (R120X) failed to decrease the active states of RAC1 and CDC42, and the missense mutant (G173V) only weakly decreased their active states (Figure 3B). In contrast, both mutants decreased the active state of RHOA (Figure 3C). This indicates that signals relayed by RAC1 and CDC42 may be more relevant to the pathogenesis of this form of SRNS than signals relayed by RHOA. In addition, we demonstrated by GST pulldown that active RAC1 is spontaneously elevated in EBV-transformed lymphoblasts from patient A1432-21 with an ARHGDIA mutation when compared with the healthy control individual (A2238-26), while the heterozygous father (A4578-11) of an individual with an ARHGDIA mutation (A4578-21) showed intermediate elevation (Supplemental Figure 3).
We then examined the effect of ARHGDIA on podocyte migration using the xCELLigence system (Roche Applied Science), which monitors cell migration in real time. We found that the increase in podocyte migration, which is induced by the addition of serum, was reduced when wild-type ARHGDIA was transfected into cultured human podocytes (Figure 3D). In contrast, the 2 ARHGDIA mutants detected in SRNS families A1432 and A4578 failed to reduce the migratory podocyte phenotype (Figure 2D). In summary, both ARHGDIA mutations from individuals with SRNS abrogated interaction with RHO GTPases and failed to regulate active GTP-bound RAC1 and CDC42, but not RHOA, indicating that RAC1 and CDC42 are more relevant than RHOA to the pathogenesis of this SRNS variant. The mutations resulted in a failure to regulate the migratory phenotype of cultured human podocytes (Supplemental Figure 4).
ARHGDIA knockdown in cultured podocytes causes an increased migratory phenotype, which was reversed by RAC1 inhibitors.
To test whether the effects of an ARHGDIA mutant in podocytes result from loss of function of ARHGDIA, we performed knockdown of ARHGDIA in cultured human podocytes. The GST pulldown for active RAC1, CDC42, and RHOA yielded results consistent with the notion that ARHGDIA loss of function increases the active states of RAC1 and CDC42 by 168% and 185%, respectively, but has no such effect on RHOA (Figure 4, A–E, and Supplement Figures 5 and 6).
Figure 4. Effects of ARHGDIA knockdown on RHO GTPase activity and podocyte migration in cultured human podocytes.

(A) Active GTP-bound RAC1 and CDC42 precipitated from podocytes transfected with scrambled (Scr) or ARHGDIA siRNA using a GST-PAK1 (CRIB) pulldown assay. Ponceau red staining at the top shows the GST proteins used. Compared with control podoctyes, podocytes transfected with ARHGDIA siRNA exhibited a significant increase in relative RAC1 and CDC42 (168% and 185%, respectively). The efficiency of knockdown by siRNA was confirmed by immunoblotting with an anti-ARHGDIA antibody (second to lowest panel). (B) Active GTP-bound RHOA precipitated from podocytes transfected with scrambled or ARHGDIA siRNA using a GST-rhotekin (RBD) pulldown assay. Cells transfected with scrambled control siRNA versus ARHGDIA siRNA exhibited no significant difference in relative RHOA activity. A and B represent 3 experiments each. (C–E) Quantification of RAC1 (C), CDC42 (D), and RHOA (E) in ARHGDIA-depleted cells compared with control cells. Error bars indicate the SEM for greater than 4 independent experiments. *P < 0.05; **P < 0.01; difference from proteins in control podocytes transfected with scrambled siRNA. (F) Effect of ARHGDIA knockdown on podocyte migration. Podocytes transfected with ARHGDIA siRNA exhibited more active migration compared with those transfected with scrambled siRNA. Increase in podocyte migration by ARHGDIA knockdown (red) in serum-induced podocytes was reduced by 2 different RAC1 inhibitors (green). Error bars are shown in only one direction for clarity and indicate SDs for more than 4 independent experiments.
Because RAC1 inhibitors were shown to improve the NS phenotype of Arhgdia–/– mice (34), we tested the effect of the 2 RAC1 inhibitors using the xCELLigence system. We found that knockdown of ARHGDIA caused an enhanced migratory phenotype of cultured human podocytes (Figure 4F, Supplemental Figure 6C, and Supplemental Figure 7A), which was reversed by overexpression of siRNA-resistant ARHGDIA (Supplemental Figure 6C and Supplemental Figure 7). Interestingly, the addition to podocyte cultures of either one of the RAC1 inhibitors reversed the migratory phenotype, confirming that the activated state of RAC1 induced by loss of ARHGDIA function mediates the migratory phenotype of cultured human podocytes and is central to the pathogenesis of SRNS due to ARHGDIA mutations. The findings suggest that RAC1 inhibitors are potentially useful therapeutics in humans with ARHGDIA loss of function.
Our findings are consistent with the concept that activation of RHO GTPases through the regulation of actin (15, 19, 35) converts the sessile cultured human podocyte phenotype into a migratory one, which interferes with glomerular slit membrane integrity, thus causing NS (Supplemental Figure 4) (36). Our data therefore emphasize the pathogenic role of RHO GTPase signaling in human SRNS.
Knockdown of arhgdia in animal models recapitulates NS and responds to treatment.
To further validate the causative roles of ARHGDIA mutations in the SRNS disease phenotypes, we performed knockdown of the ARHGDIA ortholog in zebrafish. We observed a new zebrafish nephrosis phenotype at the whole-animal level, in which the fish develop periorbital edema (“bug eye” phenotype) and total body edema, reminiscent of the signs of NS in humans (Figure 5, A and B). For zebrafish gene knockdown experiments, we used a p53 morpholino-oligonucleotide (MO) as a negative control to minimize nonspecific apoptotic MO effects (37). p53 MO injection into fertilized zebrafish eggs at the 1- to 4-cell stage did not produce any phenotype through 168 hours post fertilization (hpf) (Figure 5A). Coinjection of the p53 control MO with an MO targeting the translation initiation site of zebrafish arhgdia caused the nephrosis phenotype of periorbital and total body edema in 70% of embryos at 120 hpf (Figure 5B). Similar results were obtained with an MO targeting the splice acceptor site (Supplemental Figure 8). The proteinuric effect of the arhgdia MO was confirmed using an established zebrafish proteinuria ELISA assay (38) against a fusion protein of vitamin D–binding protein and GFP in /-fabp::VDBP-GFP transgenic zebrafish (Figure 5E). Similarly, the protein uptake assay using Drosophila pericardial nephrocytes, which share remarkable similarities to the glomerular podocytes (39), showed that knockdown of Drosophila RhoGDI (CG7823) in nephrocytes reduced the uptake ability of the secreted fusion protein of rat atrial natriuretic factor (ANF) and GFP, suggesting that CG7823 is required for nephrocyte function in Drosophila (Supplemental Figure 9).
Figure 5. Functional analysis of arhgdia knockdown in zebrafish.

(A) Control zebrafish injected with p53 MO (0.2 mM). p53 MO did not produce any phenotype until 168 hpf (n >100). Scale bars: 1 mm (×3-fold for insets) (A and B). (B) Zebrafish coinjected with an arhgdia MO (0.2 mM) targeting the translation initiation site of zebrafish arhgdia and a p53 MO. At 120 hpf, the arhgdia morphants displayed the nephrosis phenotype of periorbital edema (“bug-eye”; arrows) and total body edema in 70% of embryos (255 of 360). (C) Effects of various RAC1 and RHOA inhibitors on the zebrafish nephrosis phenotype. Drugs were applied at 48 hpf in fish water and replenished every day. The number of arhgdia morphants that showed periorbital edema was counted at 144 hpf and is represented as a percentage. Two RAC1 inhibitors and eplerenone were partially effective in reducing edema, whereas RHOA inhibitors were not. Error bars indicate SDs for greater than 3 independent experiments in C and D. (D) Dose-response curve for the RAC1 inhibitor, which shows the greatest efficacy in C. The IC50 value of the RAC1 inhibitor for reducing periorbital edema was 8.85 μM. conc., concentration. (E) Proteinuria assay by ELISA against a fusion protein of vitamin D–binding protein and GFP in /-fabp::VDBP-GFP transgenic zebrafish. Note that knockdown of arhgdia caused significant proteinuria compared with the control. The RAC1 inhibitor was partially effective in reducing proteinuria in arhgdia morphants, whereas the RHO inhibitor Y-27632 was not. Error bars indicate the SEM for more than 3 independent experiments.
We used our zebrafish model of arhgdia knockdown to further investigate the role of RHO GTPase inhibitors in the pathogenesis of SRNS due to loss of ARHGDIA function. The sign of periorbital edema provided a reproducible and obvious phenotypic readout of nephrosis (Figure 5, A and B). We tested RAC1 inhibitors and the mineralocorticoid inhibitor eplerenone because it has been described in Arhgdia–/– mice that RAC1 (but not RHOA) was increased and enhanced mineralocorticoid receptor–dependent (MR-dependent) signaling, while a RAC-specific small-molecule inhibitor diminished MR overactivity and renal damage, as did MR blockade by eplerenone (34).
We found that RAC1 inhibitors were partially effective in our zebrafish model in reducing edema from 70% in untreated arhgdia knockdown zebrafish to 47% for the RAC1 inhibitor–treated fish (Millipore), 47% for the RAC1 inhibitor II–treated fish (Millipore), and 54% for eplerenone-treated fish, whereas RHO inhibitors (RHO inhibitor 1, Y-27632, and fasudil) showed no effect (Figure 5C). A dose-response curve revealed that the 50% inhibitory coefficient (IC50) of the RAC1 inhibitor was 8.85 μM (Figure 5D). The protective effect of RAC1 inhibitor against arhgdia knockdown was also confirmed quantitatively by a proteinuria assay (Figure 5E). Our findings again confirm that RAC1 function is more important than RHOA in the pathogenesis caused by ARHGDIA mutations, as we demonstrated by RAC1 activation studies (Figures 3 and 4) and as has been suggested in an Arhgdia–/– mouse model (34). In summary, we recapitulated the NS phenotype in zebrafish upon knockdown of arhgdia and revealed a mitigating effect of eplerenone and RAC1 inhibitors in the arhgdia zebrafish model.
Discussion
We demonstrate here that WER, when combined with homozygosity mapping (24), represents a valid approach for the identification of novel causative genes in rare recessive diseases. We believe this is the first study reporting ARHGDIA mutations in humans, thereby revealing this gene’s regulatory role in RHO GTPase activity in podocytes. In summary, we found that ARHGDIA mutations in SRNS abrogate interaction with RHO GTPases, increase active GTP-bound RAC1 and CDC42, and result in a migratory phenotypic change in podocytes.
RHO GTPases control a wide range of cellular processes, including cell adhesion, migration, and proliferation, so their activity should be tightly controlled. At any given time, only a small fraction of all RHO GTPases present in the cell are in the active GTP-bound state and are associated with membranes (40). The inactive GDP-bound pool is maintained in the cytosol by associating with RHO-specific guanine nucleotide dissociation inhibitors (RHOGDIs). There are 3 genes encoding RHOGDIs in mammals (29). ARHGDIA (also known as RHOGDI1 or RHOGDIα) is the most abundant, is ubiquitously expressed, and interacts with several RHO GTPases, including RHOA, RHOC, RAC1, RAC2, and CDC42 (29). ARHGDIA was considered to passively lock RHO GTPases in an inactive state, but this view has now been changed by the finding that ARHGDIA actively controls the homeostasis of RHO GTPases (41). Recently, several works using mouse genetics have shown the importance of RHO GTPase signaling in podocytes (16–18). Wang et al. showed that transgenic mice overexpressing either constitutively active or dominant-negative Rhoa in podocytes have foot process effacement and proteinuria (18). In addition, Scott et al. showed that podocyte-specific deletion of CDC42 (but not RHOA and RAC1) in mice also results in foot process effacement and proteinuria (17). The results of inactive RHOA (Rhoa knockout versus dominant-negative Rhoa transgenic mice) seem discrepant in these 2 studies, or they may suggest that the presence of dominant-negative Rhoa, rather than the absence of RHOA, causes a more severe imbalance of RHOA and RAC1/CDC42 signaling. Previously, it was demonstrated that active RAC1 (but not RHOA) is upregulated in Arhgdia–/– mice (34). In this study, we show that the ARHGDIA mutations identified in SRNS cause the upregulation of both RAC1 and CDC42. This is congruent with the finding by Akilesh et al. that ARHGAP24 mutation was associated with FSGS and led to a podocyte phenotype very similar to that found in ARHGDIA mutations, i.e., activation of RAC1 and CDC42 (but not RHOA), increased migratory phenotype, and proteinuria (31). In conclusion, aberrant activation of RHOA, RAC1, or CDC42 seems harmful to podocytes. However, it is not clear whether activation of RAC1 or CDC42 alone is sufficient, or whether activation of both is necessary to cause foot process effacement and proteinuria. Furthermore, it is necessary to investigate how the enhanced migratory phenotype in podocytes caused by the activation of RAC1/CDC42 ultimately leads to foot process effacement.
In this study, we show that ARHGDIA mutations caused dissociation of RHOA, RAC1, and CDC42 from ARHGDIA, and that thereafter, only RAC1 and CDC42 switched to active GTP–bound forms. RHOA was not converted to active forms and still remained in the inactive GDP-bound forms. This suggests that RHOA is regulated differently after it dissociates from ARHGDIA and other factors, and that GTPase-activating proteins (GAPs), guanine nucleotide exchange factors (GEFs), or SMURF1 (32) may be involved.
ARHGDIA is expressed in most tissues, whereas the other 2 RHOGDIs (RHOGDIβ and RHOGDIγ) show tissue-specific expression (29). Considering its ubiquitous expression pattern, it is interesting that defects in ARHGDIA lead to prominent renal phenotypes in both humans and mice. Arhgdia–/– mice additionally show impaired spermatogenesis in males as well as massive proteinuria, which leads to death due to renal failure within a year (30). It is not clear whether individuals with ARHGDIA mutations also have abnormal reproductive phenotypes, however, 3 of 4 (75%) individuals from 2 independent families had neurological phenotypes including intellectual disability (Table 1). Podocytes share many cell biological characteristics with neurons. RHO GTPases also play important roles in regulating dendritic spines and branches and are vital for the maintenance and reorganization of dendritic structures in maturing neurons (42). Therefore, it is interesting to investigate whether Arhgdia–/– mice also have neurological defects or morphological abnormalities in neurons.
Our finding that RHO GTPase signaling is an important component of NS pathogenesis is emphasized by several factors: (a) The fact that single-gene recessive mutations in humans usually convey 100% penetrance and the finding that there was high penetrance in the zebrafish model of arhgdia knockdown demonstrate that ARHGDIA function is necessary to avoid the NS disease phenotype. (b) An exaggerated migratory phenotype in podocytes resulted from the activation of RAC1/CDC42 (but not RHOA) and was reversed by RAC1 inhibitors and an MR blocker. (c) Demonstration of mitigation of the zebrafish NS model by drugs that interfere with RAC1 function implicates RAC1 in the pathogenesis of SRNS via another independent route.
In summary, our findings of ARHGDIA mutation implicate the RHO small GTPase RAC1/CDC42 in the pathogenesis of human NS and permit a noninvasive, causation-based diagnosis. Furthermore, the zebrafish models we generated allow for the screening of new therapeutic options for NS.
Methods
Study participants.
Informed consent was obtained from individuals worldwide with SRNS or SSNS for the use of their clinical data and blood samples. The diagnosis of NS was made by (pediatric) nephrologists based on standardized clinical and renal histological criteria (43). Renal biopsies were evaluated by renal pathologists. Clinical data were obtained using a standardized questionnaire ( http://www.renalgenes.org).
Homozygosity mapping.
We performed homozygosity mapping of individuals with SRNS or SSNS as established previously (24). We used the Human Mapping 250k StyI array or the Affymetrix Genome-Wide Human SNP Array 6.0. Genomic DNA samples were hybridized and scanned at the University of Michigan Core Facility using the manufacturer’s standard protocol. Nonparametric lod (NPL) scores were calculated for both affected siblings together in family A1432 as described in Hildebrandt et al. (24) using ALLEGRO and assuming first-degree cousin consanguinity of the parents, regardless of actual consanguinity status. Nonparametric NPL scores (minor allele frequency >0.2) were plotted over genetic distance across the genome, where chromosomal positions are concatenated from the p arm to the q arm (left to right) (see Figure 1A). Homozygosity peaks exceeding the empirical cutoff value of 2.0 represent possible segments of homozygosity by descent, one of which (Figure 1A, black arrowhead) harbors the homozygous disease-causing gene mutation in family A1432. We set the disease allele frequency at 0.0001, and used mixed European descent marker allele frequencies.
WER.
Exome enrichment was conducted following the manufacturer’s protocol for the NimbleGen SeqCap EZ Exome v2 beads (Roche NimbleGen). The kit interrogates a total of approximately 30,000 genes (~330,000 consensus-coding DNA sequence [CCDS] exons). Massively parallel sequencing was performed largely as described in Bentley et al. (44) Briefly, 3 μg of genomic DNA was fragmented by sonication using the Covaris S2 system to achieve a uniform distribution of fragments with a mean size of 300 bp. The fragmented DNA was purified using Agencourt’s AMPure XP Solid Phase Reversible Immobilization paramagnetic (SPRI) beads, followed by polishing of the DNA ends by removing the 3′ overhangs and filling in the 5′ overhangs that resulted from sonication using T4 DNA polymerase and the Klenow fragment (New England BioLabs). Following end polishing, a single A-base was added to the 3′ end of the DNA fragments using Klenow fragment (3′ to 5′ exo minus). This prepares the DNA fragments for ligation to specialized adaptors that have a T-base overhang at their 3′ ends. The end-repaired DNA with a single A-base overhang was ligated to Illumina paired-end adaptors in a standard ligation reaction using T4 DNA ligase and a 2- to 4-μM final adaptor concentration, depending on the postpurification DNA yield after addition of the A-base (a 10-fold molar excess of adaptors was used in each reaction). Following ligation, the samples were purified using SPRI beads amplified by 6 cycles of PCR to maintain complexity and to avoid bias due to amplification and were then quality controlled by library size assessment using the Agilent Bioanalyzer and quantitated using PicoGreen reagent (Invitrogen).
One microgram of amplified, purified DNA (DNA library) was prepared for hybridization by adding to the DNA library COT1 DNA and blocking oligonucleotides, desiccating the DNA completely and resuspending the material in NimbleGen (Roche NimbleGen) hybridization buffer. The resuspended material was denatured at 95°C prior to addition of the exome capture library bait material. The DNA library and biotin-labeled capture library were then hybridized by incubation at 47°C for 68 hours. Following hybridization, streptavidin-coated magnetic beads were used to purify the DNA:DNA hybrids formed between the capture library and sequencing library during hybridization. The purified sequencing library was amplified directly from the purification beads using 8 cycles of PCR with Pfx DNA polymerase (Invitrogen). The libraries were purified following amplification, and the library size was assessed using the Agilent Bioanalyzer. A single peak between 350 to 400 bp indicates a properly constructed and amplified library that is ready for sequencing. Final quantitation of the library was performed using the Kapa Biosystems Real-time PCR assay, and appropriate amounts were loaded onto the Illumina flow cell for sequencing by paired-end 100-nt sequencing on the Illumina HiSeq 2000.
Mutation calling.
Sequence reads were mapped to the human reference genome assembly (NCBI build 36/hg18) using CLC Genomics Workbench (version 4.7.2) software (CLC bio). Mutation calling was performed with a team of geneticists and cell biologists who had knowledge of the clinical phenotypes and pedigree structure, as well as experience with homozygosity mapping and exome evaluation. Minimum length fraction of a read to match the reference sequence was set to 90%. For SNP detection, the minimum quality score of the central base as well as the minimum average quality score of surrounding bases were kept at a default score of 20 and 15, respectively. Quality assessment was performed within a window of 11 bases. Only reads that uniquely aligned to the reference genome were used for variant SNP or deletion/insertion polymorphism (DIP) calling. In patients with evidence of homozygosity by descent, the threshold for the number of reads (minor allele frequency) was set to greater than 55%. The threshold coverage for minimum valid reads (minimum variant count), which displays the variant at a given position, was set to 2 reads.
Filtering of variants from the normal reference sequence (28).
For DIPs and SNPs, we used the following a priori criteria to restrict the high number of variants from normal reference sequences (VRSs) (average of 53,272 for DIPs and 315,372 for SNPs) as follows (see Supplemental Table 1): (a) We retained exonic variants (missense, nonsense, indels) and obligatory splice site variants only. (b) We included only VRSs that are not listed in the SNP132 database of innocuous polymorphisms. (c) We evaluated exonic changes only within genomic regions, in which homozygosity mapping showed linkage for both affected siblings. (d) Variants were analyzed using the BLAT program ( http://genome.ucsc.edu/cgi-bin/hgBlat?command=start) on the UCSC Human Genome Bioinformatics Browser ( http://genome.ucsc.edu/) for the presence of paralogous genes, pseudogenes, misalignments at the ends of sequence reads, and to determine whether the variant is a known dbSNP132 with an allele frequency greater than 1% in populations of mixed European descent. In families in whom mapping demonstrated homozygosity by descent, we retained only homozygous variants and scrutinized all of them in the sequence alignments within the CLC Genomic Workbench software program for the presence of mismatches indicating potential false alignments or poor sequence quality. (e) Sanger sequencing was performed to confirm the remaining variants in original DNA samples and to test for intrafamilial segregation in a recessive mode. (f) Finally, the remaining variants were ranked by whether mutations were truncating the conceptual reading frame (nonsense, frameshift, and obligatory splice variants) or by evolutionary conservation analysis of missense variants. We also used web-based programs to predict the impact of disease candidate variants on the encoded protein or to predict whether these variants were known disease-causing mutations (see Supplemental Table 1).
Segregation analysis by Sanger sequencing.
We applied Sanger dideoxy terminator sequencing to confirm and segregate potential disease-causing variants in the respective patients, their affected siblings, and their parents. In patients in whom only 1 heterozygous mutation was detected by exome capture and massively parallel sequencing, all exons and flanking intronic sequences of the respective gene(s) were analyzed by Sanger sequencing. A touchdown PCR protocol was used as described previously (45). Sequencing was performed using a BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI 3730 XL sequencer (Applied Biosystems). Sequence traces were analyzed using Sequencher software, version 4.8 (Gene Codes Corp.).
Web-based variant analysis.
Predictions of the possible impact of an amino acid substitution on chemical change, evolutionary conservation, and protein function were obtained by using the following web-based programs: PolyPhen-2 ( http://genetics.bwh.harvard.edu/pph2/), SIFT, (Sorting Intolerant from Tolerant; http://sift.jcvi.org/), and Mutation Taster ( http://www.mutationtaster.org/). Genomic evolutionary rate profiling (GERP) calculation was performed using the following: http://snp.gs.washington.edu/SeattleSeqAnnotation137/.
Plasmids, cell culture, and transfection.
A human ARHGDIA clone was purchased from Open Biosystems (clone accession number BC016031.1). The ARHGDIA mutants were generated by a PCR-based site-directed mutagenesis method. The immortalized human podocytes (46) were maintained in RPMI plus GlutaMAX-I (Gibco) supplemented with 10% FBS, penicillin-streptomycin (50 IU/ml and 50 μg/ml, respectively), and insulin-transferrin-selenium-X. Plasmids and siRNA were transfected into podocytes grown at the permissive temperature of 33°C using Lipofectamine 2000 (Invitrogen). Podocytes were cultured for differentiation at 37°C for 14 days. The ARHGDIA-specific and control scrambled siRNAs were purchased from Dharmacon or Sigma-Aldrich. Lymphoblasts were purified from blood samples of individuals (A1432-21, A2338-26, and A4565-11 in Supplemental Figure 3) using Ficoll-Paque PLUS (GE Healthcare) according to the manufacturer’s instructions. The isolated lymphoblasts were transformed by EBV and immortalized as previously described (47).
Immunoblotting, immunoprecipitation, pulldown assay, and immunofluorescence staining.
These experiments were performed as described previously (48). The GST-PAK1 CRIB domain was purified from the BL21(DE3) E. coli strain, and GST-rhotekin RBD beads were purchased from Cytoskeleton. Anti-FLAG, anti-Myc (Cell Signaling Technology), anti-RAC1, anti-CDC42 (BD Transduction Laboratories), anti-ARHGDIA, anti-RHOA (Santa Cruz Biotechnology), and anti-synaptopodin (American Research Products) were purchased from the indicated commercial sources. Anti-podocalyxin and anti-GLEPP1 antibodies were provided by Roger C. Wiggins. Anti-PLCε1 antibody was previously described by Hinkes et al. (21) and was labeled using an APEX Alexa Fluor Labeling Kit (Invitrogen). Coimmunoprecipitation was performed using EZview Red ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich). The intensities of immunoblots were analyzed using Quantity One (BioRad). Fluorescence images were obtained with a Leica SP5X laser scanning microscope.
Podocyte migration assay.
Real-time migration assays were performed using the xCELLigence system (Roche Applied Science) in CIM-plate 16 according to the manufacturer’s instructions. Briefly, 24 hours after transfection, 4 × 104 cells were plated in serum-free media in the upper chamber. The lower chambers were filled with 10% FBS for chemoattraction or with serum-free media. The data were analyzed using RTCA software. Results are presented as the time-versus-cell index curve.
Zebrafish studies.
Zebrafish (Danio rerio) were maintained and reared as described previously (38). Approval for zebrafish research was obtained from the University Committee on the Use and Care of Animals (UCUCA) of the University of Michigan. MOs were injected into the AB* wild-type strain for phenotype analysis of morphants and drug treatment. The proteinuria assays were performed using l-fabp::VDBP-GFP transgenic fish and a GFP ELISA kit (Cell Biolabs) as described previously (38).
Statistics.
Results are presented as the means ± SEM or SD for the indicated number of experiments. Statistical analysis of continuous data was performed with a 2-tailed Student t test or a Mann-Whitney U test, as appropriate. P < 0.05 was considered statistically significant.
Study approval.
Approval for human subjects research was obtained from the IRBs of the University of Michigan, the Université Paris Descartes, and the University of Cologne.
Supplementary Material
Acknowledgments
The authors thank the families who contributed to this study. We thank Martin Griebel (KfH Kuratorium für Dialyse und Nierentransplantation e.V., Munich, Germany) for the contribution of clinical data. This research was supported by grants from the NIH (DK076683 and DK086542, to F. Hildebrandt; DK46073 and DK081943, to R.C. Wiggins; DK090917, to E.A. Otto; and DK091405, to W. Zhou). F. Hildebrandt was supported by the NephCure Foundation. W. Zhou and C. Faul are Carl W. Gottschalk Scholars, and H.Y. Gee is a Research Fellow of the American Society of Nephrology (ASN). F. Hildebrandt is a Howard Hughes Medical Institute Investigator, a Doris Duke Distinguished Clinical Scientist, and a Frederick G.L. Huetwell Professor.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(8):3243–3253. doi:10.1172/JCI69134.
References
- 1.Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA. Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant. 2007;11(4):366–373. doi: 10.1111/j.1399-3046.2007.00704.x. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrandt F, Heeringa SF. Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney Int. 2009;75(7):669–671. doi: 10.1038/ki.2008.693. [DOI] [PubMed] [Google Scholar]
- 3.Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24(4):333–335. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 4.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354(13):1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 5.Kestila M, et al. Positionally cloned gene for a novel glomerular protein — nephrin — is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1(4):575–582. doi: 10.1016/S1097-2765(00)80057-X. [DOI] [PubMed] [Google Scholar]
- 6.Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24(4):349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 7.Shih NY, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286(5438):312–315. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 8.Ozaltin F, et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am J Hum Genet. 2011;89(1):139–147. doi: 10.1016/j.ajhg.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan JM, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24(3):251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 10.Brown EJ, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42(1):72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Has C, et al. Integrin alpha3 mutations with kidney, lung, and skin disease. N Engl J Med. 2012;366(16):1508–1514. doi: 10.1056/NEJMoa1110813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasselbacher K, et al. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int. 2006;70(6):1008–1012. doi: 10.1038/sj.ki.5001679. [DOI] [PubMed] [Google Scholar]
- 13.Zenker M, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13(21):2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 14.Antignac C. Molecular basis of steroid-resistant nephrotic syndrome. Nefrologia. 2005;25(suppl 2):25–28. [PubMed] [Google Scholar]
- 15.Kistler AD, Altintas MM, Reiser J. Podocyte GTPases regulate kidney filter dynamics. Kidney Int. 2012;81(11):1053–1055. doi: 10.1038/ki.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu L, Jiang R, Aoudjit L, Jones N, Takano T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22(9):1621–1630. doi: 10.1681/ASN.2010111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott RP, et al. Podocyte-specific loss of cdc42 leads to congenital nephropathy. J Am Soc Nephrol. 2012;23(7):1149–1154. doi: 10.1681/ASN.2011121206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, et al. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012;81(11):1075–1085. doi: 10.1038/ki.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lal MA, Tryggvason K. Knocking out podocyte rho GTPases: and the winner is. J Am Soc Nephrol. 2012;23(7):1128–1129. doi: 10.1681/ASN.2012050494. [DOI] [PubMed] [Google Scholar]
- 20.Mele C, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med. 2011;365(4):295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinkes B, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38(12):1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 22.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71(12):1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 23.Hinkes BG, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics. 2007;119(4):e907–e919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 24.Hildebrandt F, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PloS Genet. 2009;5(1):31000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matejas V, et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat. 2010;31(9):992–1002. doi: 10.1002/humu.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heeringa SF, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121(5):2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otto EA, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42(10):840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12(8):493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Togawa A, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18(39):5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 31.Akilesh S, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest. 2011;121(10):4127–4137. doi: 10.1172/JCI46458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asanuma K, et al. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. 2005;115(5):1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Marzouk S, Schultz-Norton JR, Likhite VS, McLeod IX, Yates JR, Nardulli AM. Rho GDP dissociation inhibitor alpha interacts with estrogen receptor alpha and influences estrogen responsiveness. J Mol Endocrinol. 2007;39(4):249–259. doi: 10.1677/JME-07-0055. [DOI] [PubMed] [Google Scholar]
- 34.Shibata S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14(12):1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 35.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17(9):428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Faul C, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14(9):931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robu ME, et al. p53 activation by knockdown technologies. PLoS Genet. 2007;3(5):e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou W, Hildebrandt F. Inducible podocyte injury and proteinuria in transgenic zebrafish. J Am Soc Nephrol. 2012;23(6):1039–1047. doi: 10.1681/ASN.2011080776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang F, Zhao Y, Han Z. An in vivo functional analysis system for renal gene discovery in Drosophila pericardial nephrocytes. J Am Soc Nephrol. 2013;24(2):191–197. doi: 10.1681/ASN.2012080769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 41.Boulter E, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12(5):477–483. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong WT, Faulkner-Jones BE, Sanes JR, Wong RO. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. J Neurosci. 2000;20(13):5024–5036. doi: 10.1523/JNEUROSCI.20-13-05024.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.[No authors listed]. Primary nephrotic syndrome in children: clinical significance of histopathologic variants of minimal change and of diffuse mesangial hypercellularity. A Report of the International Study of Kidney Disease in Children. Kidney Int. 1981;20(6):765–771. doi: 10.1038/ki.1981.209. [DOI] [PubMed] [Google Scholar]
- 44.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otto EA, et al. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat. 2008;29(3):418–426. doi: 10.1002/humu.20669. [DOI] [PubMed] [Google Scholar]
- 46.Saleem MA, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13(3):630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 47.Amoli MM, Carthy D, Platt H, Ollier WE. EBV Immortalization of human B lymphocytes separated from small volumes of cryo-preserved whole blood. Int J Epidemiol. 2008;37(suppl 1):i41–i45. doi: 10.1093/ije/dym285. [DOI] [PubMed] [Google Scholar]
- 48.Chaki M, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150(3):533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.