

Abstract
Four decades ago, angiogenesis was recognized as a therapeutic target for blocking cancer growth. Because of its importance, VEGF has been at the center stage of antiangiogenic therapy. Now, several years after FDA approval of an anti-VEGF antibody as the first antiangiogenic agent, many patients with cancer and ocular neovascularization have benefited from VEGF-targeted therapy; however, this anticancer strategy is challenged by insufficient efficacy, intrinsic refractoriness, and resistance. Here, we examine recent discoveries of new mechanisms underlying angiogenesis, discuss successes and challenges of current antiangiogenic therapy, and highlight emerging antiangiogenic paradigms.
Introduction
The development of new blood vessels, termed angiogenesis, is a hallmark of cancer development that has long been considered an attractive therapeutic target. The signaling molecule VEGF plays a central role in angiogenesis and is frequently highly expressed in cancers. Thus, clinical efforts to develop antiangiogenic therapies have largely focused on inhibiting VEGF. VEGF blockade in neoadjuvant trials show promising benefit, but largely negative results have been obtained in the adjuvant setting. Furthermore, in certain advanced metastatic cancers, blocking VEGF alone is insufficient to prevent progression, induces resistance, and possibly even increases invasion and metastasis, although this matter remains debated. Thus, we need additional angiogenesis targets and predictive preclinical models and biomarkers, and we must better understand the context-dependent angiogenic activity of several targets. Here, we review newly discovered proangiogenic molecules and discuss emerging insights in these outstanding topics. We primarily focus on recent publications and selected angiogenic signals; more historically complete overviews are available elsewhere (1–4).
Vessel sprouting model
Several modes of vascularization exist (see below), but the vessel sprouting (angiogenesis) model has been studied most extensively (5). Key insights have been generated in the mouse retina, where vascularization occurs postnatally, thus representing a physiological angiogenesis model. Upon activation of ECs by proangiogenic molecules, cell-cell junctions (VE-cadherin, ZO-1, and others) and the basement membrane (BM) are remodeled (in part by MMPs) in tandem with pericyte detachment, allowing a tip cell to migrate in response to guidance signals (Figure 1). The sprout then elongates through proliferation of stalk cells, which form a lumen and recruit pericytes for stabilization (Figure 2). Tip cells from neighboring sprouts meet and anastomose to form a perfused branch. Upon perfusion, ECs become quiescent phalanx cells, deposit BM, establish a barrier, and are covered by mature pericytes (Figure 3). From a therapeutic viewpoint, strategies targeting tip and stalk cells would prevent neoangiogenesis, prune immature vessels, and cause vessel regression. Promoting phalanx cell formation would induce tumor vessel normalization (see below).
Figure 1. Initial steps of tip cell selection.

Vascular sprouting is initiated by proangiogenic factors (e.g., VEGF). ECs at the leading edge of the vascular sprout extend filopodia and migrate toward angiogenic signals. VEGF activates VEGFR2 to stimulate tip cell migration. The coreceptor Nrp1 complexes with and enhances VEGFR2 signaling. ECs become either the migratory vessel-leading tip cell or the proliferating stalk cell, but their phenotype is fluid; Notch regulates this specification. ECs with activated VEGFR2 signaling compete for the tip cell position by increasing their expression of DLL4, which binds to Notch receptors on neighboring ECs, releasing the transcription regulator NICD. NICD transcriptionally downregulates VEGFR2 and Nrp1 expression while increasing VEGFR1, a VEGF trap, thus enhancing the stalk cells’ unresponsiveness to VEGF. The tip cell is not a fixed position, and fluidity at the front occurs depending on the VEGFR1/VEGFR2 ratio. Tip cell migration requires BM degradation (in part due to MMP), EC junction loosening (caused by VE-cadherin, ZO-1, and others), and pericyte detachment (regulated by Ang2). VEGF increases the permeability of the vessel, allowing the extravasation of plasma proteins (e.g., fibronectin and fibrinogen) that are deposited as a provisional matrix layer while the preexisting interstitial matrix is remodeled by proteases; these events enable tip cell migration. Key molecular players discussed in this review and elsewhere (5, 132) are indicated.
Figure 2. Tip cell guidance and stalk elongation.

The growing sprout moves along a VEGF gradient. Tip cells adhere to the ECM, mediated by integrins, and migrate toward guidance signal molecules (e.g., semaphorins and ephrins). Stalk cells trail behind the tip cell and proliferate to allow sprout elongation and lumen formation. While Notch signaling inhibits proliferation, expression of Nrarp at branch points allows Wnt signaling to maintain stalk cell proliferation. This system allows vascular migration/directionality (by tip cells) and elongation of the shaft (by proliferating stalk cells). When two tip cells meet, they fuse (anastomose); this mechanism is assisted by macrophages, which accumulate at sites of vascular anastomosis to act as bridge cells by interacting with the neighboring tip cells’ filopodia. Once contact between the tip cells has been established, VE-cadherin–containing junctions further strengthen the connection. Perivascular macrophages further stimulate sprouting by producing angiogenic factors or proteolytically liberating them from the ECM. The stalk cells also deposit BM and recruit pericytes, thus stabilizing the forming vessel. Pericyte precursors are attracted to vessels by EC-expressed PDGF. Once at the vessel, these mesenchymal precursor cells differentiate to pericytes in response to TGF-β and decrease EC migration, proliferation, and vascular leakage, resulting in nascent vessel stabilization. Key molecular players discussed in this Review and elsewhere (5, 132) are indicated.
Figure 3. Maturation through resolution of quiescent phalanx cells.

Once fusion has occurred, a connected lumen is formed to allow blood flow through the new vessel. This perfuses the hypoxic tissue, and the resultant oxygen and nutrient delivery leads to decreased levels of angiogenic signals, inactivation of EC oxygen sensors, and increased proquiescent molecules that lead to EC quiescence. Establishment of the blood flow remodels vessel connections, which are regulated by the shear stress–responsive transcription factor KLF2. ECs resume a quiescent phalanx phenotype in a tightly apposed monolayer with a streamlined surface that conducts the blood flow and regulates tissue perfusion. Perfusion induces vascular maturation by reestablishment of cell-cell junctions, pericyte maturation, and BM deposition. Autocrine and paracrine signaling from ECs and surrounding support cells by VEGF, FGF, Ang1, and Notch, among others, maintain a quiescent EC phenotype and protect the vessel from environmental stresses. Reduced growth factor signaling can lead to vessel retraction and EC apoptosis. Once stabilized and matured, the vessel forms a barrier between the blood and surrounding tissue, controlling the exchange of fluids and solutes. Key molecular players discussed in this Review and elsewhere (5, 132) are indicated.
Tip cell selection: cross-talk between VEGFR2 and Notch
Vessel sprouting requires coordination between migrating tip cells and proliferative stalk cells. ECs at the leading edge extend filopodia and migrate toward angiogenic signals. At the forefront, where VEGF levels are highest, VEGF activates VEGF receptor 2 (VEGFR2) to stimulate tip cell migration (Figure 1). VEGFR2 internalization and activation of ERK1/2 signaling are important for sprouting, likely because rapid receptor turnover and signaling is essential for ECs at the vascular front to respond strongly and quickly to angiogenic signals (6). Such dynamic responses necessitate rapid clearing of activated receptors to finely tune the speed and direction of vessel branching. Signaling through VEGFR2 (and VEGFR3; see below) is enhanced by the coreceptor neuropilin-1 (Nrp1), which promotes tip cell function (7). Although the Nrp1 cytoplasmic domain (and signaling) is dispensable for angiogenesis, it is essential for separation of arteries and veins (8).
The specification between tip cells and stalk cells is regulated by Notch (9). ECs with activated VEGFR2 compete for the tip position by increasing expression of the Notch ligand Delta-like 4 (DLL4), which binds to Notch receptors on neighboring ECs and releases the Notch intracellular domain (NICD). NICD acts as a transcriptional regulator, decreasing Vegfr2 and Nrp1 expression while increasing the levels of VEGFR1, which traps VEGF (10, 11) and renders stalk cells less responsive to VEGF (Figure 1). Hence, Notch blockade induces vessel hyperbranching, while gain of function causes the opposite effect (9). In addition to regulation by VEGF/VEGFR2 signaling, initial evidence suggests that cellular or matrix components may also ensure DLL4 expression (12). The tip cell position is fluid: EC interchange occurs at the leading edge, with ECs with the highest VEGFR2 and lowest VEGFR1 levels migrating to the tip position (11). Competition and position exchange couple VEGFR levels to leadership, ensuring that the tip cell is optimally equipped to sense the VEGF gradient. Tumor ECs produce elevated DLL4 levels, and pharmacological blockage of DLL4 reduces tumor growth because it leads to supernumerary hypoperfused tumor vessels (13), but also causes hemangiomas (14).
Role of VEGF-C/VEGFR3 in tip cell formation
VEGF-C binds VEGFR3 (and weakly binds VEGFR2, but not VEGFR1) and induces tip cell activity, though less potently than VEGF (Figure 1). The sprouting activity of VEGF-C/VEGFR3 is more pronounced when VEGFR2 is blocked. Pharmacological VEGFR3 or VEGF-C blockade studies suggest that VEGFR3 activation by VEGF-C promotes tip cell formation (15). However, Vegfr3 gene deficiency increases tip cell formation (16). These discrepant results are reconciled by a model whereby VEGFR3 has a ligand-dependent (“active”) proangiogenic signaling mode and a ligand-independent (“passive”) signaling branch that activates Notch, which explains why VEGFR3 deficiency causes hyperbranching. The passive signaling operates by phosphorylation of the intracellular VEGFR3 domain via matrix-dependent activation of Src kinase (16). VEGF-C–producing macrophages that localize to vessel branch points activate Notch target genes, independently of Notch ligands, thereby decreasing the sensitivity to VEGF and facilitating vascular loop assembly. Hence, VEGFR3 regulates the conversion of tip cells to stalk cells at points of sprout fusion, where tip cells of opposing branches anastomose (16). Furthermore, Benedito et al. (12) reported that Notch downregulates expression of VEGFR3, but not of VEGFR2 (in contrast to ref. 9), and that low Notch signaling induces VEGFR3-driven angiogenesis independent of VEGFR2 signaling (12). Inhibition of VEGFR3’s kinase activity, but not ligand binding, suppressed EC sprouting, which suggests that VEGFR3 has ligand-independent activity in low-Notch conditions (12). Future work is required to reconcile these divergent findings on the roles of VEGFR2, VEGFR3, and Notch in a unifying model. Regardless of the mechanisms, VEGFR3 levels are upregulated in tumor vessels, and inhibitors blocking VEGFR3 homodimerization, VEGFR3/VEGFR2 heterodimerization, or VEGF-C binding inhibit tumor angiogenesis in culture and in mice (17).
Role of Ang2/Tie2 in tip cell formation
Angiopoietin1 (Ang1) and Ang2 bind Tie2, a tyrosine kinase receptor expressed in stalk and phalanx cells. Perivascular cell expression of Ang1 stabilizes and tightens the EC barrier by recruiting complexes between Tie2 and the phosphotyrosine vascular endothelial protein tyrosine phosphatase (VE-PTP) to cell-cell junctions and by preventing VEGFR2-induced internalization of the junctional molecule VE-cadherin (18). Ang1-Tie2 complexes assemble in trans at EC-EC junctions, promoting EC-EC adhesion and EC survival. Ang1 also promotes collective directional migration of ECs by relocating atypical PKCζ to the leading EC edge, where it forms a complex with β-catenin that interacts with polarity proteins at adherens junctions (19). In atypical PKCζ morphant zebrafish, tip cells, after initial sprouting from the aorta, separate from the secondary connector stalk cells and lose polarity cues by extending filopodia more randomly (Figure 2). In ischemic tissues, Ang1 promotes vessel growth and enlargement, but without inducing vessel leakage (as VEGF does), making it a potential target for therapeutic angiogenesis (20).
EC-expressed Ang2 antagonizes Ang1 activity and thereby stimulates vessel destabilization and sensitizes ECs to proangiogenic signals (Figure 1 and ref. 21). In this case, Tie2 translocates to cell-matrix contacts. However, Ang2 also stimulates angiogenesis by activating Tie2. Indeed, Ang2 attenuates Ang1-Tie2 activation in the presence of Ang1 (in mature tumor supply vessels), but activates Tie2 signaling when Ang1 is absent (in immature pericyte-deprived tumor vessels), which suggests that Ang2 is a partial agonist (22). Ang2 also stimulates tip cell migration by activating integrins independently of Tie2 (Figure 1 and ref. 23). Tie1, an orphan receptor homologous to Tie2, heterodimerizes with Tie2 and regulates Ang2 activity. In the presence of Tie1, Ang2 is unable to activate Tie2; however, loss of Tie1 reveals agonist capabilities of Ang2.
Tumor ECs express elevated Ang2 levels, and an increased Ang2/Ang1 ratio correlates with tumor angiogenesis and poor prognosis in many cancers, making Ang2 an attractive therapeutic target. Anti-Ang2 antibodies inhibit tumor angiogenesis and growth and improve the antiangiogenic efficacy of VEGF blockers in xenograft models (22), while a combination of angiopoietin blockers and cytotoxic drugs increases the progression-free survival (PFS) of patients with ovarian cancer (24). Simultaneous targeting of angiopoietins and VEGF by the chimeric decoy receptor double antiangiogenic protein (DAAP) also inhibits tumor angiogenesis and growth in preclinical models (25). Nonetheless, the effects of Ang2 on tumor progression upon over- or underexpression are complex and often divergent (26). Because of its pleiotropic effects in angiogenic, lymphatic, and macrophage biology as well as the complexities in receptor use and contextual localization, a better understanding of the tumor-specific expression of the Tie receptors and their ligands is needed to improve Ang/Tie-targeted therapy.
Tip cell guidance
ECs express guidance receptors that probe the environment (Figure 2). Nrp1, alone or complexed to plexin family members, interacts with semaphorins (Sema). Sema6a regulates VEGFR2 expression and its downstream signaling (27), while Sema3E activates PlexinD1 in tip cells and maintains the tip cell/stalk cell balance by regulating VEGF activity and DLL4 expression (28). Another guidance class involves SLIT proteins, which are ligands for roundabout receptors (Robo). Robo4 is expressed in ECs, but its role remains debated, as it has been attributed both chemoattractant and repellant activity (29, 30). However, when binding to Unc5B, a netrin receptor, Robo4 increases vessel integrity and reduces angiogenesis by inhibiting VEGF signaling (31). VEGF also promotes the expression of the transcription factor Hlx1, which increases expression of Unc5, plexin 5, and Sema3G, suggesting feedback with Robo4 (32). Hlx1 is expressed in sprouting ECs, where it maintains the stalk cell phenotype by regulating repulsive signals (33).
Ephrins activate Eph receptor tyrosine kinases to regulate developmental vessel morphogenesis (34). In zebrafish, angioblasts form a precursor vessel that segregates into discrete arterial and venous vessels. Ephrin-B2–expressing ECs, fated to form arterial vessels, segregate from EphB4-expressing ECs, which become venous vessels due to repulsive cues (35). Ephrin-B2 activates Eph receptors in a positive feedback loop and has its own reverse signaling activity, which is important for EC morphology and motility (36). Deletion of Ephrin-B2’s PDZ binding motif results in fewer tip cells and filopodia, an effect due to its regulation of VEGFR2/VEGFR3 internalization and trafficking (37, 38). Furthermore, antibody blockade of Ephrin-B2 inhibits tumor angiogenesis in preclinical studies (39).
Macrophages orchestrate vessel fusion and formation
When tip cells of adjacent vessels meet via filopodia, they connect and anastomoze (Figure 2). Imaging in zebrafish reveals that cell junctions at the site of contact expand into rings, generating an interface of apical membrane compartments (40). Macrophages act as “bridge cells” between anastomosing tip cells by releasing angiogenic factors (41). In disease, macrophages have contextual effects. In ischemia they promote collateral vessel growth (42), while in tumors M1-polarized macrophages are tumoricidal, but M2-polarized macrophages promote tumor vascularization by producing proangiogenic factors (43). It is unclear whether tumors “educate” macrophages to these phenotypes or whether tumors recruit distinct monocyte populations. Targeting myeloid cells is gaining increasing attention for blocking tumor angiogenesis and growth (44). Possible targets include placental growth factor (PlGF), which promotes M2 polarization (45), or Ang2, which increases macrophage association with tumor blood vessels (46, 47). The oxygen sensor HIF-prolyl hydroxylase domain protein 2 (PHD2) also modulates the macrophage phenotype and regulates collateral vessel growth in ischemia (42).
Stalk cell proliferation and maintenance
Stalk cells elongate the sprout shaft (Figure 2). In vitro, Notch inhibits EC proliferation; however, stalk cells must proliferate to elongate the shaft in vivo. To overcome this, stalk cells express the Notch target Notch-regulated ankyrin repeat protein (Nrarp), which limits Notch signaling at branch points while allowing continued Wnt signaling to promote EC proliferation and vessel stability (48). Because of the pro–stalk cell activity of Notch, posttranslational modifications finely tune its activity to prevent excessive signaling. The NICD is acetylated, which stabilizes the protein against ubiquitylation-dependent proteasomal degradation. Sirtuin-1, a NAD+-dependent deacetylase, associates with NICD and reduces its deacetylation levels (49). Interestingly, sirtuin-1 is more active during fuel and energy stress, which suggests that it promotes vessel branching via Notch inactivation to guide ECs to fuel-rich areas (49). How targeting these stalk cell signals can be used therapeutically for cancer remains to be determined.
Other modulators of tip/stalk cell balance
Activin receptor–like kinase 1 (Alk1), an EC-specific member of the TGF-β/BMP9 receptor superfamily, is inactivated in patients with hereditary hemorrhagic telangiectasia (HHT) (50). Smad1/5, effectors of Alk1 signaling, also orchestrate the balance between tip and stalk cells in the mouse retina (Figure 2). Loss of Smad1/5 impairs Notch signaling and increases tip cells at the expense of stalk cells, possibly because the interdependence between Notch and Smad1/5 results in oscillatory fluctuations of tip/stalk cell targets that determine the dynamic shuffling of ECs at the tip (51). In agreement with this model, Alk1 inhibits retinal angiogenesis by cooperating with Notch: combined blockade of Alk1 and Notch exacerbates hypervascularization, while activation of Alk1 by BMP9 rescues hypersprouting by Notch inhibition (52). However, Alk1’s activity is context dependent; in preclinical tumor models, Alk1 is proangiogenic in the presence of VEGF, and an anti-Alk1 antibody and BMP9 trap inhibit tumor angiogenesis (53).
Vessel maturation by pericyte recruitment
Mural pericytes reduce EC proliferation, migration, and vessel leakage, thereby stabilizing nascent vessels (Figure 2 and refs. 54, 55). TGF-β1 promotes the differentiation of precursor cells to pericytes (56). PDGFR-β–expressing pericytes migrate in response to PDGF-B from ECs and surround newly formed vessels. Ang1 was previously suggested to promote pericyte coverage of blood vessels (57). However, conditional global Ang1 gene inactivation studies showed that early Ang1 deficiency causes vascular morphogenesis defects, which are caused by cardiac defects and secondary flow disturbance, without affecting pericyte recruitment (58). In postnatal angiogenic conditions, Ang1 deficiency accelerated angiogenesis, which suggests that Ang1 is dispensable for quiescent vessels but modulates the vascular response after injury (58).
Pericyte-expressed sphingosine-1-phosphate (S1P) regulates EC barrier properties by upregulating N-cadherin between ECs and pericytes while downregulating Ang2 in ECs (59). S1P receptor (S1PR) signaling acts as a vascular stabilization mechanism by impairing sprouting via inhibition of VEGF signaling and stabilization of VE-cadherin junctions (60, 61). Reduced pericyte coverage is associated with metastasis in patients, and overexpression of PDGF-B increases pericyte coverage that results in tumor growth inhibition. Concerns have been raised regarding pericyte targeting, as this increases epithelial-to-mesenchymal transition and metastasis because of a reduced barrier for tumor cells to intravasate (62).
Resuming quiescence
Phalanx ECs line quiescent vessels (Figure 3). Once the hypoxic tissue is perfused by neovessels, levels of angiogenic signals are reduced, and proquiescent molecules are increased. Autocrine signals, including VEGF, Ang1, FGF, and Notch, maintain ECs in quiescence (63). Ang1 induces DLL4 expression and NICD signaling (64). Furthermore, deposition of a BM around quiescent ECs promotes vessel stabilization, partly because the BM component laminin-α4 in tip cells limits their number by inducing Notch signaling (65, 66). Phalanx cells in a tightly apposed monolayer optimize conduction of blood flow, establish tissue barriers, and form intercellular junctions to tighten the EC barrier. An oxygen-sensing system ensures that ECs normalize abnormalities in structure and function of ECs to readapt oxygen supply to tissue needs. Indeed, via stabilization of HIF-2α, haplodeficiency of the PHD2 oxygen sensor promotes phalanx differentiation, thereby tightening the EC barrier and reducing tumor cell intravasation (67). Accordingly, endothelial HIF-1α increased leakage and tumor cell intravasation and extravasation, while HIF-2α had opposite effects (68).
Pathological angiogenesis: distinct from vascular development?
It has been postulated that the molecular players and vascular branching model in pathological angiogenesis are parallel to developmental angiogenesis, but have dysregulated expression. However, some molecules have different functions during physiological and pathological angiogenesis. For example, VEGFR1 and its ligands, PlGF and VEGF-B, are dispensable for development, yet they regulate angiogenesis in disease (69, 70). In developmental angiogenesis, VEGFR1 has a negative role by trapping VEGF (71), but this model does not explain its disease-restricted proangiogenic activity (72, 73). Stromal cell PlGF production, induced by contact with tumor cells, not only promotes angiogenesis in the leukemic bone marrow or medulloblastoma, but also stimulates tumor cell proliferation via Nrp1 signaling (74, 75). Although it is superfluous for vascular development, VEGF-B promotes contextual enlargement of myocardial capillaries (76) or growth of coronary vessels (77). Another example is ataxia teleangectasia mutated (ATM), which only regulates angiogenesis in disease, not in health (78). These examples (and others) suggest that part of the molecular basis of pathological angiogenesis is different from that in vascular development. Moreover, insights obtained from developmental angiogenesis models may not completely recapitulate the mechanisms that drive human pathological angiogenesis. To further our understanding of antiangiogenic medicines, it is therefore essential that sufficient feedback about the mechanism of pathological angiogenesis is provided by bedside-to-bench research.
Successes and challenges of antiangiogenic drugs
The field has focused on developing VEGF and VEGFR inhibitors (VEGFIs and VEGFRIs, respectively) (1, 79). While low autocrine VEGF signaling maintains quiescent vessel integrity, increased VEGF/VEGFR2 signaling induces angiogenesis, thereby creating a therapeutic window for antiangiogenic therapy. Current VEGFI/VEGFRI-based therapies prolong PFS and/or overall survival (OS) in a fraction of cancer patients (ref. 1, Table 1, and Supplemental Table 1; available online with this article; doi: 10.1172/JCI70212DS1). Bevacizumab also shows efficacy in the neoadjuvant setting in breast cancer (80).
Table 1.
Clinical results obtained for anti-VEGF agents
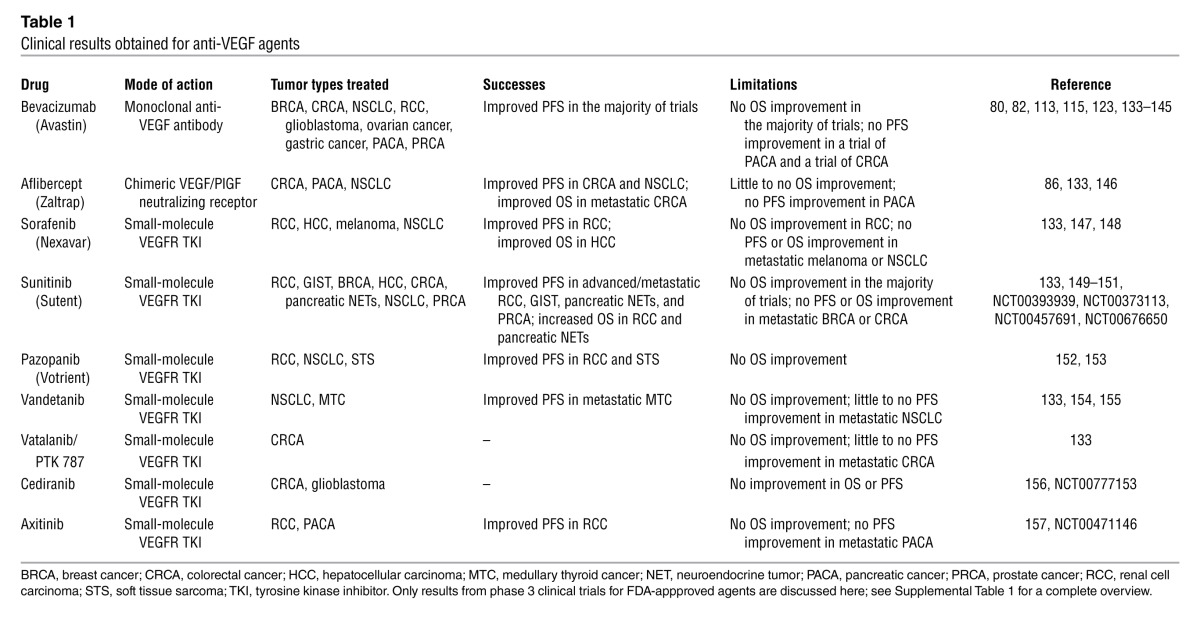
Despite the success of antiangiogenic drugs, several questions warrant further research to improve anticancer treatment. First, some cancers are resistant; even in responsive patients, antiangiogenic drugs generally prolong survival only in the order of months. The FDA revoked the approval of bevacizumab for metastatic breast cancer (81). In general, clinical efficacy is lower than that observed in preclinical cancer models (79). These models often represent rapidly growing ectopic tumors that do not reflect the heterogeneous human cancers developing over years in situ. Even transgenic models do not fully reflect the multistep carcinogenesis that occurs in humans. Another concern is that the majority of preclinical studies were undertaken in the neoadjuvant setting, which is a poor model for human metastatic cancer (79). Moreover, many drug combinations that proved ineffective were not studied preclinically (82).
One mechanism underlying resistance is that tumors produce multiple proangiogenic molecules in addition to VEGF, including PlGF, FGFs, interleukin-8, and others. Tumor ECs engineered to overexpress DLL4 develop enlarged mature vessels that are resistant against VEGF blockade, while inhibition of Notch signaling restores the sensitivity to antiangiogenic drugs in a xenograft model (83). Resistance can result from activation of FGF2-FGFR and EphB4–Ephrin-B2 pathways or from decreased levels of VEGFR2 (83). Several proangiogenic molecules become upregulated under selective pressure by VEGFIs/VEGFRIs (79, 84). PlGF and FGF2 plasma levels increased prior to progression of colorectal cancer patients treated with bevacizumab and chemotherapy (85). A phase III trial reported the efficacy of aflibercept, which blocks VEGF and PlGF, in patients who progressed on bevacizumab therapy (86).
Cancers also switch between different modes of vascularization, further explaining the resistance to VEGF blockade (Figure 4). Besides sprouting angiogenesis, they use vessel cooption (by growing around preexisting vessels), vascular mimicry (replacement of ECs by tumor cells), and vasculogenesis (vessel growth from bone marrow–derived progenitor cells), although the clinical relevance of these mechanisms remains unclear (87–89). For instance, metastases of melanoma and lung cancer grow in an angiogenesis-independent manner around existing vessels or switch to vessel cooption upon treatment with bevacizumab (90). Furthermore, cancer stem cell–like cells differentiate to ECs that exhibit reduced sensitivity to VEGF blockade (88, 91). Skin cancer stem cells in vascular niches express Nrp1 and produce VEGF, which might promote stemness independently of VEGF’s activity (92). Resistance in certain cancers is associated with pericyte-covered vessels, while tortuous uncovered vessels are observed in primary resistance (93).
Figure 4. Tumor vascularization modes.

After development, the vasculature rarely extends, but does so in tumor formation. Tumor vascularization occurs via a number of potential mechanisms. While angiogenesis is the most investigated, and the focus of this Review, other mechanisms have been observed. Endothelial progenitor cells (EPCs), which can either reside in the vascular wall or migrate from bone marrow in response to chemoattractants from the tumor cell, can differentiate into ECs and contribute to vessel formation. Vascular mimicry can also occur, whereby tumor cells can act as replacement cells for ECs. Another possibility is that chromosomal abnormalities in putative cancer stems cells allows tumor cells to differentiate into ECs. Other mechanisms by which tumor cells can obtain a blood flow include vessel cooption, whereby the tumor cell arises near to (or migrates toward) a preexisting blood vessel, or the process of intussusception, whereby a preformed vessel splits into two daughter vessels by the insertion of a tissue pillar.
CD11b+Gr1+ myeloid-derived suppressor cells (MDSCs) confer resistance to initially sensitive tumors (94). G-CSF plays a role in mobilization of MDSCs from the bone marrow (94). Strategies aimed at reducing mobilization and/or tumor infiltration of MDSCs might help to reduce resistance. Antiangiogenic drugs induce a systemic proinflammatory and proangiogenic burst in tumor-bearing healthy mice by upregulating PlGF, G-CSF, and osteopontin (95), which induce mobilization of resistance-conferring MDSCs (96, 97). The tumor microenvironment can also cause refractoriness. For instance, pancreatic adenocarcinomas have high interstitial fluid pressure due to abundant deposition of hyaluronic acid, which impairs perfusion and drug distribution (98). Notably, disaggregation of hyaluronic acid by enzymatic treatment improved perfusion (98).
Another hypothesis to explain the lower than expected efficacy of VEGF-targeted antiangiogenic drugs is that these treatments increase, rather than reduce, tumor malignancy. Indeed, certain preclinical studies show enhanced metastasis in tumor-bearing mice treated with VEGF-blocking drugs, such as sunitinib (79, 84, 99, 100). However, these findings remain debated because other preclinical studies did not detect increased metastasis (101, 102), and large meta-analyses have not shown more metastatic dissemination in patients (79, 103). Strategies combining antiangiogenesis with inhibition of metastasis might be useful to increase therapeutic efficacy. For instance, VEGF suppresses HGF-dependent c-MET phosphorylation and tumor cell migration, which explains why VEGF blockade promoted invasiveness (104) and combined VEGF/c-MET inhibition reduced metastasis (105, 106). c-MET is upregulated in bevacizumab-resistant glioblastomas (107). VEGF inhibitors also cause class-specific side effects (thromboembolic events, hypertension, gastrointestinal perforation, impaired wound healing, renal toxicity, and congestive heart failure) by depriving quiescent ECs of VEGF’s prosurvival effect (108).
Mode of action and schedule of application
The precise mode of action of antiangiogenic drugs in cancer patients remains incompletely understood. Antiangiogenic drugs can block angiogenesis, inhibit recruitment of proangiogenic bone marrow–derived cells (5, 109), induce vessel regression, and promote sensitization to radio- and chemotherapy by depriving ECs of VEGF’s prosurvival effect (5, 109). Antiangiogenic drugs also inhibit VEGFR-expressing tumor cells (5, 109) or deprive cancer stem cells from EC-derived angiocrine signals (110). Moreover, VEGF inhibitors reduce the number and self-renewal capability of cancer stem cells (92). Alternatively, antiangiogenic drugs block excess VEGF levels secreted by tumor cells. This uncoordinated secretion of VEGF induces a chaotic proangiogenic response characterized by hyperbranching and lack of maturation, which renders tumor vessels dysfunctional. Consequently, delivery of chemo- and radiotherapy is impaired. Anti-VEGF normalizes tumor vessels and restores delivery of radio- and/or chemotherapy in tumors, although this subject remains debated (4, 45, 111). Nonetheless, vessel normalization can explain why bevacizumab works better when combined with chemotherapy. The challenge in the clinic is to identify agents that cause permanent tumor vessel normalization.
Another question is the optimal duration and dose of antiangiogenic drug application. Certain preclinical studies indicate rapid revascularization after cessation of treatment (112); the clinical relevance of this observation requires future study. Adjuvant application of bevacizumab for 12 months had a beneficial effect on PFS of early-stage colorectal cancer patients when analyzed after 15 months, but this effect was lost after 36 months (113); additionally, the outcome of patients with metastatic colorectal cancer was better when bevacizumab was given beyond progression (114). A phase III trial in colon cancer showed a modest OS advantage for patients treated with bevacizumab beyond progression (115). Bevacizumab beyond progression is currently being investigated in other cancer types.
Contextual mode of action
The efficacy of antiangiogenic therapies differs among cancer types. Antiangiogenic drugs are more efficient in well-vascularized cancers (e.g., clear cell renal cancer), in which bevacizumab is effective without chemotherapy (116, 117). In contrast, antiangiogenic agents are less effective in less vascularized cancers (e.g., pancreatic adenocarcinoma and gastric cancer) (118, 119). Nonetheless, pharmacological VEGF blockers can have dose- and drug class–dependent effects (120). Indeed, in mouse models of metastasis, an anti-VEGF antibody did not promote metastasis, in contrast to small-molecule receptor tyrosine kinase inhibitors. One of those, sunitinib, enhanced metastasis and lung permeability and promoted tumor cell extravasation by inhibiting tyrosine phosphorylation of proteins, important for EC-EC junctions (120). Another concern is the context-dependent efficacy of antiangiogenic agents in micro- versus macrometastatic disease. Two phase III studies failed to show benefit of bevacizumab combined with chemotherapy in early-stage (stage II and III) colorectal cancer in the adjuvant setting (113, 121). Similarly, in breast and ovarian cancer, bevacizumab lacks efficacy in the adjuvant setting (122, 123). The reasons remain elusive, but may be due to rebound angiogenesis, although this has not been observed when using tyrosine kinase inhibitors (124). Another possibility is that micrometastases grow in an angiogenesis-independent fashion and might survive antiangiogenic treatment in a dormant state, or use other vascularization principles, such as vessel cooption, which is less sensitive to VEGF inhibition (125). In vivo imaging documented that anti-VEGF treatment prolonged the dormancy of micrometastatic tumor cell aggregates by blocking angiogenesis (90). Alternatively, vessels in micrometastases might be less abnormal, because proangiogenic factors are less abundant and the chemosensitizing effect of bevacizumab would therefore be less important. Given that more than 20,000 cancer patients are being enrolled in trials testing anti-VEGF therapy in the adjuvant setting, understanding the modes and mechanisms of vascularization of micrometastatic disease is a center-stage priority.
Conclusion and perspectives
Research in angiogenesis continues to yield new molecular insights about vessel branching. Emerging avenues include studying the role of metabolism in ECs (126), the molecular determinants of cellular shape and size in patterning connectivity, the role of pericytes in neurovascular disorders, the mechanistic basis of EC polarity, the different modes of tumor vascularization, and the genetic basis of tumor vessel normalization. Another intriguing area of research is how ECs feedback on cancer (stem) cells by providing angiocrine signals (110, 127). Still, much needs to be clarified about the context-dependent activity of many angiogenic factors. The challenge is to translate this knowledge into improving therapy (Figure 2). Numerous areas of clinical research are of high priority, including the optimization of drug regimes, the use of predictive biomarkers to identify putative responders versus nonresponders (as illustrated by a VEGFR1 genetic locus, ref. 128; and short VEGF isoform, ref. 129), the development of antiangiogenic treatment of pediatric tumors (such as anti-PlGF therapy of medulloblastoma, ref. 75), the development of vessel normalization drugs, and the development of VEGF-independent antiangiogenic drugs that can be used in combination with existing antiangiogenic therapies. The development of allosteric antiangiogenic inhibitors, which offer a superior advantage of safety, specificity, and efficacy over current orthosteric antiangiogenic antagonists, is also commendable (130, 131). Finally, more bedside-to-bench studies are needed to provide the necessary feedback needed to further improve the overall efficiency of antiangiogenesis therapy.
Supplementary Material
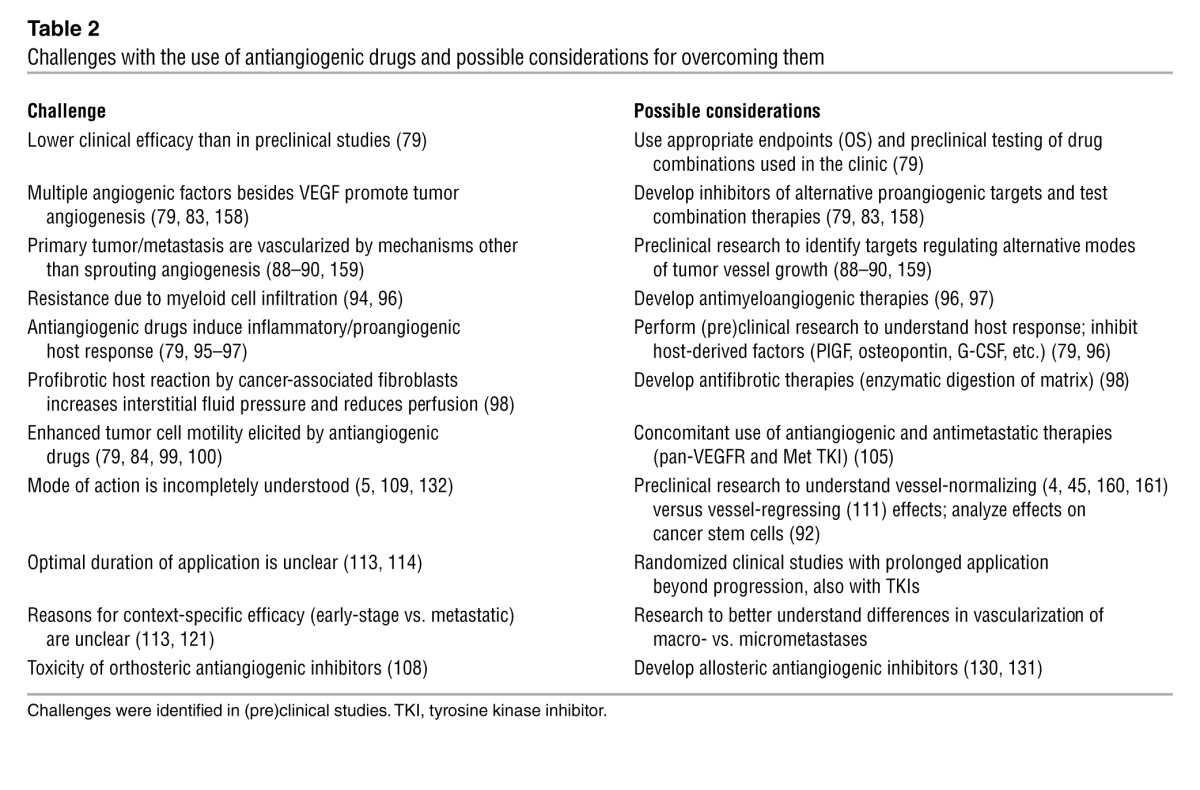
Table 2.
Challenges with the use of antiangiogenic drugs and possible considerations for overcoming them
Acknowledgments
The authors thank K. Alitalo, M. Mazzone, R. Benedito, and H. Gerhardt for constructive discussions. J. Welti is funded by EU-Marie Curie PIEF Funding. S. Loges is funded by the Max-Eder group leader program of the Deutsche Krebshilfe, by the Deutsche Forschungsgemeinschaft (DFG), by the Hamburg Excellence Initiative (LEXI), by the Hamburger Krebsgesellschaft, and by the Roggenbuck Stiftung. The work of P. Carmeliet is funded by long-term structural funding — Methusalem Funding by the Flemish Government, by Leducq Transatlantic Network, and by Belgian Science Policy IAP7. S. Dimmeler is supported by the DFG (SFB834 and Exc147-1) and an ERC advanced grant “Angiomir.” We apologize to those authors whose work could not be cited due to space and citation restrictions.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(8):3190–3200. doi:10.1172/JCI70212.
References
- 1.Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- 2.Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol. 2010;22(5):617–625. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Saharinen P, Eklund L, Pulkki K, Bono P, Alitalo K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol Med. 2011;17(7):347–362. doi: 10.1016/j.molmed.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 5.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Nakayama M, et al. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15(3):249–260. doi: 10.1038/ncb2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fantin A, et al. NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood. 2013;121(12):2352–2362. doi: 10.1182/blood-2012-05-424713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fantin A, Schwarz Q, Davidson K, Normando EM, Denti L, Ruhrberg C. The cytoplasmic domain of neuropilin 1 is dispensable for angiogenesis, but promotes the spatial separation of retinal arteries and veins. Development. 2011;138(19):4185–4191. doi: 10.1242/dev.070037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geudens I, Gerhardt H. Coordinating cell behaviour during blood vessel formation. Development. 2011;138(21):4569–4583. doi: 10.1242/dev.062323. [DOI] [PubMed] [Google Scholar]
- 10.Krueger J, et al. Flt1 acts as a negative regulator of tip cell formation and branching morphogenesis in the zebrafish embryo. Development. 2011;138(10):2111–2120. doi: 10.1242/dev.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakobsson L, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12(10):943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 12.Benedito R, et al. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484(7392):110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 13.Thurston G, Noguera-Troise I, Yancopoulos GD. The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7(5):327–331. doi: 10.1038/nrc2130. [DOI] [PubMed] [Google Scholar]
- 14.Yan M, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010;463(7282):E6–E7. doi: 10.1038/nature08751. [DOI] [PubMed] [Google Scholar]
- 15.Tammela T, et al. Blocking VEGFR-3 suppresses angiogenic sprouting vascular network formation. Nature. 2008;454(7204):656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 16.Tammela T, et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol. 2011;13(10):1202–1213. doi: 10.1038/ncb2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tvorogov D, et al. Effective suppression of vascular network formation by combination of antibodies blocking VEGFR ligand binding receptor dimerization. Cancer Cell. 2010;18(6):630–640. doi: 10.1016/j.ccr.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319(9):1271–1280. doi: 10.1016/j.yexcr.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Oubaha M, et al. Formation of a PKCzeta/beta-catenin complex in endothelial cells promotes angiopoietin-1-induced collective directional migration angiogenic sprouting. Blood. 2012;120(16):3371–3381. doi: 10.1182/blood-2012-03-419721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh GY. Orchestral actions of angiopoietin-1 in vascular regeneration. Trends Mol Med. 2013;19(1):31–39. doi: 10.1016/j.molmed.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Gerald D, Chintharlapalli S, Augustin HG, Benjamin LE. Angiopoietin-2: an attractive target for improved antiangiogenic tumor therapy. Cancer Res. 2013;73(6):1649–1657. doi: 10.1158/0008-5472.CAN-12-4697. [DOI] [PubMed] [Google Scholar]
- 22.Daly C, et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013;73(1):108–118. doi: 10.1158/0008-5472.CAN-12-2064. [DOI] [PubMed] [Google Scholar]
- 23.Felcht M, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 integrin signaling. J Clin Invest. 2012;122(6):1991–2005. doi: 10.1172/JCI58832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlan BY, et al. Randomized, double-blind, placebo-controlled phase II study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian cancer. J Clin Oncol. 2012;30(4):362–371. doi: 10.1200/JCO.2010.34.3178. [DOI] [PubMed] [Google Scholar]
- 25.Koh YJ, et al. Double antiangiogenic protein DAAP, vascular leakage. Cancer Cell. 2010;18(2):171–184. doi: 10.1016/j.ccr.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. 2013;328(1):18–26. doi: 10.1016/j.canlet.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Segarra M, et al. Semaphorin 6A regulates angiogenesis by modulating VEGF signaling. Blood. 2012;120(19):4104–4115. doi: 10.1182/blood-2012-02-410076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Oh WJ, Gaiano N, Yoshida Y, Gu C. Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011;25(13):1399–1411. doi: 10.1101/gad.2042011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.London NR, Li DY. Robo4-dependent Slit signaling stabilizes the vasculature during pathologic angiogenesis and cytokine storm. Curr Opin Hematol. 2011;18(3):186–190. doi: 10.1097/MOH.0b013e328345a4b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones CA, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis endothelial hyperpermeability. Nat Med. 2008;14(4):448–453. doi: 10.1038/nm1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koch AW, et al. Robo4 maintains vessel integrity inhibits angiogenesis by interacting with UNC5B. Dev Cell. 2011;20(1):33–46. doi: 10.1016/j.devcel.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Testori J, et al. The VEGF-regulated transcription factor HLX controls the expression of guidance cues negatively regulates sprouting of endothelial cells. Blood. 2011;117(9):2735–2744. doi: 10.1182/blood-2010-07-293209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herbert SP, Cheung JY, Stainier DY. Determination of endothelial stalk versus tip cell potential during angiogenesis by H2.0-like homeobox-1. Curr Biol. 2012;22(19):1789–1794. doi: 10.1016/j.cub.2012.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pitulescu ME, Adams RH. Eph/ephrin molecules — a hub for signaling and endocytosis. Genes Dev. 2010;24(22):2480–2492. doi: 10.1101/gad.1973910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salvucci O, Tosato G. Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis. Adv Cancer Res. 2012;114:21–57. doi: 10.1016/B978-0-12-386503-8.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bochenek ML, Dickinson S, Astin JW, Adams RH, Nobes CD. Ephrin-B2 regulates endothelial cell morphology motility independently of Eph-receptor binding. J Cell Sci. 2010;123(pt 8):1235–1246. doi: 10.1242/jcs.061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawamiphak S, et al. Ephrin-B2 regulates VEGFR2 function in developmental tumour angiogenesis. Nature. 2010;465(7297):487–491. doi: 10.1038/nature08995. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, et al. Ephrin-B2 controls VEGF-induced angiogenesis lymphangiogenesis. Nature. 2010;465(7297):483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 39.Abéngozar MA, et al. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood. 2012;119(19):4565–4576. doi: 10.1182/blood-2011-09-380006. [DOI] [PubMed] [Google Scholar]
- 40.Herwig L, et al. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr Biol. 2011;21(22):1942–1948. doi: 10.1016/j.cub.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 41.Fantin A, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116(5):829–840. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeda Y, et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479(7371):122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33(3):119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rolny C, et al. HRG inhibits tumor growth metastasis by inducing macrophage polarization vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Mazzieri R, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–526. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 47.De Palma M, Naldini L. Angiopoietin-2 TIEs up macrophages in tumor angiogenesis. Clin Cancer Res. 2011;17(16):5226–5232. doi: 10.1158/1078-0432.CCR-10-0171. [DOI] [PubMed] [Google Scholar]
- 48.Phng LK, et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell. 2009;16(1):70–82. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guarani V, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473(7346):234–238. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storkebaum E, Quaegebeur A, Vikkula M, Carmeliet P. Cerebrovascular disorders: molecular insights therapeutic opportunities. Nat Neurosci. 2011;14(11):1390–1397. doi: 10.1038/nn.2947. [DOI] [PubMed] [Google Scholar]
- 51.Moya IM, et al. Stalk cell phenotype depends on integration of notch smad1/5 signaling cascades. Dev Cell. 2012;22(3):501–514. doi: 10.1016/j.devcel.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Larrivée B, et al. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell. 2012;22(3):489–500. doi: 10.1016/j.devcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu-Lowe DD, et al. Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res. 2011;71(4):1362–1373. doi: 10.1158/0008-5472.CAN-10-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron. 2011;71(3):406–424. doi: 10.1016/j.neuron.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 55.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murakami M. Signaling required for blood vessel maintenance: molecular basis and pathological manifestations. Int J Vasc Med. 2012;2012:293641. doi: 10.1155/2012/293641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saharinen P, Alitalo K. The yin, the yang, and the angiopoietin-1. J Clin Invest. 2011;121(6):2157–2159. doi: 10.1172/JCI58196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeansson M, et al. Angiopoietin-1 is essential in mouse vasculature during development in response to injury. J Clin Invest. 2011;121(6):2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGuire PG, Rangasamy S, Maestas J, Das A. Pericyte-derived sphingosine 1-phosphate induces the expression of adhesion proteins and modulates the retinal endothelial cell barrier. Arterioscler Thromb Vasc Biol. 2011;31(12):e107–e115. doi: 10.1161/ATVBAHA.111.235408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ben Shoham A, et al. S1P1 inhibits sprouting angiogenesis during vascular development. Development. 2012;139(20):3859–3869. doi: 10.1242/dev.078550. [DOI] [PubMed] [Google Scholar]
- 61.Gaengel K, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin VEGFR2. Dev Cell. 2012;23(3):587–599. doi: 10.1016/j.devcel.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 62.Cooke VG, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition metastasis mediated by met signaling pathway. Cancer Cell. 2012;21(1):66–81. doi: 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eichmann A, Simons M. VEGF signaling inside vascular endothelial cells and beyond. Curr Opin Cell Biol. 2012;24(2):188–193. doi: 10.1016/j.ceb.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, et al. Angiopoietin-1/Tie2 signal augments basal Notch signal controlling vascular quiescence by inducing delta-like 4 expression through AKT-mediated activation of β-catenin. J Biol Chem. 2011;286(10):8055–8066. doi: 10.1074/jbc.M110.192641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kitajewski J. Endothelial laminins underlie the tip cell microenvironment. EMBO Rep. 2011;12(11):1087–1088. doi: 10.1038/embor.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stenzel D, et al. Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 2011;12(11):1135–1143. doi: 10.1038/embor.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mazzone M, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Branco-Price C, et al. Endothelial cell HIF-1alpha HIF-2alpha differentially regulate metastatic success. Cancer Cell. 2012;21(1):52–65. doi: 10.1016/j.ccr.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 its ligands VEGFB PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8(12):942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 70.Dewerchin M, Carmeliet P. PlGF, a multi-tasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med. 2012;2(8):a011056. doi: 10.1101/cshperspect.a011056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chappell JC, Taylor SM, Ferrara N, Bautch VL. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell. 2009;17(3):377–386. doi: 10.1016/j.devcel.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee HK, Chauhan SK, Kay E, Dana R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7-dependent pathway. Blood. 2011;117(21):5762–5771. doi: 10.1182/blood-2010-09-306928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boscolo E, Mulliken JB, Bischoff J. VEGFR-1 mediates endothelial differentiation and formation of blood vessels in a murine model of infantile hemangioma. Am J Pathol. 2011;179(5):2266–2277. doi: 10.1016/j.ajpath.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmidt T, et al. Loss or inhibition of stromal-derived PlGF prolongs survival of mice with imatinib-resistant Bcr-Abl1(+) leukemia. Cancer Cell. 2011;19(6):740–753. doi: 10.1016/j.ccr.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 75.Snuderl M, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth spread of medulloblastoma. Cell. 2013;152(5):1065–1076. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Serpi R, et al. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation capillary dilatation in rats. Cardiovasc Res. 2011;89(1):204–213. doi: 10.1093/cvr/cvq267. [DOI] [PubMed] [Google Scholar]
- 77.Bry M, et al. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122(17):1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 78.Okuno Y, Nakamura-Ishizu A, Otsu K, Suda T, Kubota Y. Nat Med. Pathological neoangiogenesis depends on oxidative stress regulation by ATM [published online ahead of print July 15, 2012]. doi: 10.1038/nm.2846 . [DOI] [PubMed] [Google Scholar]
- 79.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.von Minckwitz G, et al. Neoadjuvant chemotherapy bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012;366(4):299–309. doi: 10.1056/NEJMoa1111065. [DOI] [PubMed] [Google Scholar]
- 81.Mackey JR, et al. Controlling angiogenesis in breast cancer: A systematic review of anti-angiogenic trials. Cancer Treat Rev. 2012;38(6):673–688. doi: 10.1016/j.ctrv.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 82.Tol J, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360(6):563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 83.Li JL, et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res. 2011;71(18):6073–6083. doi: 10.1158/0008-5472.CAN-11-1704. [DOI] [PubMed] [Google Scholar]
- 84.Loges S, Mazzone M, Hohensinner P, Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15(3):167–170. doi: 10.1016/j.ccr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 85.Kopetz S, et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol. 2010;28(3):453–459. doi: 10.1200/JCO.2009.24.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Cutsem E, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30(28):3499–3506. doi: 10.1200/JCO.2012.42.8201. [DOI] [PubMed] [Google Scholar]
- 87.Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18(10):2726–2732. doi: 10.1158/1078-0432.CCR-11-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang R, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468(7325):829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 89.Ricci-Vitiani L, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468(7325):824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 90.Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16(1):116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 91.Soda Y, et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011;108(11):4274–4280. doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beck B, et al. A vascular niche a VEGF-Nrp1 loop regulate the initiation stemness of skin tumours. Nature. 2011;478(7369):399–403. doi: 10.1038/nature10525. [DOI] [PubMed] [Google Scholar]
- 93.Cascone T, et al. Upregulated stromal EGFR vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest. 2011;121(4):1313–1328. doi: 10.1172/JCI42405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 95.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent correlate with antitumor efficacy. Proc Natl Acad Sci U S A. 2007;104(43):17069–17074. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320(2):130–137. doi: 10.1016/j.canlet.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 97.Shojaei F, et al. G-CSF-initiated myeloid cell mobilization angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pàez-Ribes M, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Padera TP, et al. Differential response of primary tumor versus lymphatic metastasis to VEGFR-2 VEGFR-3 kinase inhibitors cediranib vandetanib. Mol Cancer Ther. 2008;7(8):2272–2279. doi: 10.1158/1535-7163.MCT-08-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Singh M, et al. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J Pathol. 2012;227(4):417–430. doi: 10.1002/path.4053. [DOI] [PubMed] [Google Scholar]
- 103.Miles D, et al. Disease course patterns after discontinuation of bevacizumab: pooled analysis of randomized phase III trials. J Clin Oncol. 2011;29(1):83–88. doi: 10.1200/JCO.2010.30.2794. [DOI] [PubMed] [Google Scholar]
- 104.Lu KV, et al. VEGF inhibits tumor cell invasion mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.You WK, et al. VEGF c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71(14):4758–4768. doi: 10.1158/0008-5472.CAN-10-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sennino B, et al. Suppression of tumor invasion metastasis by concurrent inhibition of c-Met VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2(3):270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jahangiri A, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of anti-angiogenic therapy resistance. Clin Cancer Res. 2013;19(7):1773–1783. doi: 10.1158/1078-0432.CCR-12-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hayman SR, Leung N, Grande JP, Garovic VD. VEGF inhibition, hypertension, and renal toxicity. Curr Oncol Rep. 2012;14(4):285–294. doi: 10.1007/s11912-012-0242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 110.Lu J, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 2013;23(2):171–185. doi: 10.1016/j.ccr.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van der Veldt AA, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21(1):82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 112.Mancuso MR, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116(10):2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Allegra CJ, et al. Phase III trial assessing bevacizumab in stages II III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol. 2011;29(1):11–16. doi: 10.1200/JCO.2010.30.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grothey A, et al. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol. 2008;26(33):5326–5334. doi: 10.1200/JCO.2008.16.3212. [DOI] [PubMed] [Google Scholar]
- 115.Bennouna J, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14(1):29–37. doi: 10.1016/S1470-2045(12)70477-1. [DOI] [PubMed] [Google Scholar]
- 116.Yang JC, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349(5):427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Escudier B, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 118.Kindler HL. Pancreatic cancer: an update. Curr Oncol Rep. 2007;9(3):170–176. doi: 10.1007/s11912-007-0018-z. [DOI] [PubMed] [Google Scholar]
- 119.Ohtsu A, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol. 2011;29(30):3968–3976. doi: 10.1200/JCO.2011.36.2236. [DOI] [PubMed] [Google Scholar]
- 120.Chung AS, et al. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J Pathol. 2012;227(4):404–416. doi: 10.1002/path.4052. [DOI] [PubMed] [Google Scholar]
- 121.Rousseau B, et al. Stage II stage III colon cancer: treatment advances future directions. Cancer J. 2010;16(3):202–209. doi: 10.1097/PPO.0b013e3181ddc5bf. [DOI] [PubMed] [Google Scholar]
- 122.Cameron D, et al. Primary results of BEATRICE, a randomized phase III trial evaluating adjuvant bevacizumab-containing therapy in triple-negative breast cancer. Cancer Res. 2012;72(24):supplement 3. [Google Scholar]
- 123.Perren TJ, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 124.di Tomaso E, et al. Glioblastoma recurrence after cediranib therapy in patients: lack of “rebound” revascularization as mode of escape. Cancer Res. 2011;71(1):19–28. doi: 10.1158/0008-5472.CAN-10-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim ES, et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc Natl Acad Sci U S A. 2002;99(17):11399–11404. doi: 10.1073/pnas.172398399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.De Bock K, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. doi: 10.1016/j.cell.2013.06.037. In press. [DOI] [PubMed] [Google Scholar]
- 127.Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. 2010;10(2):138–146. doi: 10.1038/nrc2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lambrechts D, et al. VEGF pathway genetic variants as biomarkers of treatment outcome with bevacizumab: an analysis of data from the AViTA AVOREN randomised trials. Lancet Oncol. 2012;13(7):724–733. doi: 10.1016/S1470-2045(12)70231-0. [DOI] [PubMed] [Google Scholar]
- 129.Lambrechts D, Lenz HJ, de Haas S, Carmeliet P, Scherer SJ. Markers of response for the antiangiogenic agent bevacizumab. J Clin Oncol. 2013;31(9):1219–1230. doi: 10.1200/JCO.2012.46.2762. [DOI] [PubMed] [Google Scholar]
- 130.Bono F, et al. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell. 2013;23(4):477–488. doi: 10.1016/j.ccr.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 131.Herbert C, et al. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell. 2013;23(4):489–501. doi: 10.1016/j.ccr.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 132.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. 2011;473(7347):298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9(7):378–390. doi: 10.1038/nrclinonc.2012.64. [DOI] [PubMed] [Google Scholar]
- 134.Miller K, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 135.Miles DW, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010;28(20):3239–3247. doi: 10.1200/JCO.2008.21.6457. [DOI] [PubMed] [Google Scholar]
- 136.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 137.Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med. 2012;2(10):a006577. doi: 10.1101/cshperspect.a006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saltz L, et al. Phase III trial of cetuximab, bevacizumab, and 5-fluorouracil/leucovorin vs. FOLFOX-bevacizumab in colorectal cancer. Clin Colorectal Cancer. 2012;11(2):101–111. doi: 10.1016/j.clcc.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 139.Reck M, et al. Overall survival with cisplatin–gemcitabine bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL). Ann Oncol. 2010;21(9):1804–1809. doi: 10.1093/annonc/mdq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Herbst RS, et al. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet. 2011;377(9780):1846–1854. doi: 10.1016/S0140-6736(11)60545-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rini BI, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28(13):2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Friedman HS, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 143.Burger RA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 144.Aghajanian C, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kindler HL, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ramlau R, et al. Aflibercept and Docetaxel versus Docetaxel alone after platinum failure in patients with advanced or metastatic non-small-cell lung cancer: a randomized, controlled phase III trial. J Clin Oncol. 2012;30(29):3640–3647. doi: 10.1200/JCO.2012.42.6932. [DOI] [PubMed] [Google Scholar]
- 147.Escudier B, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27(20):3312–3318. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 148.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 149.Motzer RJ, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27(22):3584–3590. doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Goodman VL, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–1373. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 151.Raymond E, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 152.Sternberg CN, et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur J Cancer. 2013;49(6):1287–1296. doi: 10.1016/j.ejca.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 153.van der Graaf WT, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 154.Herbst RS, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11(7):619–626. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wells SA, Jr, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schmoll HJ, et al. Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first-line treatment for patients with advanced colorectal cancer: a double-blind, randomized phase III study (HORIZON III). J Clin Oncol. 2012;30(29):3588–3595. doi: 10.1200/JCO.2012.42.5355. [DOI] [PubMed] [Google Scholar]
- 157.Motzer RJ, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14(6):552–562. doi: 10.1016/S1470-2045(13)70093-7. [DOI] [PubMed] [Google Scholar]
- 158.Loges S, Schmidt T, Carmeliet P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer. 2010;1(1):12–25. doi: 10.1177/1947601909356574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bautch VL. Cancer: Tumour stem cells switch sides. Nature. 2010;468(7325):770–771. doi: 10.1038/468770a. [DOI] [PubMed] [Google Scholar]
- 160.Claes A, Wesseling P, Jeuken J, Maass C, Heerschap A, Leenders WP. Antiangiogenic compounds interfere with chemotherapy of brain tumors due to vessel normalization. Mol Cancer Ther. 2008;7(1):71–78. doi: 10.1158/1535-7163.MCT-07-0552. [DOI] [PubMed] [Google Scholar]
- 161.Goel S, Wong AH, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med. 2012;2(3):a006486. doi: 10.1101/cshperspect.a006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.